

Clinical Cases and the Molecular Profiling of a Novel Childhood Encephalopathy-Causing GNAO1 Mutation P170R
 , ,
, , 
Abstract
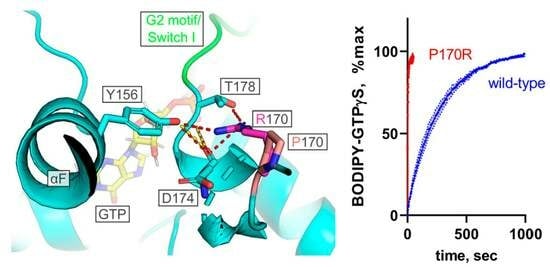
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
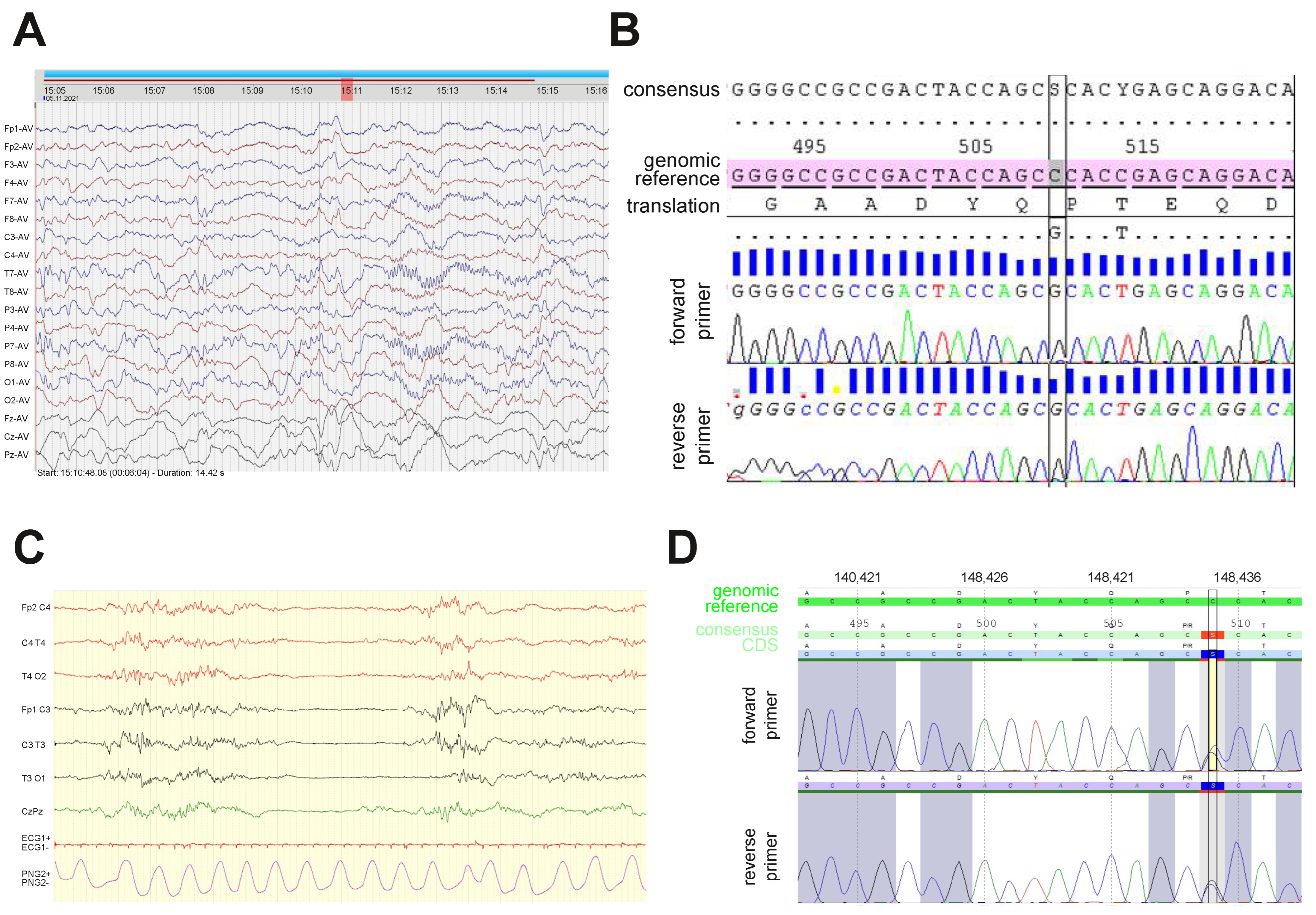
2.2. Genetic Investigation
2.3. Antibodies
2.4. Plasmids and Molecular Cloning
2.5. Fluorescence-Based Test for GTP Binding and Hydrolysis
2.6. Cell Line and Culture Conditions
2.7. Co-Immunoprecipitation
2.8. Molecular Modeling
2.9. Determination of Cellular Stability of Gαo
2.10. Immunofluorescence and Microscopy
3. Results
3.1. Early Onset Encephalopathy and a Developmental Delay Are Driven by c.509C>G Substitution in GNAO1 Gene
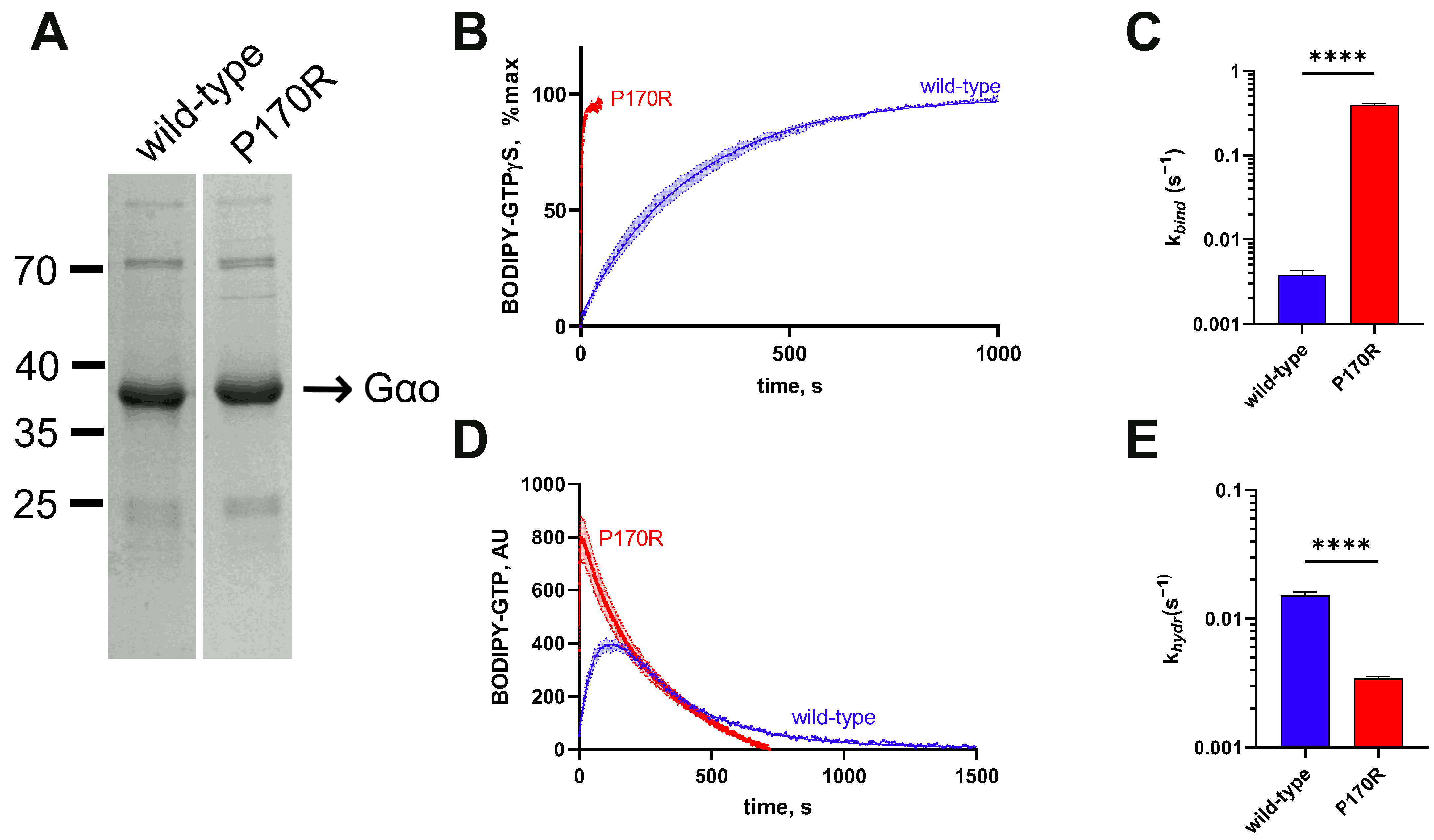
3.2. Biochemical Characterization of the Purified P170R Mutant Reveals Highly Accelerated GTP Uptake with a Mild Impairment of Hydrolysis
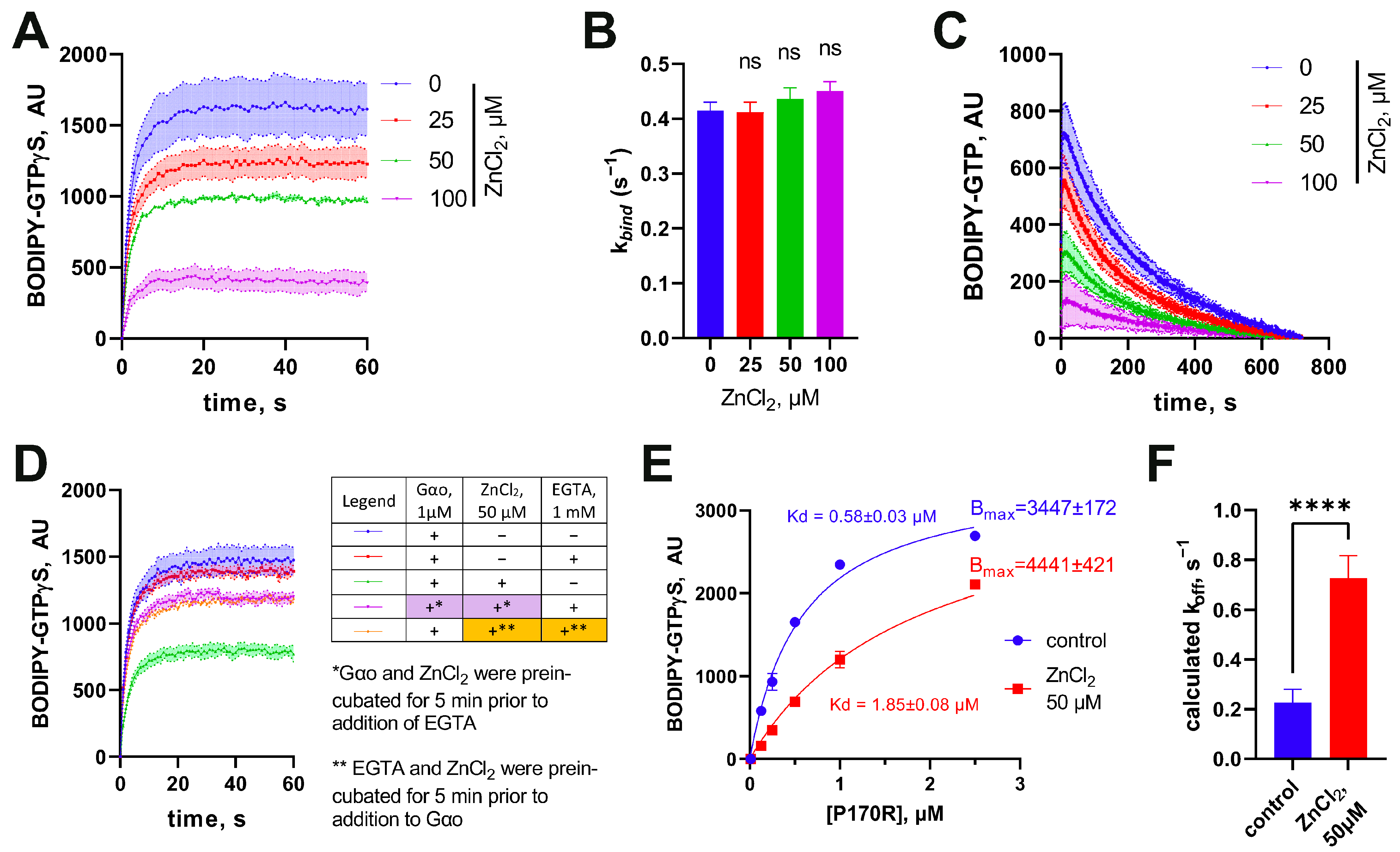
3.3. Zn2+ Ions Elicit a Unique Effect on P170R, Kinetically Promoting It to Lose Bound GTP
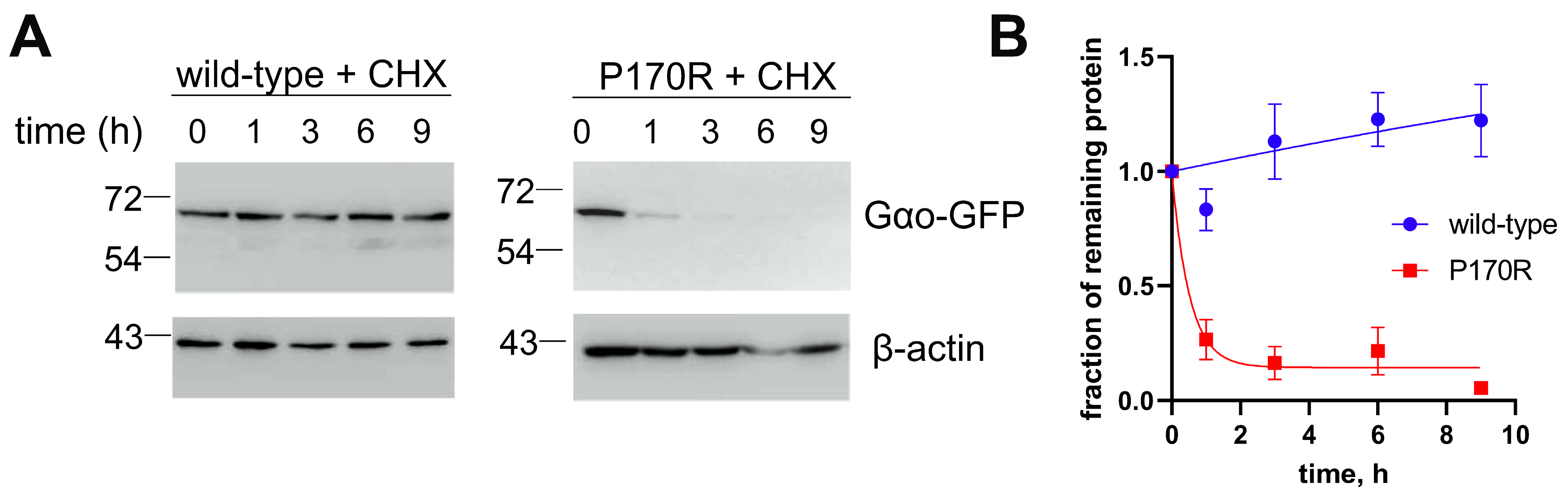
3.4. The P170R Substitution Perturbs the Protein’s Structure, Stability, Cellular Interactions and Localization
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nakamura, K.; Kodera, H.; Akita, T.; Shiina, M.; Kato, M.; Hoshino, H.; Terashima, H.; Osaka, H.; Nakamura, S.; Tohyama, J.; et al. De Novo mutations in GNAO1, encoding a Galphao subunit of heterotrimeric G proteins, cause epileptic encephalopathy. Am. J. Hum. Genet. 2013, 93, 496–505. [Google Scholar] [CrossRef]
