

Dissecting the Role of Autophagy-Related Proteins in Cancer Metabolism and Plasticity



Abstract
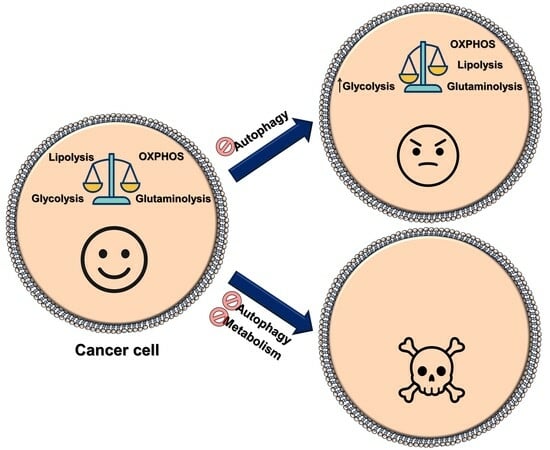
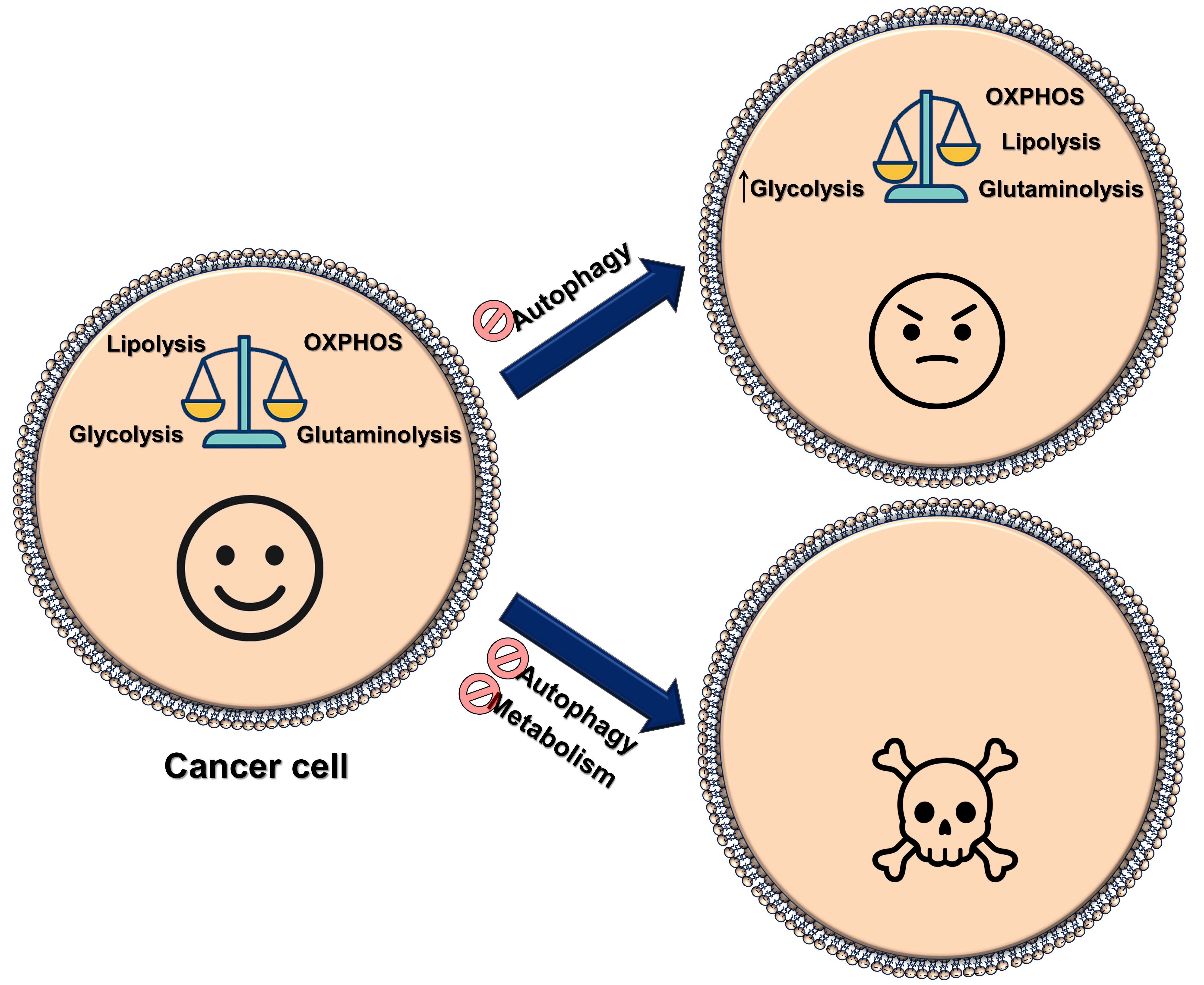
:
1. Introduction
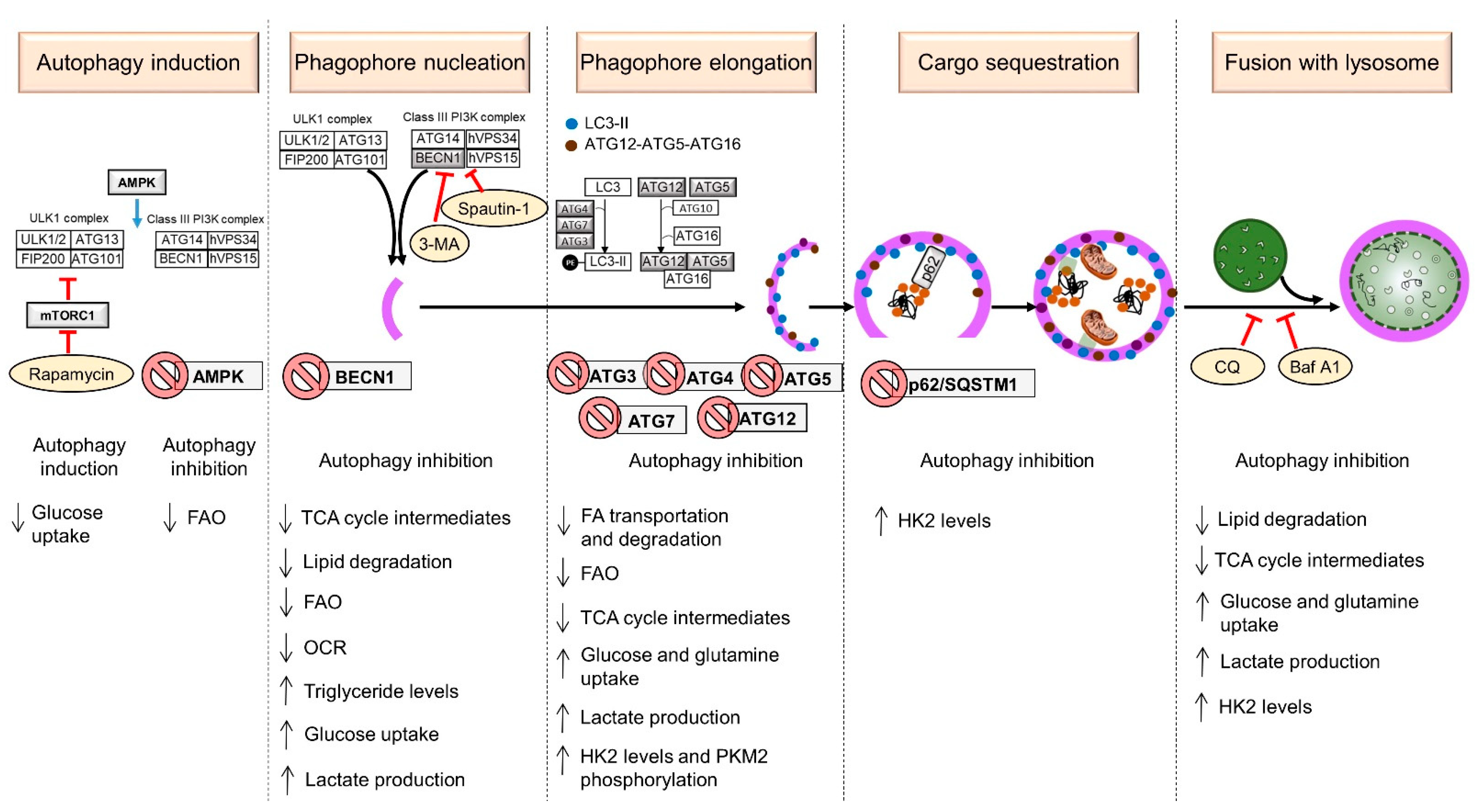
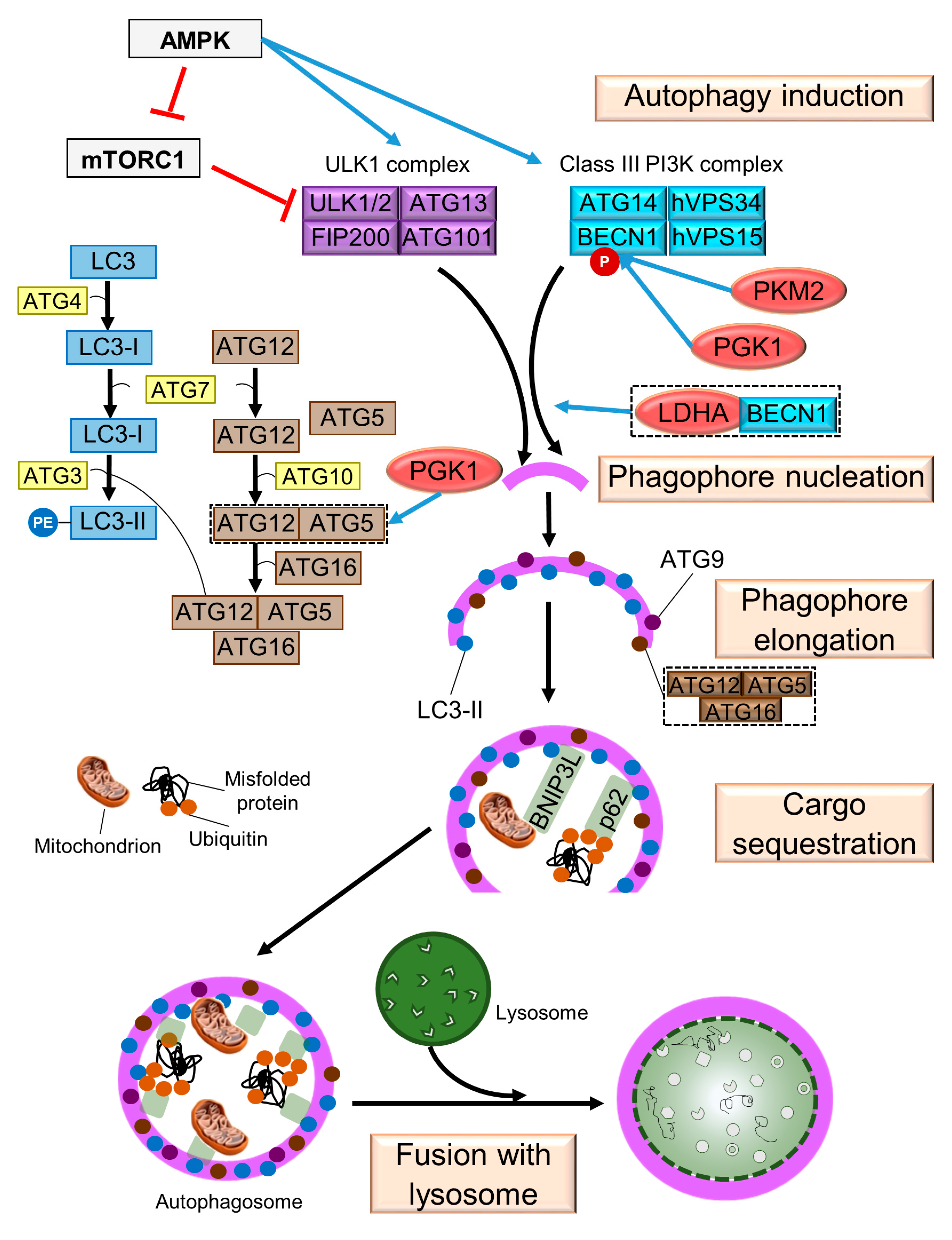
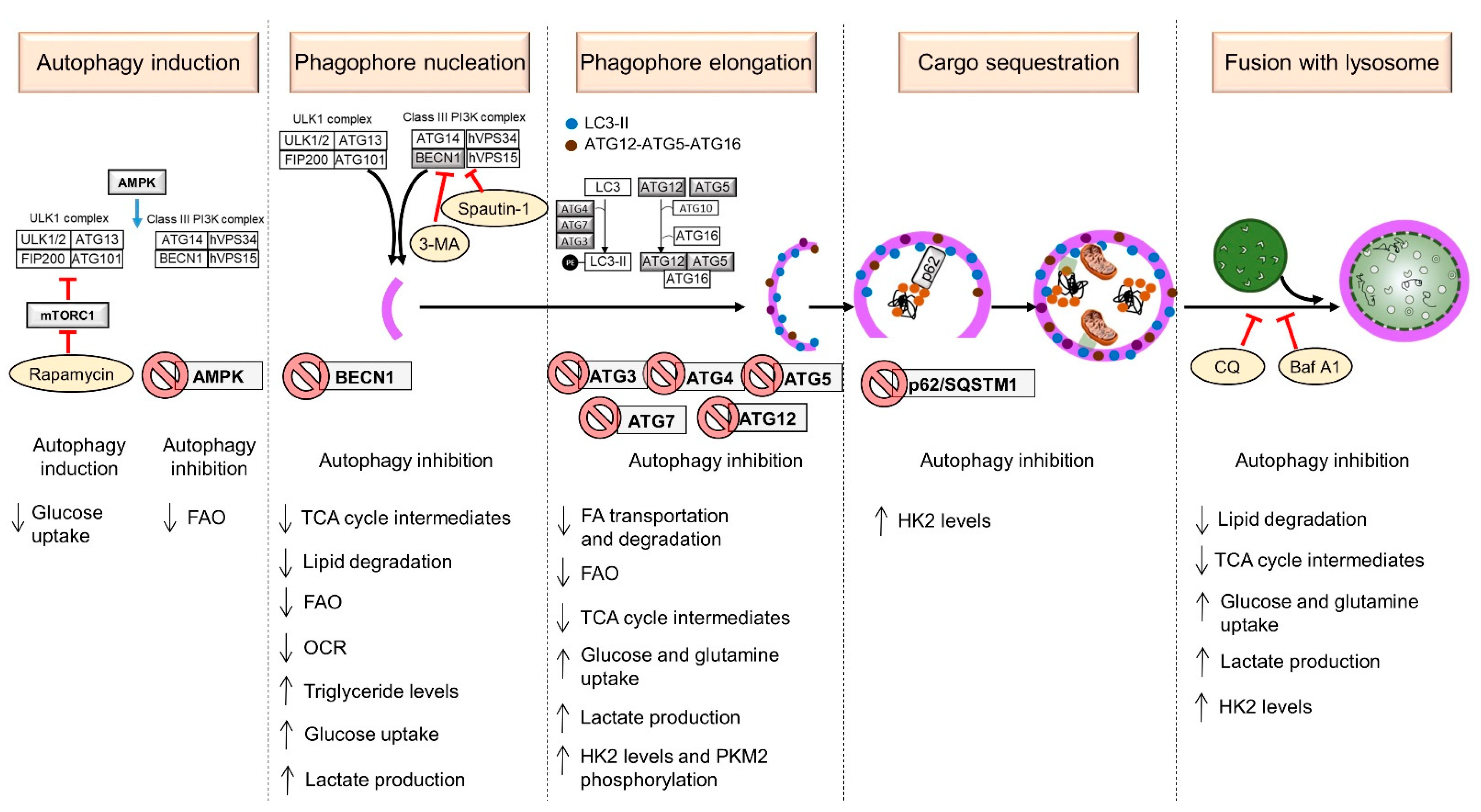
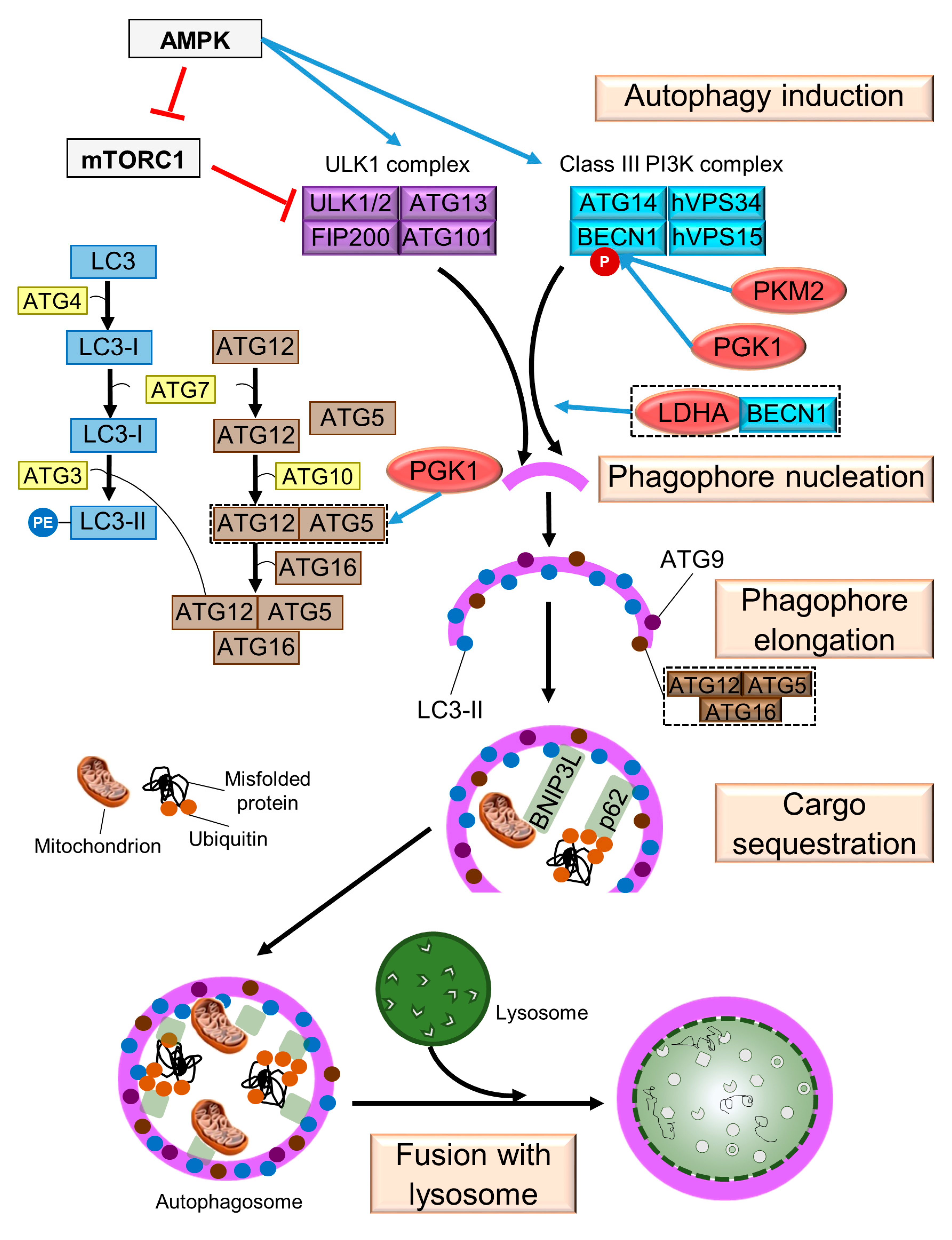
2. Mechanism of Autophagy in Mammals
2.1. Induction
2.2. Phagophore Nucleation
2.3. Phagophore Elongation
2.4. Cargo Sequestration
2.5. Fusion with Lysosome
3. Cancer Progression under the Control of ATGs
3.1. ATG Status in Different Types of Cancer Cells May Vary
3.2. ATG Regulation Is Related to Cell Metabolism
3.3. ATG Status in Cancer Microenvironment
4. Metabolic Changes Caused by Modulation of Autophagy in Cancer Cells
4.1. Metabolic Changes Caused by Autophagy Induction
4.2. Metabolic Changes Caused by Knockdown or Inhibition of ATGs
4.3. The Blockade of the Autophagic Flux Alters the Metabolism of Human Cancer Cells

{kind=link}
{kind=link}
{kind=link}
| Autophagy Step | Strategy | Model * | Effect on Metabolism | Reference |
|---|---|---|---|---|
| Autophagy induction | Rapamycin | U937 and NB4 cell lines (acute myeloid leukemia) | Decreased glucose uptake | [102] |
| (−) AMPKα1 | TAM-resistant MCF7 cell line (breast adenocarcinoma) | Decreased expression of proteins that promote FAO through estrogen receptor | [118] | |
| Initiation and phagophore nucleation | (−) BECN1 | DLD1 cell line (colon cancer) | Attenuated lipid degradation | [86] |
| (−) BECN1 | SGC C-7901 and MGC-803 cell lines (gastric cancer) | Increased glucose uptake and lactate secretion Reduced citrate and fumarase level Shift from OXPHOS to glycolysis | [87] | |
| (−) BECN1 | MOLM14 cell line (acute myeloid leukemia) | Attenuated lipid degradation Decreased basal OCR and ATP-linked OCR | [85] | |
| 3-MA | Huh7 cell line (hepatocellular carcinoma) | Decreased intracellular ATP and β-hydroxybutyrate levels Down-regulation of genes involved in FAO and fatty acid transportation | [109] | |
| 3-MA | MOLM14 and U937 cell lines (acute myeloid leukemia) | Attenuated lipid degradation Increased triglyceride levels Reduced FAO Decreased basal OCR and ATP-linked OCR | [85] | |
| Spautin-1 | A549 cell line (lung cancer) | Decreased HK2 levels and glucose uptake | [114] | |
| Spautin-1 | HT1080 cell line (fibrosarcoma) | Reduced OCR and suppressed mitochondrial complex I activity | [108] | |
| Phagophore elongation | (−) ATG3 | THP-1 and MV4-11 cell lines (acute myeloid leukemia) | Increased levels of fumarate and succinate Increased basal OCR and ATP-linked OCR Increased glucose uptake, glucose consumption and lactate production and decreased lactate excretion | [110] |
| Inactive ATG4 (ATG4BC74A) | Huh7 cell line (hepatocellular carcinoma) | Decreased intracellular ATP and β-hydroxybutyrate levels Down-regulation of genes involved in FAO and fatty acid transportation | [109] | |
| (−) ATG5 | SMMC7721 cell line (hepatocellular carcinoma) | Enhanced glucose consumption and lactate production Increased HK2 levels | [111] | |
| (−) ATG7 | MDA-MB-231 cell line (breast cancer) | Decreased LDH activity, decrease in glycolytic capacity | [115] | |
| (−) ATG7 | 8988 T cell line (pancreatic cancer) | Increased glutamine consumption Decreased intracellular glutamine levels under glutamine deprivation conditions Decreased TCA cycle intermediates | [113] | |
| (−) ATG7 | SMMC7721 cell line (hepatocellular carcinoma) | Increased glucose uptake and lactate production | [111] | |
| (−) ATG7 | HeLa cell line (cervical carcinoma) | Increased PKM2 phosphorylation Increased glucose consumption and lactate production | [80] | |
| (+) ATG7 | HeLa cell line (cervical carcinoma) | Reduced PKM2 phosphorylation Inhibition of the Warburg effect | [80] | |
| (−) ATG7 | K562 cell line (chronic myeloid leukemia) | Decreased glucose uptake and lactate production Increased extracellular glutamate from transamination of α-ketoglutarate | [116] | |
| (−) ATG12 | MOLM14 cell line (acute myeloid leukemia) | Reduced FAO | [85] | |
| Cargo sequestration | (−) p62/SQSTM1 | SMMC7721 cell line (hepatocellular carcinoma) | Increased HK2 levels | [111] |
| Fusion with lysosome | CQ | SW480 and DLD1 cell lines (colon cancer) | Attenuated lipid degradation | [86] |
| CQ | 8988 T and MIAPaCa2 cell lines (pancreatic cancer) | Increased glutamine consumption Decreased intracellular glutamine levels under glutamine deprivation conditions Decreased TCA cycle intermediates | [113] | |
| CQ | QBC939 cell line (cholangiocarcinoma) | Decreased glucose-6-phosphate dehydrogenase activity | [122] | |
| CQ | Primary chronic myeloid leukemia CD34+ cells | Increased levels of the TCA cycle intermediates (α-ketoglutarate and glutamate) | [116] | |
| Baf A1 | BEL-7402; Huh7/SMMC7721 cell lines (hepatocellular carcinoma) | Increased glucose uptake and lactate production Increased HK2 levels | [111] | |
| Baf A1 | BEL-7402 and HO-8910 cell lines (hepatocellular carcinoma and ovarian carcinoma, respectively) | Pathways related to glucose or lipid metabolism were altered | [123] | |
| Baf A1 | Patient-derived AML blasts and MOLM-13 cell line (acute myeloid leukemia) | Decreased basal and maximal OCR | [124] |
5. Therapeutic Implications of Autophagy and Metabolism
5.1. How Cross-Talk between Autophagy and Cell Metabolism Affects the Drug Sensitivity of Tumor Cells
| Autophagy Step Altered | Strategy | Metabolic Pathway Altered | Strategy | Model * | Biological Effects | Reference |
|---|---|---|---|---|---|---|
| Autophagy induction | Rapamycin | Glycolysis | 3-BrPA | SH-SY5Y and SK-N-SH cell lines (neuroblastoma) | Decreased cell proliferation Increased cell death (apoptosis) | [128] |
| Rapamycin | Glycolysis | 2-DG | 1420 and SKBR3 cell lines (pancreatic and breast cancer, respectively) | Decreased 2-DG-induced cell death | [126] | |
| Rapamycin | Glycolysis | 2-DG | SK-N-BE and RKO cell lines (neuroblastoma and colon carcinoma, respectively) | Prevention of 2-DG-induced apoptosis | [127] | |
| Rapamycin | OXPHOS | Rotenone | SH-SY5Y cells (neuroblastoma) | Decreased rotenone-induced apoptosis | [142] | |