- Kelly, M.; Park, M.; Mihalek, I.; Rochtus, A.; Gramm, M.; Pérez-Palma, E.; Axeen, E.T.; Hung, C.Y.; Olson, H.; Swanson, L.; et al. Spectrum of neurodevelopmental disease associated with the GNAO1 guanosine triphosphate–binding region. Epilepsia 2019, 60, 406–418. [Google Scholar] [CrossRef]
- Novelli, M.; Galosi, S.; Zorzi, G.; Martinelli, S.; Capuano, A.; Nardecchia, F.; Granata, T.; Pollini, L.; Di Rocco, M.; Marras, C.E.; et al. GNAO1-related movement disorder: An update on phenomenology, clinical course, and response to treatments. Parkinsonism Relat. Disord. 2023, 111, 105405. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef] [PubMed]
- Sternweis, P.C.; Robishaw, J.D. Isolation of two proteins with high affinity for guanine nucleotides from membranes of bovine brain. J. Biol. Chem. 1984, 259, 13806–13813. [Google Scholar] [CrossRef] [PubMed]
- Oldham, W.M.; Hamm, H.E. Heterotrimeric G protein activation by G-protein-coupled receptors. Nat. Rev. Mol. Cell Biol. 2008, 9, 60–71. [Google Scholar] [CrossRef]
- Ross, E.M.; Wilkie, T.M. GTPase-activating proteins for heterotrimeric G proteins: Regulators of G protein signaling (RGS) and RGS-like proteins. Annu. Rev. Biochem. 2000, 69, 795–827. [Google Scholar] [CrossRef] [PubMed]
- Wootten, D.; Christopoulos, A.; Marti-Solano, M.; Babu, M.M.; Sexton, P.M. Mechanisms of signalling and biased agonism in G protein-coupled receptors. Nat. Rev. Mol. Cell Biol. 2018, 19, 638–653. [Google Scholar] [CrossRef]
- Jiang, M.; Gold, M.S.; Boulay, G.; Spicher, K.; Peyton, M.; Brabet, P.; Srinivasan, Y.; Rudolph, U.; Ellison, G.; Birnbaumer, L. Multiple neurological abnormalities in mice deficient in the G protein Go. Proc. Natl. Acad. Sci. USA 1998, 95, 3269–3274. [Google Scholar] [CrossRef]