| Rapamycin | OXPHOS | Paraquat | SK-N-SH cell line (neuroblastoma) | Decreased paraquat-induced necrosis | [143] | |
| Rapamycin | OXPHOS | MPP+ | SK-N-SH cell line (neuroblastoma) | Increased MPP+-induced cell death | [143] | |
| Rapamycin | OXPHOS | Doxycycline | U251 and U373 cell lines (glioblastoma) | Decreased cell proliferation | [145] | |
| (−) AMPKα | Glycolysis/ OXPHOS | Metformin | Daudi and Jurkat cell lines (B-lymphoma and T-lymphoma, respectively) | Decreased metformin-mediated growth inhibition and cell cycle arrest | [138] | |
| AMPK inhibitor compound C | Glycolysis/ OXPHOS | Metformin | Daudi and Jurkat cell lines (B-lymphoma and T-lymphoma, respectively) | Decreased metformin-mediated growth inhibition and cell cycle arrest | [138] | |
| Initiation and phagophore nucleation | 3-MA | Glycolysis | 3-BrPA | MDA-MB-231 and MDA-MB-435 cell lines (breast cancer) | Decreased cell viability | [129] |
| 3-MA | Glycolysis | DCA | LoVo cell line (colon adenocarcinoma) | Enhanced DCA-induced necrosis Decreased cell viability and proliferation | [130] | |
| 3-MA | Glycolysis | 2-DG | CNE-1 and CNE-2 cell lines (nasopharyngeal carcinoma) | Decreased cell viability and colony formation and promotion of apoptosis | [131] | |
| 3-MA | Glycolysis | 2-DG | 1420 and MDA-MB-435 cell lines (pancreatic cancer and melanoma, respectively) | Increased 2-DG-induced cell death and sensitization to 2-DG | [126] | |
| 3-MA | Glycolysis/ OXPHOS | Metformin | Ishikawa cell line (endometrial adenocarcinoma) | Reduced metformin-mediated cytotoxicity (viability and apoptosis) | [141] | |
| 3-MA | Glycolysis/ OXPHOS | Metformin | Daudi and Jurkat cell lines (B-lymphoma and T-lymphoma, respectively) | Decreased metformin-mediated growth inhibition | [138] | |
| 3-MA | Glycolysis/ OXPHOS | Metformin | AGS cell line (gastric cancer) | Decreased metformin-induced loss of cell viability | [139] | |
| 3-MA | Lipolysis | Orlistat | PANC-1 cell line (pancreatic cancer) | Reduced cell viability Increased apoptosis (caspase-3) | [150] | |
| 3-MA | Lipolysis | Orlistat | SKOV3 and A2780 cell lines (epithelial ovarian cancer) | Decreased orlistat-induced loss of cell viability | [151] | |
| Spautin-1 | Glycolysis | 2-DG | HT-29 and HT1080 cell lines (adenocarcinoma and fibrosarcoma, respectively) | Decreased cell viability | [108] | |
| Spautin-1 | Glycolysis/ OXPHOS | Metformin | BRCA1-deficient mammary tumor cells | Enhanced metformin-mediated inhibition of colony formation | [140] | |
| Spautin-1 | Glycolysis | 2-DG | GBM8 cell line (glioblastoma) | Enhanced 2-DG-induced loss of cell viability Increased apoptosis | [133] | |
| (−) BECN1 | Glycolysis/ OXPHOS | Metformin | Ishikawa cell line (endometrial adenocarcinoma) | Reduced metformin-mediated apoptosis | [141] | |
| Phagophore elongation | (−) ATG7 | Glycolysis | 3-BrPA | MDA-MB-231 and MDA-MB-435 cell lines (breast cancer) | Decreased cell viability | [129] |
| (−) ATG7 | Glycolysis | DCA | LoVo cell line (colon adenocarcinoma) | Enhanced DCA-induced necrosis Decreased cell viability and proliferation | [130] | |
| (−) ATG7 | Glycolysis | 2-DG | 1420 cell line (pancreatic cancer) | Increased 2-DG-induced cell death | [126] | |
| (−) ATG5 | Glycolysis | 2-DG 3-BrPA Lonidamine | SMMC7721 cell line (hepatocellular carcinoma) | Decreased cell proliferation | [111] | |
| dnATG5 | OXPHOS | Paraquat MPP+ | SK-N-SH cell line (neuroblastoma) | Increased paraquat/MPP+-induced cell death | [143] | |
| Fusion with lysosome | CQ | Glycolysis | 3-BrPA | MDA-MB-231 and MDA-MB-435 cell lines (breast cancer) | Decreased cell viability Induction of apoptosis/necrosis Enhanced pro-apoptotic Bax and Bak expression | [129] |
| CQ | Glycolysis | Silibinin (Silybin) | A172 and SR cell lines (glioblastoma) | Enhanced silibinin-induced apoptosis | [134] | |
| CQ | Glycolysis | 2-DG | U251 cell line (glioblastoma) | Induction of cytotoxicity | [135] | |
| CQ | Glycolysis/ OXPHOS | Metformin | Ishikawa cell line (endometrial adenocarcinoma) | Reduced metformin-mediated cytotoxicity (viability) | [141] | |
| CQ | Glutaminolysis | BPTES | 8988 T and MIAPaCa2 cell lines (pancreatic cancer) | Decreased cell viability Induction of apoptosis | [113] | |
| CQ | OXPHOS | Mito-Lonidamine | H2030BrM3 cells (from brain metastases of H2030 cell line, lung adenocarcinoma) | Reduced Mito-Lonidamine-induced necrosis | [146] | |
| CQ | Lipolysis | Etomoxir | Primary ovarian tumor tissue | Disruption of spheroid structure and cell death | [154] | |
| CQ | Lipolysis | Emodin | HepG2 cell line (hepatocellular carcinoma) | Reduced emodin-mediated inhibition of migration and invasion | [153] | |
| CQ | Glutaminolysis | GA inhibitor-968 | H1299 cell line (non-small cell lung cancer) | Enhanced 968-mediated cell growth inhibition | [147] | |
| CQ | Glutaminolysis | GA inhibitor-968 | SW480 and SW620 cell lines (colorectal cancer) | Decreased cell viability Induction of apoptosis | [149] | |
| CQ | Glutaminolysis | GA inhibitor-968 | MDA-MB-231 and HCC38 cell lines (breast cancer); NCI-H1838 cell line (non-small cell lung cancer) | Decreased cell viability | [148] | |
| Baf A1 | Glutaminolysis | GA inhibitor-968 | MDA-MB-231 cell line (breast cancer) | Decreased cell viability | [148] |
5.2. Combination of Drugs Targeting Autophagy and Metabolic Pathways as a Strategy That Can Improve Chemotherapeutic Protocols
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- White, E.; Lattime, E.C.; Guo, J.Y. Autophagy Regulates Stress Responses, Metabolism, and Anticancer Immunity. Trends Cancer 2021, 7, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, V.; Hawkins, W.D.; Klionsky, D.J. Watch What You (Self-) Eat: Autophagic Mechanisms that Modulate Metabolism. Cell Metab. 2019, 29, 803–826. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, C.M.; Dass, C.R. Metabolic stress and cancer: Is autophagy the common denominator and a feasible target? J. Pharm. Pharmacol. 2014, 66, 597–614. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.; Yadav, P.; Shukla, S. Unfolding the role of autophagy in the cancer metabolism. Biochem. Biophys. Rep. 2021, 28, 101158. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Cheng, S.; Sheng, X.; Dai, H.; He, K.; Du, Y. The role of autophagy in regulating metabolism in the tumor microenvironment. Genes Dis. 2021, 10, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Cheong, H.; Lu, C.; Lindsten, T.; Thompson, C.B. Therapeutic targets in cancer cell metabolism and autophagy. Nat. Biotechnol. 2012, 30, 671–678. [Google Scholar] [CrossRef]