- Cha, H.L.; Choi, J.-M.; Oh, H.-H.; Bashyal, N.; Kim, S.-S.; Birnbaumer, L.; Suh-Kim, H. Deletion of the α subunit of the heterotrimeric Go protein impairs cerebellar cortical development in mice. Mol. Brain 2019, 12, 57. [Google Scholar] [CrossRef]
- Silachev, D.; Koval, A.; Savitsky, M.; Padmasola, G.; Quairiaux, C.; Thorel, F.; Katanaev, V.L. Mouse models characterize GNAO1 encephalopathy as a neurodevelopmental disorder leading to motor anomalies: From a severe G203R to a milder C215Y mutation. Acta Neuropathol. Commun. 2022, 10, 9. [Google Scholar] [CrossRef] [PubMed]
- Larasati, Y.A.; Savitsky, M.; Koval, A.; Solis, G.P.; Valnohova, J.; Katanaev, V.L. Restoration of the GTPase activity and cellular interactions of Gαo mutants by Zn2+ in GNAO1 encephalopathy models. Sci. Adv. 2022, 8, eabn9350. [Google Scholar] [CrossRef] [PubMed]
- Solis, G.P.; Koval, A.; Valnohova, J.; Savitsky, M.; Katanaev, V.L. Ric8 proteins as the neomorphic partners of Gαo in GNAO1 encephalopathies. bioRxiv 2023. [Google Scholar] [CrossRef]
- Savitsky, M.; Solis, G.P.; Kryuchkov, M.; Katanaev, V.L. Humanization of Drosophila Galphao to Model GNAO1 Paediatric Encephalopathies. Biomedicines 2020, 8, 395. [Google Scholar] [CrossRef] [PubMed]
- Członkowska, A.; Litwin, T.; Dusek, P.; Ferenci, P.; Lutsenko, S.; Medici, V.; Rybakowski, J.K.; Weiss, K.H.; Schilsky, M.L. Wilson disease. Nat. Rev. Dis. Primers 2018, 4, 21. [Google Scholar] [CrossRef] [PubMed]
- Grabrucker, A.M.; Rowan, M.; Garner, C.C. Brain-Delivery of Zinc-Ions as Potential Treatment for Neurological Diseases: Mini Review. Drug Deliv. Lett. 2011, 1, 13–23. [Google Scholar]
- Koval, A.; Larasati, Y.A.; Savitsky, M.; Solis, G.P.; Good, J.M.; Quinodoz, M.; Rivolta, C.; Superti-Furga, A.; Katanaev, V.L. In-depth molecular profiling of an intronic GNAO1 mutant as the basis for personalized high-throughput drug screening. Med 2023, 4, 311–325.e7. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Solis, G.P.; Kozhanova, T.V.; Koval, A.; Zhilina, S.S.; Mescheryakova, T.I.; Abramov, A.A.; Ishmuratov, E.V.; Bolshakova, E.S.; Osipova, K.V.; Ayvazyan, S.O.; et al. Pediatric Encephalopathy: Clinical, Biochemical and Cellular Insights into the Role of Gln52 of GNAO1 and GNAI1 for the Dominant Disease. Cells 2021, 10, 2749. [Google Scholar] [CrossRef]
- Lin, C.; Koval, A.; Tishchenko, S.; Gabdulkhakov, A.; Tin, U.; Solis, G.P.; Katanaev, V.L. Double suppression of the Galpha protein activity by RGS proteins. Mol. Cell 2014, 53, 663–671. [Google Scholar] [CrossRef]
- Solis, G.P.; Bilousov, O.; Koval, A.; Luchtenborg, A.M.; Lin, C.; Katanaev, V.L. Golgi-Resident Galphao Promotes Protrusive Membrane Dynamics. Cell 2017, 170, 939–955. [Google Scholar] [CrossRef] [PubMed]
- Katoh, Y.; Nozaki, S.; Hartanto, D.; Miyano, R.; Nakayama, K. Architectures of multisubunit complexes revealed by a visible immunoprecipitation assay using fluorescent fusion proteins. J. Cell Sci. 2015, 128, 2351–2362. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Thiel, M.; Bamborschke, D.; Janzarik, W.G.; Assmann, B.; Zittel, S.; Patzer, S.; Auhuber, A.; Opp, J.; Matzker, E.; Bevot, A.; et al. Genotype-phenotype correlation and treatment effects in young patients with GNAO1-associated disorders. J. Neurol. Neurosurg. Psychiatry 2023, 94, 806–815. [Google Scholar] [CrossRef] [PubMed]