- Raj, S.; Chandel, V.; Kumar, A.; Kesari, K.K.; Asthana, S.; Ruokolainen, J.; Kamal, M.A.; Kumar, D. Molecular mechanisms of interplay between autophagy and metabolism in cancer. Life Sci. 2020, 259, 118184. [Google Scholar] [CrossRef]
- Fujikake, N.; Shin, M.; Shimizu, S. Association between autophagy and neurodegenerative diseases. Front. Neurosci. 2018, 12, 255. [Google Scholar] [CrossRef]
- Schiattarella, G.G.; Hill, J.A. Therapeutic targeting of autophagy in cardiovascular disease. J. Mol. Cell. Cardiol. 2016, 95, 86–93. [Google Scholar] [CrossRef]
- Bravo-San Pedro, J.M.; Kroemer, G.; Galluzzi, L. Autophagy and Mitophagy in Cardiovascular Disease. Circ. Res. 2017, 120, 1812–1824. [Google Scholar] [CrossRef]
- Yin, H.; Wu, H.; Chen, Y.; Zhang, J.; Zheng, M.; Chen, G.; Li, L.; Lu, Q. The Therapeutic and Pathogenic Role of Autophagy in Autoimmune Diseases. Front. Immunol. 2018, 9, 1512. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.Y.; Wang, E.J.; Feng, D.; Li, M.; Ye, R.D.; Lu, J.H. Pharmacological insights into autophagy modulation in autoimmune diseases. Acta Pharm. Sin. B 2021, 11, 3364–3378. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy Levy, J.M.; Thorburn, A. Autophagy in cancer: Moving from understanding mechanism to improving therapy responses in patients. Cell Death Differ. 2020, 27, 843–857. [Google Scholar] [CrossRef] [PubMed]
- Vakifahmetoglu-Norberg, H.; Xia, H.G.; Yuan, J. Pharmacologic agents targeting autophagy. J. Clin. Investig. 2015, 125, 5–13. [Google Scholar] [CrossRef]
- Evangelisti, C.; Evangelisti, C.; Chiarini, F.; Lonetti, A.; Buontempo, F.; Neri, L.M.; McCubrey, J.A.; Martelli, A.M. Autophagy in acute leukemias: A double-edged sword with important therapeutic implications. Biochim. Biophys. Acta 2015, 1853, 14–26. [Google Scholar] [CrossRef]
- Li, X.; He, S.; Ma, B. Autophagy and autophagy-related proteins in cancer. Mol. Cancer 2020, 19, 12. [Google Scholar] [CrossRef]
- Lim, S.M.; Mohamad Hanif, E.A.; Chin, S.F. Is targeting autophagy mechanism in cancer a good approach? The possible double-edge sword effect. Cell Biosci. 2021, 11, 56. [Google Scholar] [CrossRef]
- Seo, W.; Silwal, P.; Song, I.C.; Jo, E.K. The dual role of autophagy in acute myeloid leukemia. J. Hematol. Oncol. 2022, 15, 51. [Google Scholar] [CrossRef]
- Maycotte, P.; Thorburn, A. Autophagy and cancer therapy. Cancer Biol. Ther. 2011, 11, 127–137. [Google Scholar] [CrossRef]
- Galluzzi, L.; Pietrocola, F.; Levine, B.; Kroemer, G. Metabolic control of autophagy. Cell 2014, 159, 1263–1276. [Google Scholar] [CrossRef]
- Yin, Z.; Pascual, C.; Klionsky, D.J. Autophagy: Machinery and regulation. Microb. Cell 2016, 3, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Jose, C.; Bellance, N.; Rossignol, R. Choosing between glycolysis and oxidative phosphorylation: A tumor’s dilemma? Biochim. Biophys. Acta 2011, 1807, 552–561. [Google Scholar] [CrossRef]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.S.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef]
- Ma, Y.; Temkin, S.M.; Hawkridge, A.M.; Guo, C.; Wang, W.; Wang, X.Y.; Fang, X. Fatty acid oxidation: An emerging facet of metabolic transformation in cancer. Cancer Lett. 2018, 435, 92–100. [Google Scholar] [CrossRef]
- Srivastava, A.; Srivastava, P.; Mathur, S.; Abbas, S.; Rai, N.; Tiwari, S.; Tiwari, M.; Sharma, L. Lipid Metabolism and Mitochondria: Cross Talk in Cancer. Curr. Drug Targets 2022, 23, 606–627. [Google Scholar] [CrossRef]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Jung, S.; Jeong, H.; Yu, S.W. Autophagy as a decisive process for cell death. Exp. Mol. Med. 2020, 52, 921–930. [Google Scholar] [CrossRef]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Klionsky, D.J. Autophagy and disease: Unanswered questions. Cell Death Differ. 2020, 27, 858–871. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 Association with the ULK1–Atg13–FIP200 Complex Required for Autophagy. Mol. Biol. Cell. 2009, 20, 1981–1991. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK Phosphorylation of Raptor Mediates a Metabolic Checkpoint. Mol. Cell. 2008, 30, 214–226. [Google Scholar] [CrossRef]
- Kim, J.; Kim, Y.C.; Fang, C.; Russell, R.C.; Kim, J.H.; Fan, W.; Liu, R.; Zhong, Q.; Guan, K.L. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell 2013, 152, 290–303. [Google Scholar] [CrossRef]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-Activated Protein Kinase Connects Energy Sensing to Mitophagy. Science 2011, 331, 456–461. [Google Scholar] [CrossRef]
- Mack, H.I.D.; Zheng, B.; Asara, J.M.; Thomas, S.M. AMPK-dependent phosphorylation of ULK1 regulates ATG9 localization. Autophagy 2012, 8, 1197–1214. [Google Scholar] [CrossRef]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Sawa-Makarska, J.; Baumann, V.; Coudevylle, N.; von Bülow, S.; Nogellova, V.; Abert, C.; Schuschnig, M.; Graef, M.; Hummer, G.; Martens, S. Reconstitution of autophagosome nucleation defines Atg9 vesicles as seeds for membrane formation. Science 2020, 369, eaaz7714. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Ma, K.; Gao, R.; Mu, C.; Chen, L.; Liu, Q.; Luo, Q.; Feng, D.; Zhu, Y.; Chen, Q. Regulation of mATG9 trafficking by Src- and ULK1-mediated phosphorylation in basal and starvation-induced autophagy. Cell Res. 2017, 27, 184–201. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef]
- Pengo, N.; Agrotis, A.; Prak, K.; Jones, J.; Ketteler, R. A reversible phospho-switch mediated by ULK1 regulates the activity of autophagy protease ATG4B. Nat. Commun. 2017, 8, 294. [Google Scholar] [CrossRef]
- Walczak, M.; Martens, S. Dissecting the role of the Atg12-Atg5-Atg16 complex during autophagosome formation. Autophagy 2013, 9, 424–425. [Google Scholar] [CrossRef]
- Lee, Y.K.; Lee, J.A. Role of the mammalian ATG8/LC3 family in autophagy: Differential and compensatory roles in the spatiotemporal regulation of autophagy. BMB Rep. 2016, 49, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Shen, T.; Wang, L.; Lu, K. Oligomerization of selective autophagy receptors for the targeting and degradation of protein aggregates. Cells 2021, 10, 1989. [Google Scholar] [CrossRef]
- Li, W.; He, P.; Huang, Y.; Li, Y.F.; Lu, J.; Li, M.; Kurihara, H.; Luo, Z.; Meng, T.; Onishi, M.; et al. Selective autophagy of intracellular organelles: Recent research advances. Theranostics 2021, 11, 222–256. [Google Scholar] [CrossRef]
- Kumar, A.V.; Mills, J.; Lapierre, L.R. Selective Autophagy Receptor p62/SQSTM1, a Pivotal Player in Stress and Aging. Front. Cell Dev. Biol. 2022, 10, 793328. [Google Scholar] [CrossRef]
- Yu, L.; Mcphee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010, 465, 942–946. [Google Scholar] [CrossRef] [PubMed]
- Lőrincz, P.; Juhász, G. Autophagosome-Lysosome Fusion. J. Mol. Biol. 2020, 432, 2462–2482. [Google Scholar] [CrossRef]
- Lee, J.W.; Jeong, E.G.; Lee, S.H.; Yoo, N.J.; Lee, S.H. Somatic mutations of BECN1, an autophagy-related gene, in human cancers. APMIS 2007, 115, 750–756. [Google Scholar] [CrossRef]
- Laddha, S.V.; Ganesan, S.; Chan, C.S.; White, E. Mutational Landscape of the Essential Autophagy Gene BECN1 in Human Cancers. Mol. Cancer Res. 2014, 12, 485–490. [Google Scholar] [CrossRef]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [PubMed]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar] [CrossRef]
- Vega-Rubín-de-Celis, S. The role of Beclin 1-dependent autophagy in cancer. Biology 2020, 9, 4. [Google Scholar] [CrossRef]
- Nicotra, G.; Mercalli, F.; Peracchio, C.; Castino, R.; Follo, C.; Valente, G.; Isidoro, C. Autophagy-active beclin-1 correlates with favourable clinical outcome in non-Hodgkin lymphomas. Mod. Pathol. 2010, 23, 937–950. [Google Scholar] [CrossRef]
- Sivridis, E.; Koukourakis, M.I.; Mendrinos, S.E.; Karpouzis, A.; Fiska, A.; Kouskoukis, C.; Giatromanolaki, A. Beclin-1 and LC3A expression in cutaneous malignant melanomas: A biphasic survival pattern for beclin-1. Melanoma Res. 2011, 21, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Shao, Z.; Xiong, L.; Che, B.; Deng, C.; Xu, W. Expression of Beclin1 in osteosarcoma and the effects of down-regulation of autophagy on the chemotherapeutic sensitivity. J. Huazhong Univ. Sci. Technol. Med. Sci. 2009, 29, 737–740. [Google Scholar] [CrossRef]
- Miracco, C.; Cosci, E.; Oliveri, G.; Luzi, P.; Pacenti, L.; Monciatti, I.; Mannucci, S.; De Nisi, M.C.; Toscano, M.; Malagnino, V.; et al. Protein and mRNA expression of autophagy gene Beclin 1 in human brain tumours. Int. J. Oncol. 2007, 30, 429–436. [Google Scholar]
- Liu, J.; Lin, Y.; Yang, H.; Deng, Q.; Chen, G.; He, J. The expression of p33(ING1), p53, and autophagy-related gene Beclin1 in patients with non-small cell lung cancer. Tumour Biol. 2011, 32, 1113–1121. [Google Scholar] [CrossRef]
- Jin, J.; Britschgi, A.; Schläfli, A.M.; Humbert, M.; Shan-Krauer, D.; Batliner, J.; Federzoni, E.A.; Ernst, M.; Torbett, B.E.; Yousefi, S.; et al. Low autophagy (ATG) gene expression is associated with an immature AML blast cell phenotype and can be restored during AML differentiation therapy. Oxid. Med. Cell Longev. 2018, 2018, 1482795. [Google Scholar] [CrossRef]
- Trocoli, A.; Bensadoun, P.; Richard, E.; Labrunie, G.; Merhi, F.; Schläfli, A.M.; Brigger, D.; Souquere, S.; Pierron, G.; Pasquet, J.M.; et al. p62/SQSTM1 upregulation constitutes a survival mechanism that occurs during granulocytic differentiation of acute myeloid leukemia cells. Cell Death Differ. 2014, 21, 1852–1861. [Google Scholar] [CrossRef] [PubMed]
- Lazarini, M.; Machado-Neto, J.A.; Duarte, A.S.S.; Pericole, F.V.; Ferro, K.P.; de Melo Campos, P.; Traina, F.; Saad, S.T.O. Nix (BNIP3L) Is Downregulated in High-Risk Myelodysplastic Syndromes and Acute Myeloid Leukemia and Its Silencing Enhances Decitabine-Mediated Apoptosis. Blood 2014, 124, 3239. [Google Scholar] [CrossRef]
- Liang, P.Q.; Miao, M.; Hu, R.; Liu, Z.G.; Jiang, H.N.; Li, C.; Ma, S.Y. Expression of autophagy genes in acute myeloid leukemia: Associations with clinical characteristics and prognosis. Neoplasma 2018, 65, 807–814. [Google Scholar] [CrossRef]
- Tandel, P.; Ranjbaran, R.; Ebrahimi, E.; Rezvani, A.; Ramzi, M.; Tamaddon, G. Decreased expression of autophagy-related genes in the complete remission phase of acute myeloid leukemia. Mol. Genet. Genom. Med. 2022, 10, e1872. [Google Scholar] [CrossRef]
- Kong, Y.L.; Huang, Y.; Wu, J.Z.; Cao, X.; Liang, J.H.; Xia, Y.; Wu, W.; Cao, L.; Zhu, H.Y.; Wang, L.; et al. Expression of autophagy related genes in chronic lymphocytic leukemia is associated with disease course. Leuk. Res. 2018, 66, 8–14. [Google Scholar] [CrossRef]
- Lian, Y.; Xie, Y.; Hong, M.; Zhu, Y.; Zhao, H.; Zhao, X.; Zhu, H.; Qiao, C.; Li, J.; Qian, S. Clinical significance of BECLIN1 and ATG5 expression in AML patients. Int. J. Clin. Exp. Pathol. 2018, 11, 1529–1537. [Google Scholar] [PubMed]
- Rothe, K.; Lin, H.; Lin, K.B.L.; Leung, A.; Wang, H.M.; Malekesmaeili, M.; Brinkman, R.R.; Forrest, D.L.; Gorski, S.M.; Jiang, X. The core autophagy protein ATG4B is a potential biomarker and therapeutic target in CML stem/progenitor cells. Blood 2014, 123, 3622–3634. [Google Scholar] [CrossRef]
- Sarang, Z.; Gyurina, K.; Scholtz, B.; Kiss, C.; Szegedi, I. Altered expression of autophagy-related genes might contribute to glucocorticoid resistance in precursor B-cell-type acute lymphoblastic leukemia. Eur. J. Haematol. 2016, 97, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Piya, S.; Kornblau, S.M.; Ruvolo, V.R.; Mu, H.; Ruvolo, P.P.; McQueen, T.; Davis, R.E.; Hail, N.; Kantarjian, H.; Andreeff, M.; et al. Atg7 suppression enhances chemotherapeutic agent sensitivity and overcomes stroma-mediated chemoresistance in acute myeloid leukemia. Blood 2016, 128, 126–1269. [Google Scholar] [CrossRef] [PubMed]
- Ahn, C.H.; Jeong, E.G.; Lee, J.W.; Kim, M.S.; Kim, S.H.; Kim, S.S.; Yoo, N.J.; Lee, S.H. Expression of beclin-1, an autophagy-related protein, in gastric and colorectal cancers. APMIS 2007, 115, 1344–1349. [Google Scholar] [CrossRef]