- Kopein, D.; Katanaev, V.L. Drosophila GoLoco-protein pins is a target of Galpha(o)-mediated G protein-coupled receptor signaling. Mol. Biol. Cell 2009, 20, 3865–3877. [Google Scholar] [CrossRef]
- Egger-Adam, D.; Katanaev, V.L. The trimeric G protein Go inflicts a double impact on axin in the Wnt/frizzled signaling pathway. Dev. Dyn. 2010, 239, 168–183. [Google Scholar] [CrossRef]
- McEwen, D.P.; Gee, K.R.; Kang, H.C.; Neubig, R.R. Fluorescent BODIPY-GTP analogs: Real-time measurement of nucleotide binding to G proteins. Anal. Biochem. 2001, 291, 109–117. [Google Scholar] [CrossRef]
- Larrivee, C.L.; Feng, H.; Quinn, J.A.; Shaw, V.S.; Leipprandt, J.R.; Demireva, E.Y.; Xie, H.; Neubig, R.R. Mice with GNAO1 R209H Movement Disorder Variant Display Hyperlocomotion Alleviated by Risperidone. J. Pharmacol. Exp. Ther. 2020, 373, 24–33. [Google Scholar] [CrossRef]
- Jameson, E.E.; Roof, R.A.; Whorton, M.R.; Mosberg, H.I.; Sunahara, R.K.; Neubig, R.R.; Kennedy, R.T. Real-time detection of basal and stimulated G protein GTPase activity using fluorescent GTP analogues. J. Biol. Chem. 2005, 280, 7712–7719. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Han, Z.; Huang, G.; Li, R.; Liu, Y.; Lu, J.; Liu, L.; Miao, R. Structural comparison of unconventional G protein YchF with heterotrimeric G protein and small G protein. Plant Signal Behav. 2022, 17, 2024405. [Google Scholar] [CrossRef]