- Huang, G.; Lu, Z.; Rao, Y.; Gao, H.; Lv, X. Screening and identification of autophagy-related biomarkers for oral squamous cell carcinoma (OSCC) via integrated bioinformatics analysis. J. Cell. Mol. Med. 2021, 25, 4444–4454. [Google Scholar] [CrossRef]
- Cao, Q.; Liu, F.; Yang, Z.; Fu, X.; Yang, Z.; Liu, Q.; Wang, L. Prognostic value of autophagy related proteins ULK1, LC3B and p62 / SQSTM1 in gastric cancer. Am. J. Transl. Res. 2016, 8, 3831–3847. [Google Scholar]
- Wei, Y.; Sinha, S.; Levine, B. Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy 2008, 4, 949–951. [Google Scholar] [CrossRef]
- Roberts, D.J.; Tan-Sah, V.P.; Ding, E.Y.; Smith, J.M.; Miyamoto, S. Hexokinase-II positively regulates glucose starvation-induced autophagy through TORC1 inhibition. Mol. Cell 2014, 53, 521–533. [Google Scholar] [CrossRef]
- Feng, Y.; Liu, J.; Guo, W.; Guan, Y.; Xu, H.; Guo, Q.; Song, X.; Yi, F.; Liu, T.; Zhang, W.; et al. Atg7 inhibits warburg effect by suppressing PKM2 phosphorylation resulting reduced epithelial-mesenchymal transition. Int. J. Biol. Sci. 2018, 14, 755–783. [Google Scholar] [CrossRef]
- Wang, L.; Yang, L.; Yang, Z.; Tang, Y.; Tao, Y.; Zhan, Q.; Lei, L.; Jing, Y.; Jiang, X.; Jin, H.; et al. Glycolytic enzyme PKM2 mediates autophagic activation to promote cell survival in NPM1-mutated leukemia. Int. J. Biol. Sci. 2019, 15, 882–894. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Li, X.; Cai, Q.; Zhang, C.; Yu, Q.; Jiang, Y.; Lee, J.H.; Hawke, D.; Wang, Y.; Xia, Y.; et al. Phosphoglycerate Kinase 1 Phosphorylates Beclin1 to Induce Autophagy. Mol. Cell 2017, 65, 917–931.e6. [Google Scholar] [CrossRef] [PubMed]
- Das, C.K.; Parekh, A.; Parida, P.K.; Bhutia, S.K.; Mandal, M. Lactate dehydrogenase A regulates autophagy and tamoxifen resistance in breast cancer. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1004–1018. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Wang, S.; Jin, L.; Weng, M.; Zhou, D.; Wang, J.; Tang, Z.; Quan, Z. Long non-coding RNA GBCDRlnc1 induces chemoresistance of gallbladder cancer cells by activating autophagy. Mol. Cancer 2019, 18, 82. [Google Scholar] [CrossRef] [PubMed]
- Bosc, C.; Broin, N.; Fanjul, M.; Saland, E.; Farge, T.; Courdy, C.; Batut, A.; Masoud, R.; Larrue, C.; Skuli, S.; et al. Autophagy regulates fatty acid availability for oxidative phosphorylation through mitochondria-endoplasmic reticulum contact sites. Nat. Commun. 2020, 11, 4056. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.A.; Xing, X.; Harris, J.W.; Zaytseva, Y.Y.; Mitov, M.I.; Napier, D.L.; Weiss, H.L.; Mark Evers, B.; Gao, T. Adipocytes activate mitochondrial fatty acid oxidation and autophagy to promote tumor growth in colon cancer. Cell Death Dis. 2017, 8, e2593. [Google Scholar] [CrossRef]
- Qin, W.; Li, C.; Zheng, W.; Guo, Q.; Zhang, Y.; Kang, M.; Zhang, B.; Yang, B.; Li, B.; Yang, H.; et al. Inhibition of autophagy promotes metastasis and glycolysis by inducing ROS in gastric cancer cells. Oncotarget 2015, 6, 39839–39854. [Google Scholar] [CrossRef]
- Pereira, O.; Teixeira, A.; Sampaio-Marques, B.; Castro, I.; Girão, H.; Ludovico, P. Signalling mechanisms that regulate metabolic profile and autophagy of acute myeloid leukaemia cells. J. Cell. Mol. Med. 2018, 22, 4807–4817. [Google Scholar] [CrossRef]
- Suganuma, K.; Miwa, H.; Imai, N.; Shikami, M.; Gotou, M.; Goto, M.; Mizuno, S.; Takahashi, M.; Yamamoto, H.; Hiramatsu, A.; et al. Energy metabolism of leukemia cells: Glycolysis versus oxidative phosphorylation. Leuk. Lymphoma 2010, 51, 2112–2119. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Mahapatra, K.K.; Praharaj, P.P.; Patil, S.; Bhutia, S.K. Recent progress of autophagy signaling in tumor microenvironment and its targeting for possible cancer therapeutics. Semin. Cancer Biol. 2022, 85, 196–208. [Google Scholar] [CrossRef]
- Folkerts, H.; Hilgendorf, S.; Vellenga, E.; Bremer, E.; Wiersma, V.R. The multifaceted role of autophagy in cancer and the microenvironment. Med. Res. Rev. 2018, 39, 517–560. [Google Scholar] [CrossRef]
- New, J.; Arnold, L.; Ananth, M.; Alvi, S.; Thornton, M.; Werner, L.; Tawfik, O.; Dai, H.; Shnayder, Y.; Kakarala, K.; et al. Secretory autophagy in cancer-associated fibroblasts promotes head and neck cancer progression and offers a novel therapeutic target. Cancer Res. 2017, 77, 6679–6691. [Google Scholar] [CrossRef] [PubMed]
- Sousa, C.M.; Biancur, D.E.; Wang, X.; Halbrook, C.J.; Sherman, M.H.; Zhang, L.; Kremer, D.; Hwang, R.F.; Witkiewicz, A.K.; Ying, H.; et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 2016, 536, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Maynard, R.S.; Hellmich, C.; Bowles, K.M.; Rushworth, S.A. Acute Myeloid Leukaemia Drives Metabolic Changes in the Bone Marrow Niche. Front. Oncol. 2022, 12, 924567. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jiang, Y.; Cheng, J.; Ma, J.; Li, Q.; Pang, T. ATG5 regulates mesenchymal stem cells differentiation and mediates chemosensitivity in acute myeloid leukemia. Biochem. Biophys. Res. Commun. 2020, 525, 398–405. [Google Scholar] [CrossRef]
- Nomura, H.; Uzawa, K.; Yamano, Y.; Fushimi, K.; Ishigami, T.; Kouzu, Y.; Koike, H.; Siiba, M.; Bukawa, H.; Yokoe, H.; et al. Overexpression and altered subcellular localization of autophagy-related 16-like 1 in human oral squamous-cell carcinoma: Correlation with lymphovascular invasion and lymph-node metastasis. Hum. Pathol. 2009, 40, 83–91. [Google Scholar] [CrossRef]
- Jo, Y.K.; Kim, S.C.; Park, I.J.; Park, S.J.; Jin, D.H.; Hong, S.W.; Cho, D.H.; Kim, J.C. Increased Expression of ATG10 in Colorectal Cancer Is Associated with Lymphovascular Invasion and Lymph Node Metastasis. PLoS ONE 2012, 7, e52705. [Google Scholar] [CrossRef]
- Thomas, D.; Majeti, R. Biology and relevance of human acute myeloid leukemia stem cells. Blood 2017, 129, 1577–1585. [Google Scholar] [CrossRef]
- Peng, M.; Huang, Y.; Zhang, L.; Zhao, X.; Hou, Y. Targeting Mitochondrial Oxidative Phosphorylation Eradicates Acute Myeloid Leukemic Stem Cells. Front. Oncol. 2022, 12, 899502. [Google Scholar] [CrossRef]
- Jones, C.L.; Stevens, B.M.; D’Alessandro, A.; Reisz, J.A.; Culp-Hill, R.; Nemkov, T.; Pei, S.; Khan, N.; Adane, B.; Ye, H.; et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell 2018, 34, 724–740.e4. [Google Scholar] [CrossRef]
- Marlein, C.R.; Zaitseva, L.; Piddock, R.E.; Robinson, S.D.; Edwards, D.R.; Shafat, M.S.; Zhou, Z.; Lawes, M.; Bowles, K.M.; Rushworth, S.A. NADPH oxidase-2 derived superoxide drives mitochondrial transfer from bone marrow stromal cells to leukemic blasts. Blood 2017, 130, 1649–1660. [Google Scholar] [CrossRef]
- Liu, L.L.; Long, Z.J.; Wang, L.X.; Zheng, F.M.; Fang, Z.G.; Yan, M.; Xu, D.F.; Chen, J.J.; Wang, S.W.; Lin, D.J.; et al. Inhibition of mTOR pathway sensitizes acute myeloid leukemia cells to Aurora inhibitors by suppression of glycolytic metabolism. Mol. Cancer Res. 2013, 11, 1326–1336. [Google Scholar] [CrossRef]
- Liu, L.; Gong, L.; Zhang, Y.; Li, N. Glycolysis in Panc-1 human pancreatic cancer cells is inhibited by everolimus. Exp. Ther. Med. 2012, 5, 338–342. [Google Scholar] [CrossRef]
- Liu, J.; Xia, H.; Kim, M.; Xu, L.; Li, Y.; Zhang, L.; Cai, Y.; Norberg, H.V.; Zhang, T.; Furuya, T.; et al. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell 2011, 147, 223–234. [Google Scholar] [CrossRef]
- Cicchini, M.; Karantza, V.; Xia, B. Molecular pathways: Autophagy in cancer-A matter of timing and context. Clin. Cancer Res. 2015, 21, 498–504. [Google Scholar] [CrossRef]
- Limpert, A.S.; Lambert, L.J.; Bakas, N.A.; Bata, N.; Brun, S.N.; Shaw, R.J.; Cosford, N.D.P. Autophagy in Cancer: Regulation by Small Molecules. Trends Pharmacol. Sci. 2018, 39, 1021–1032. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.T.; Tan, H.L.; Shui, G.; Bauvy, C.; Huang, Q.; Wenk, M.R.; Ong, C.N.; Codogno, P.; Shen, H.M. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J Biol. Chem. 2010, 285, 10850–10861. [Google Scholar] [CrossRef] [PubMed]
- Kunimasa, K.; Ikeda-Ishikawa, C.; Tani, Y.; Tsukahara, S.; Sakurai, J.; Okamoto, Y.; Koido, M.; Dan, S.; Tomida, A. Spautin-1 inhibits mitochondrial complex I and leads to suppression of the unfolded protein response and cell survival during glucose starvation. Sci Rep. 2022, 12, 11533. [Google Scholar] [CrossRef] [PubMed]
- Toshima, T.; Shirabe, K.; Matsumoto, Y.; Yoshiya, S.; Ikegami, T.; Yoshizumi, T.; Soejima, Y.; Ikeda, T.; Maehara, Y. Autophagy enhances hepatocellular carcinoma progression by activation of mitochondrial β-oxidation. J. Gastroenterol. 2014, 49, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Baker, F.; Polat, I.H.; Abou-el-ardat, K.; Alshamleh, I.; Thoelken, M.; Hymon, D.; Gubas, A.; Koschade, S.E.; Vischedyk, J.B.; Kaulich, M.; et al. Metabolic rewiring is essential for AML cell survival to overcome autophagy inhibition by loss of ATG3. Cancers 2021, 13, 6142. [Google Scholar] [CrossRef] [PubMed]
- Jiao, L.; Zhang, H.L.; Li, D.D.; Yang, K.L.; Tang, J.; Li, X.; Ji, J.; Yu, Y.; Wu, R.Y.; Ravichandran, S.; et al. Regulation of glycolytic metabolism by autophagy in liver cancer involves selective autophagic degradation of HK2 (hexokinase 2). Autophagy 2018, 14, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Hitosugi, T.; Kang, S.; Heiden, M.G.V.; Chung, T.; Lythgoe, K.; Dong, S.; Lonial, S.; Wang, X.; Chen, G.Z.; Xie, J.; et al. Tyrosine Phosphorylation Inhibits PKM2 to Promote the Warburg Effect and Tumor Growth. Sci. Signal. 2009, 2, ra73. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.W.; Choi, J.; Lee, S.Y.; Sung, S.; Yoo, H.J.; Kang, M.J.; Cheong, H.; Son, J. Autophagy is required for PDAC glutamine metabolism. Sci. Rep. 2016, 6, 37594. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.R.; Wu, S.Y.; Chen, R.Y.; Lin, Y.S.; Yeh, T.M.; Liu, H.S. Regulation of autophagy, glucose uptake, and glycolysis under dengue virus infection. Kaohsiung J. Med. Sci. 2020, 36, 911–919. [Google Scholar] [CrossRef]
- Lock, R.; Roy, S.; Kenific, C.M.; Su, J.S.; Salas, E.; Ronen, S.M.; Debnath, J. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol. Biol. Cell. 2011, 22, 165–178. [Google Scholar] [CrossRef]
- Karvela, M.; Baquero, P.; Kuntz, E.M.; Mukhopadhyay, A.; Mitchell, R.; Allan, E.K.; Chan, E.; Kranc, K.R.; Calabretta, B.; Salomoni, P.; et al. ATG7 regulates energy metabolism, differentiation and survival of Philadelphia-chromosome-positive cells. Autophagy 2016, 12, 936–948. [Google Scholar] [CrossRef]
- Wei, H.; Wei, S.; Gan, B.; Peng, X.; Zou, W.; Guan, J.L. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev. 2011, 25, 1510–1527. [Google Scholar] [CrossRef]
- Duan, L.; Calhoun, S.; Shim, D.; Perez, R.E.; Blatter, L.A.; Maki, C.G. Fatty acid oxidation and autophagy promote endoxifen resistance and counter the effect of AKT inhibition in ER-positive breast cancer cells. J. Mol. Cell Biol. 2021, 13, 433–444. [Google Scholar] [CrossRef]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Mauvezin, C.; Neufeld, T.P. Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome fusion. Autophagy 2015, 11, 1437–1438. [Google Scholar] [CrossRef]
- Xia, H.G.; Najafov, A.; Geng, J.; Galan-Acosta, L.; Han, X.; Guo, Y.; Shan, B.; Zhang, Y.; Norberg, E.; Zhang, T.; et al. Degradation of HK2 by chaperone-mediated autophagy promotes metabolic catastrophe and cell death. J. Cell Biol. 2015, 210, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Sheng, J.; Shen, L.; Su, J.; Xu, Y.; Xie, Q.; Wu, Y.; Zhang, X.; Sun, L. Autophagy inhibitor chloroquine increases sensitivity to cisplatin in QBC939 cholangiocarcinoma cells by mitochondrial ROS. PLoS ONE 2017, 12, e0173712. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Chen, L.; Chen, Y.; Shao, Q.; Qin, W. Bafilomycin A1 inhibits the growth and metastatic potential of the BEL-7402 liver cancer and HO-8910 ovarian cancer cell lines and induces alterations in their microRNA expression. Exp. Ther. Med. 2015, 10, 1829–1834. [Google Scholar] [CrossRef]
- Dykstra, K.M.; Fay, H.R.S.; Massey, A.C.; Yang, N.; Johnson, M.; Portwood, S.; Guzman, M.L.; Wang, E.S. Inhibiting autophagy targets human leukemic stem cells and hypoxic AML blasts by disrupting mitochondrial homeostasis. Blood Adv. 2021, 5, 2087–2100. [Google Scholar] [CrossRef]