- Fu, Y.; Zhong, H.; Nanamori, M.; Mortensen, R.M.; Huang, X.; Lan, K.; Neubig, R.R. RGS-insensitive G-protein mutations to study the role of endogenous RGS proteins. Methods Enzymol. 2004, 389, 229–243. [Google Scholar] [PubMed]
- Solis, G.P.; Kazemzadeh, A.; Abrami, L.; Valnohova, J.; Alvarez, C.; van der Goot, F.G.; Katanaev, V.L. Local and substrate-specific S-palmitoylation determines subcellular localization of Gαo. Nat. Commun. 2022, 13, 2072. [Google Scholar] [CrossRef] [PubMed]
- Katanaev, V.L.; Valnohova, J.; Silachev, D.N.; Larasati, Y.; Koval, A. Pediatric GNAO1 encephalopathies: From molecular etiology of the disease to drug discovery. Neural Regen. Res. 2023, 18, 2188–2189. [Google Scholar] [CrossRef]
- Muntean, B.S.; Masuho, I.; Dao, M.; Sutton, L.P.; Zucca, S.; Iwamoto, H.; Patil, D.N.; Wang, D.; Birnbaumer, L.; Blakely, R.D.; et al. Gαo is a major determinant of cAMP signaling in the pathophysiology of movement disorders. Cell Rep. 2021, 34, 108718. [Google Scholar] [CrossRef]
- Higashijima, T.; Ferguson, K.M.; Sternweis, P.C.; Ross, E.M.; Smigel, M.D.; Gilman, A.G. The effect of activating ligands on the intrinsic fluorescence of guanine nucleotide-binding regulatory proteins. J. Biol. Chem. 1987, 262, 752–756. [Google Scholar] [CrossRef]
- Azpiazu, I.; Gautam, N. A fluorescence resonance energy transfer-based sensor indicates that receptor access to a G protein is unrestricted in a living mammalian cell. J. Biol. Chem. 2004, 279, 27709–27718. [Google Scholar] [CrossRef]
- Olsen, R.H.J.; DiBerto, J.F.; English, J.G.; Glaudin, A.M.; Krumm, B.E.; Slocum, S.T.; Che, T.; Gavin, A.C.; McCorvy, J.D.; Roth, B.L.; et al. TRUPATH, an open-source biosensor platform for interrogating the GPCR transducerome. Nat. Chem. Biol. 2020, 16, 841–849. [Google Scholar] [CrossRef]