- Pajak, B.; Siwiak, E.; Sołtyka, M.; Priebe, A.; Zieliński, R.; Fokt, I.; Ziemniak, M.; Jaśkiewicz, A.; Borowski, R.; Domoradzki, T.; et al. 2-Deoxy-D-Glucose and its analogs: From diagnostic to therapeutic agents. Int. J. Mol. Sci. 2020, 21, 234. [Google Scholar] [CrossRef] [PubMed]
- Xi, H.; Kurtoglu, M.; Liu, H.; Wangpaichitr, M.; You, M.; Liu, X.; Savaraj, N.; Lampidis, T.J. 2-Deoxy-d-glucose activates autophagy via endoplasmic reticulum stress rather than ATP depletion. Cancer Chemother. Pharmacol. 2011, 67, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Maximchik, P.; Abdrakhmanov, A.; Inozemtseva, E.; Tyurin-Kuzmin, P.A.; Zhivotovsky, B.; Gogvadze, V. 2-Deoxy-D-glucose has distinct and cell line-specific effects on the survival of different cancer cells upon antitumor drug treatment. FEBS J. 2018, 285, 4590–4601. [Google Scholar] [CrossRef]
- Gan, L.; Ren, Y.; Lu, J.; Ma, J.; Shen, X.; Zhuang, Z. Synergistic effect of 3-bromopyruvate in combination with rapamycin impacted neuroblastoma metabolism by inhibiting autophagy. Onco Targets Ther. 2020, 13, 11125–11137. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, Y.; Zhang, P.; Chao, Z.; Xia, F.; Jiang, C.; Zhang, X.; Jiang, Z.; Liu, H. Hexokinase II inhibitor, 3-BrPA induced autophagy by stimulating ROS formation in human breast cancer cells. Genes Cancer 2014, 5, 100–112. [Google Scholar] [CrossRef]
- Gong, F.; Peng, X.; Sang, Y.; Qiu, M.; Luo, C.; He, Z.; Zhao, X.; Tong, A. Dichloroacetate induces protective autophagy in LoVo cells: Involvement of cathepsin D/thioredoxin-like protein 1 and Akt-mTOR-mediated signaling. Cell Death Dis. 2013, 4, e913. [Google Scholar] [CrossRef]
- Song, K.; Li, M.; Xu, X.; Xuan, L.; Huang, G.; Liu, Q. Resistance to chemotherapy is associated with altered glucose metabolism in acute myeloid leukemia. Oncol. Lett. 2016, 12, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Prasad, V.V.T.S.; Gopalan, R.O.G. Continued use of MDA-MB-435, a melanoma cell line, as a model for human breast cancer, even in year, 2014. npj Breast Cancer 2015, 1, 15002. [Google Scholar] [CrossRef] [PubMed]
- Martell, E.; Kuzmychova, H.; Senthil, H.; Kaul, E.; Chokshi, C.R.; Venugopal, C.; Anderson, C.M.; Singh, S.K.; Sharif, T. Compensatory cross-talk between autophagy and glycolysis regulates senescence and stemness in heterogeneous glioblastoma tumor subpopulations. Acta Neuropathol. Commun. 2023, 11, 110. [Google Scholar] [CrossRef] [PubMed]
- Bai, Z.L.; Tay, V.; Guo, S.Z.; Ren, J.; Shu, M.G. Silibinin Induced Human Glioblastoma Cell Apoptosis Concomitant with Autophagy through Simultaneous Inhibition of mTOR and YAP. Biomed. Res. Int. 2018, 2018, 6165192. [Google Scholar] [CrossRef]
- Kosic, M.; Arsikin-Csordas, K.; Paunovic, V.; Firestone, R.A.; Ristic, B.; Mircic, A.; Petricevic, S.; Bosnjak, M.; Zogovic, N.; Mandic, M.; et al. Synergistic anticancer action of lysosomal membrane permeabilization and glycolysis inhibition. J. Biol. Chem. 2016, 291, 22936–22948. [Google Scholar] [CrossRef]
- Salani, B.; Del Rio, A.; Marini, C.; Sambuceti, G.; Cordera, R.; Maggi, D. Metformin, cancer and glucose metabolism. Endocr. Relat. Cancer 2014, 21, R461–R471. [Google Scholar] [CrossRef]
- Fontaine, E. Metformin-Induced Mitochondrial Complex I Inhibition: Facts, Uncertainties, and Consequences. Front. Endocrinol. 2018, 9, 23–28. [Google Scholar] [CrossRef]
- Shi, W.Y.; Xiao, D.; Wang, L.; Dong, L.H.; Yan, Z.X.; Shen, Z.X.; Chen, S.J.; Chen, Y.; Zhao, W.L. Therapeutic metformin/AMPK activation blocked lymphoma cell growth via inhibition of mTOR pathway and induction of autophagy. Cell Death Dis. 2012, 3, e275. [Google Scholar] [CrossRef]
- Fang, C.W.; Yang, J.S.; Chiang, J.H.; Shieh, P.C.; Tsai, F.J.; Tsai, C.W.; Chang, W.S. Metformin induces autophagy of cisplatin-resistant human gastric cancer cells in addition to apoptosis. BioMedicine 2023, 13, 13–23. [Google Scholar] [CrossRef]
- Yeo, S.K.; Paul, R.; Haas, M.; Wang, C.; Guan, J.L. Improved efficacy of mitochondrial disrupting agents upon inhibition of autophagy in a mouse model of BRCA1-deficient breast cancer. Autophagy 2018, 14, 1214–1225. [Google Scholar] [CrossRef]
- Takahashi, A.; Kimura, F.; Yamanaka, A.; Takebayashi, A.; Kita, N.; Takahashi, K.; Murakami, T. Metformin impairs growth of endometrial cancer cells via cell cycle arrest and concomitant autophagy and apoptosis. Cancer Cell Int. 2014, 14, 53. [Google Scholar] [CrossRef] [PubMed]
- Pan, T.; Rawal, P.; Wu, Y.; Xie, W.; Jankovic, J.; Le, W. Rapamycin protects against rotenone-induced apoptosis through autophagy induction. Neuroscience 2009, 164, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Garcia, A.; Anandhan, A.; Burns, M.; Chen, H.; Zhou, Y.; Franco, R. Impairment of Atg5-dependent autophagic flux promotes paraquat- and MPP+-induced apoptosis but not rotenone or 6-hydroxydopamine toxicity. Toxicol. Sci. 2013, 136, 166–182. [Google Scholar] [CrossRef] [PubMed]
- Moullan, N.; Mouchiroud, L.; Wang, X.; Ryu, D.; Williams, E.G.; Mottis, A.; Jovaisaite, V.; Frochaux, M.V.; Quiros, P.M.; Deplancke, B.; et al. Tetracyclines disturb mitochondrial function across eukaryotic models: A call for caution in biomedical research. Cell Rep. 2015, 10, 1681–1691. [Google Scholar] [CrossRef]
- Petővári, G.; Hujber, Z.; Krencz, I.; Dankó, T.; Nagy, N.; Tóth, F.; Raffay, R.; Mészáros, K.; Rajnai, H.; Vetlényi, E.; et al. Targeting cellular metabolism using rapamycin and/or doxycycline enhances anti-tumour effects in human glioma cells. Cancer Cell Int. 2018, 18, 211. [Google Scholar] [CrossRef]
- Cheng, G.; Zhang, Q.; Pan, J.; Lee, Y.; Ouari, O.; Hardy, M.; Zielonka, M.; Myers, C.R.; Zielonka, J.; Weh, K.; et al. Targeting lonidamine to mitochondria mitigates lung tumorigenesis and brain metastasis. Nat. Commun. 2019, 10, 2205. [Google Scholar] [CrossRef]
- Han, T.; Guo, M.; Zhang, T.; Gan, M.; Xie, C.; Wang, J. Bin. A novel glutaminase inhibitor-968 inhibits the migration and proliferation of non-small cell lung cancer cells by targeting EGFR/ERK signaling pathway. Oncotarget 2017, 8, 28063–28073. [Google Scholar] [CrossRef]
- Halama, A.; Kulinski, M.; Dib, S.S.; Zaghlool, S.B.; Siveen, K.S.; Iskandarani, A.; Zierer, J.; Prabhu, K.S.; Satheesh, N.J.; Bhagwat, A.M.; et al. Accelerated lipid catabolism and autophagy are cancer survival mechanisms under inhibited glutaminolysis. Cancer Lett. 2018, 430, 133–147. [Google Scholar] [CrossRef]
- Li, J.; Song, P.; Zhu, L.; Aziz, N.; Zhou, Q.; Zhang, Y.; Xu, W.; Feng, L.; Chen, D.; Wang, X.; et al. Synthetic lethality of glutaminolysis inhibition, autophagy inactivation and asparagine depletion in colon cancer. Oncotarget 2017, 8, 42664–42672. [Google Scholar] [CrossRef]
- Sharma, V.; Ramachandran, R.; Dhawan, S.; Kaur, J. Orlistat Induced Endoplasmic Reticulum Stress Mediated Apoptosis and Protective Autophagy in PANC-1 Cells: The Key Role of JNK and Mitochondrial Dependent Signalling. Acta Sci. Pharm. Sci. 2023, 7, 2581–5423. [Google Scholar]
- Peng, H.; Wang, Q.; Qi, X.; Wang, X.; Zhao, X. Orlistat induces apoptosis and protective autophagy in ovarian cancer cells: Involvement of Akt-mTOR-mediated signaling pathway. Arch Gynecol Obstet. 2018, 298, 597–605. [Google Scholar] [CrossRef]
- Lee, K.H.; Lee, M.S.; Cha, E.Y.; Sul, J.Y.; Lee, J.S.; Kim, J.S.; Park, J.B.; Kim, J.Y. Inhibitory effect of emodin on fatty acid synthase, colon cancer proliferation and apoptosis. Mol. Med. Rep. 2017, 15, 2163–2173. [Google Scholar] [CrossRef]
- Qin, B.; Zeng, Z.; Xu, J.; Shangwen, J.; Ye, Z.J.; Wang, S.; Wu, Y.; Peng, G.; Wang, Q.; Gu, W.; et al. Emodin inhibits invasion and migration of hepatocellular carcinoma cells via regulating autophagy-mediated degradation of snail and β-catenin. BMC Cancer 2022, 22, 671. [Google Scholar] [CrossRef]
- Kumar, S.; Swamy, S.N.; Devaraj, V.R.; Premalathac, C.S.; Pallavi, V.R.; Sagar, B.C.; Shinde, D.; Gawari, R. Metabolic Reprogramming and Lipophagy Mediates Survival of Ascites Derived Metastatic Ovarian Cancer Cells. Asian Pac. J. Cancer Prev. 2022, 23, 1699–1709. [Google Scholar] [CrossRef]
- Shiratori, R.; Furuichi, K.; Yamaguchi, M.; Miyazaki, N.; Aoki, H.; Chibana, H.; Ito, K.; Aoki, S. Glycolytic suppression dramatically changes the intracellular metabolic profile of multiple cancer cell lines in a mitochondrial metabolism-dependent manner. Sci. Rep. 2019, 9, 18699. [Google Scholar] [CrossRef] [PubMed]
- Olivas-Aguirre, M.; Pérez-Chávez, J.A.; Torres-López, L.; Hernández-Cruz, A.; Pottosin, I.; Dobrovinskaya, O. Dexamethasone-Induced Fatty Acid Oxidation and Autophagy/Mitophagy Are Essential for T-ALL Glucocorticoid Resistance. Cancers 2023, 15, 445. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.H.; Israelsen, W.J.; Lee, D.; Yu, V.W.C.; Jeanson, N.T.; Clish, C.B.; Cantley, L.C.; Vander Heiden, M.G.; Scadden, D.T. Cell-state-specific metabolic dependency in hematopoiesis and leukemogenesis. Cell 2014, 158, 1309–1323. [Google Scholar] [CrossRef]
- Zhu, S.; Guo, Y.; Zhang, X.; Liu, H.; Yin, M.; Chen, X.; Peng, C. Pyruvate kinase M2 (PKM2) in cancer and cancer therapeutics. Cancer Lett. 2021, 503, 240–248. [Google Scholar] [CrossRef]
- Li, Y.J.; Fahrmann, J.F.; Aftabizadeh, M.; Zhao, Q.; Tripathi, S.C.; Zhang, C.; Yuan, Y.; Ann, D.; Hanash, S.; Yu, H. Fatty acid oxidation protects cancer cells from apoptosis by increasing mitochondrial membrane lipids. Cell Rep. 2022, 39, 110870. [Google Scholar] [CrossRef] [PubMed]
- You, L.; Wang, Z.; Li, H.; Shou, J.; Jing, Z.; Xie, J.; Sui, X.; Pan, H.; Han, W. The role of STAT3 in autophagy. Autophagy 2015, 11, 729–739. [Google Scholar] [CrossRef]
- Li, J.; Zhao, S.; Zhou, X.; Zhang, T.; Zhao, L.; Miao, P.; Song, S.; Sun, X.; Liu, J.; Zhao, X.; et al. Inhibition of lipolysis by mercaptoacetate and etomoxir specifically sensitize drug-resistant lung adenocarcinoma cell to paclitaxel. PLoS ONE 2013, 8, e74623. [Google Scholar] [CrossRef] [PubMed]
- Terabe, T.; Uchida, F.; Nagai, H.; Omori, S.; Ishibashi-Kanno, N.; Hasegawa, S.; Yamagata, K.; Gosho, M.; Yanagawa, T.; Bukawa, H. Expression of autophagy-related markers at the surgical margin of oral squamous cell carcinoma correlates with poor prognosis and tumor recurrence. Hum. Pathol. 2018, 73, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Yan, F.; Zhang, X.; Tan, R.; Li, M.; Xiao, Z.; Wang, H.; Zhang, Z.; Ma, Z.; Liu, Z. Autophagic flux in cancer cells at the invasive front in the tumorstroma border. Aging 2021, 13, 20229–20245. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.Y.; Zhang, X.Y.; Wang, X.F.; Sun, B.C. Autophagy enhances the aggressiveness of human colorectal cancer cells and their ability to adapt to apoptotic stimulus. Cancer Biol. Med. 2012, 9, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Nihira, K.; Miki, Y.; Ono, K.; Suzuki, T.; Sasano, H. An inhibition of p62/SQSTM1 caused autophagic cell death of several human carcinoma cells. Cancer Sci. 2014, 105, 568–575. [Google Scholar] [CrossRef]
- Abdul Rahim, S.A.; Dirkse, A.; Oudin, A.; Schuster, A.; Bohler, J.; Barthelemy, V.; Muller, A.; Vallar, L.; Janji, B.; Golebiewska, A.; et al. Regulation of hypoxia-induced autophagy in glioblastoma involves ATG9A. Br. J. Cancer 2017, 117, 813–825. [Google Scholar] [CrossRef]
- Polak, R.; Bierings, M.B.; van der Leije, C.S.; Sanders, M.A.; Roovers, O.; Marchante, J.R.M.; Boer, J.M.; Cornelissen, J.J.; Pieters, R.; Den Boer, M.L.; et al. Autophagy inhibition as a potential future targeted therapy for ETV6-RUNX1-driven B-cell precursor acute lymphoblastic leukemia. Haematologica 2019, 104, 738–748. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torres-López, L.; Dobrovinskaya, O. Dissecting the Role of Autophagy-Related Proteins in Cancer Metabolism and Plasticity. Cells 2023, 12, 2486. https://doi.org/10.3390/cells12202486
Torres-López L, Dobrovinskaya O. Dissecting the Role of Autophagy-Related Proteins in Cancer Metabolism and Plasticity. Cells. 2023; 12(20):2486. https://doi.org/10.3390/cells12202486
Chicago/Turabian StyleTorres-López, Liliana, and Oxana Dobrovinskaya. 2023. "Dissecting the Role of Autophagy-Related Proteins in Cancer Metabolism and Plasticity" Cells 12, no. 20: 2486. https://doi.org/10.3390/cells12202486
APA StyleTorres-López, L., & Dobrovinskaya, O. (2023). Dissecting the Role of Autophagy-Related Proteins in Cancer Metabolism and Plasticity. Cells, 12(20), 2486. https://doi.org/10.3390/cells12202486