- Schihada, H.; Shekhani, R.; Schulte, G. Quantitative assessment of constitutive G protein-coupled receptor activity with BRET-based G protein biosensors. Sci. Signal. 2021, 14, eabf1653. [Google Scholar] [CrossRef]
- Katanaev, V.L. Gln52 mutations in GNAO1-related disorders and personalized drug discovery. Epilepsy Behav. Rep. 2023. [Google Scholar] [CrossRef]
- Di Rocco, M.; Galosi, S.; Lanza, E.; Tosato, F.; Caprini, D.; Folli, V.; Friedman, J.; Bocchinfuso, G.; Martire, A.; Di Schiavi, E.; et al. Caenorhabditis elegans provides an efficient drug screening platform for GNAO1-related disorders and highlights the potential role of caffeine in controlling dyskinesia. Hum. Mol. Genet. 2022, 31, 929–941. [Google Scholar] [CrossRef] [PubMed]
- Di Rocco, M.; Galosi, S.; Follo, F.; Lanza, E.; Folli, V.; Martire, A.; Leuzzi, V.; Martinelli, S. Phenotypic Assessment of Pathogenic Variants in GNAO1 and Response to Caffeine in C. elegans Models of the Disease. Genes 2023, 14, 319. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, K.M.; Higashijima, T.; Smigel, M.D.; Gilman, A.G. The influence of bound GDP on the kinetics of guanine nucleotide binding to G proteins. J. Biol. Chem. 1986, 261, 7393–7399. [Google Scholar] [CrossRef] [PubMed]
- McEwen, D.P.; Gee, K.R.; Kang, H.C.; Neubig, R.R. Fluorescence approaches to study G protein mechanisms. Methods Enzymol. 2002, 344, 403–420. [Google Scholar]
- Katanaev, V.L.; Chornomorets, M. Kinetic diversity in G-protein-coupled receptor signalling. Biochem. J. 2007, 401, 485–495. [Google Scholar] [CrossRef]
- Connors, K.A. Chemical Kinetics: The Study of Reaction Rates in Solution; VCA Publishers, Inc.: New York, NY, USA, 1990; p. 480. [Google Scholar]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Larasati, Y.A.; Solis, G.P.; Koval, A.; Griffiths, S.T.; Berentsen, R.; Aukrust, I.; Lesca, G.; Chatron, N.; Ville, D.; Korff, C.M.; et al. Clinical Cases and the Molecular Profiling of a Novel Childhood Encephalopathy-Causing GNAO1 Mutation P170R. Cells 2023, 12, 2469. https://doi.org/10.3390/cells12202469
Larasati YA, Solis GP, Koval A, Griffiths ST, Berentsen R, Aukrust I, Lesca G, Chatron N, Ville D, Korff CM, et al. Clinical Cases and the Molecular Profiling of a Novel Childhood Encephalopathy-Causing GNAO1 Mutation P170R. Cells. 2023; 12(20):2469. https://doi.org/10.3390/cells12202469
Chicago/Turabian StyleLarasati, Yonika A., Gonzalo P. Solis, Alexey Koval, Silja T. Griffiths, Ragnhild Berentsen, Ingvild Aukrust, Gaetan Lesca, Nicolas Chatron, Dorothée Ville, Christian M. Korff, and et al. 2023. "Clinical Cases and the Molecular Profiling of a Novel Childhood Encephalopathy-Causing GNAO1 Mutation P170R" Cells 12, no. 20: 2469. https://doi.org/10.3390/cells12202469
APA StyleLarasati, Y. A., Solis, G. P., Koval, A., Griffiths, S. T., Berentsen, R., Aukrust, I., Lesca, G., Chatron, N., Ville, D., Korff, C. M., & Katanaev, V. L. (2023). Clinical Cases and the Molecular Profiling of a Novel Childhood Encephalopathy-Causing GNAO1 Mutation P170R. Cells, 12(20), 2469. https://doi.org/10.3390/cells12202469