

Mitochondrial Dysfunction and Decreased Cytochrome c in Cell and Animal Models of Machado–Joseph Disease

 , , ,
, , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Cell Culture
2.2.2. Animals
2.2.3. Isolation of Neuronal and Non-Neuronal Mitochondria
2.2.4. Sample Preparation and Western Blotting
Total Fractions
Mitochondrial and Cytoplasmic-Enriched Fractions
Preparation of Mitochondrial Extracts from Isolated Mitochondria
Western Blotting
2.2.5. Measurement of O2 Consumption Using Seahorse Analyzer
2.2.6. Measurement of Total Levels of Adenine Nucleotides
2.2.7. Mitochondrial Membrane Potential
2.2.8. Mitochondrial Ca2+ Uptake
2.2.9. Intracellular Ca2+ Recordings
2.2.10. H2O2 Levels
2.2.11. Analysis of mRNA Levels
- Cyt c—F: CCAAATCTCCACGGTCTGTTC; R: ATCAGGGTATCCTCTCCCCAG.
- Ucp2—F: AGCCCACGGATGTGGTAAAG; R: CTCTCGGGCAATGGTCTTGT.
- Ucp4—F: CCTGGACACCTCCAATCCAC; R: TCCTGACCTGACCTCTCTCG.
- Ucp5—F: GTAAGCGGACATCAGAAAAGTTCC; R: GCCGAACTCGGCAACAATAG.
- β-2M—F: CCTTCAGCAAGGACTGGTCT; R: TCTCGATCCCAGTAGACGGT.
2.2.12. Transmission Electron Microscopy (TEM) Analysis
2.2.13. Analysis of mtDNA Copy Number
- mtDNA—F: CGACCTCGATGTTGGATCA; R: AGAGGATTTGAACCTCTGG.
- CCP2—F: CCAAATCTCCACGGTCTGTTC; R: ATCAGGGTATCCTCTCCCCAG.
2.2.14. Statistical Analysis
3. Results
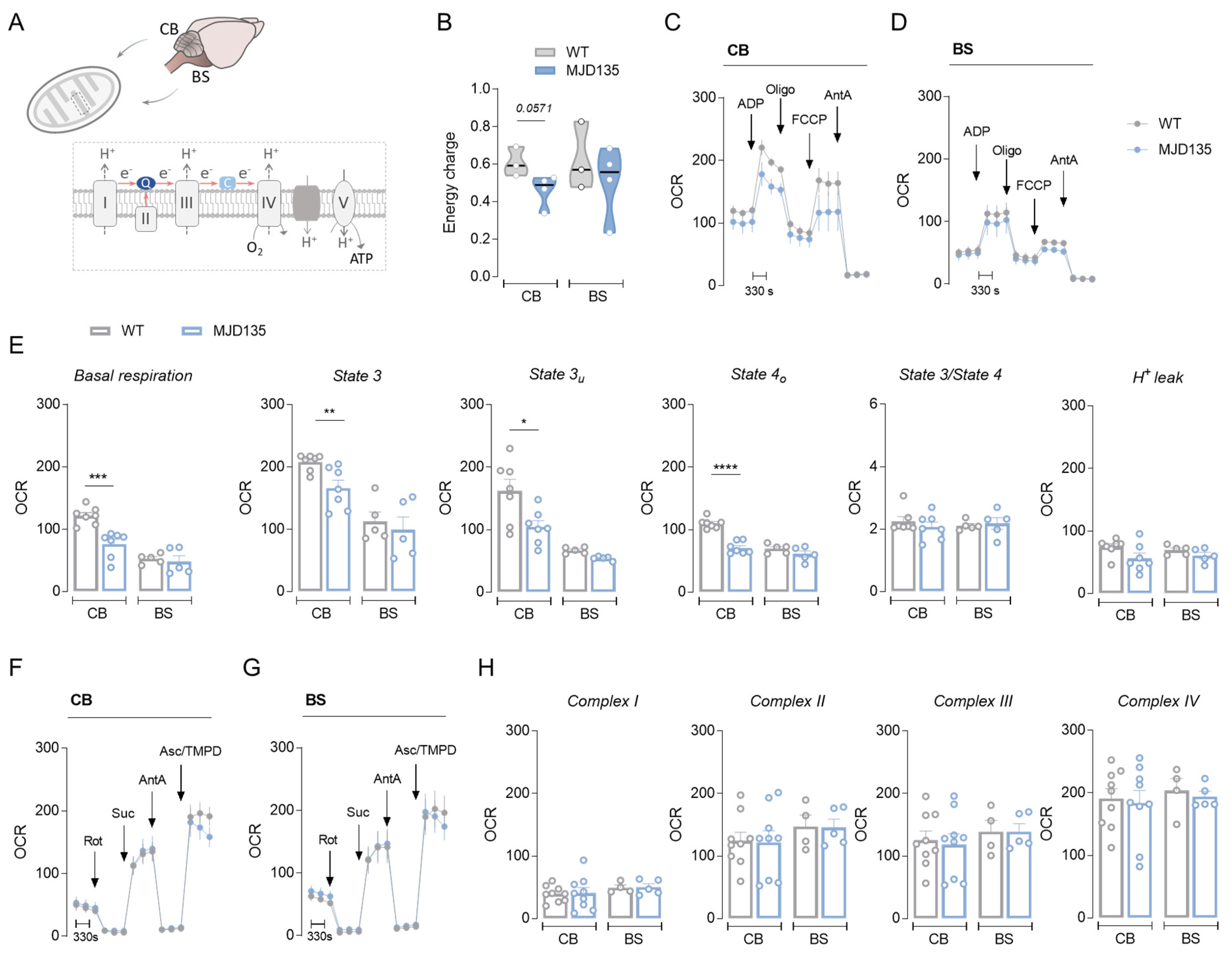
3.1. MJD135 Mice Exhibit Decreased Cerebellar Mitochondrial Respiration and Reduced ATP Production
3.2. PC6-3 Q108 Cells Show Decreased Mitochondrial Respiration and ATP Production and Enhanced Glycolysis Capacity
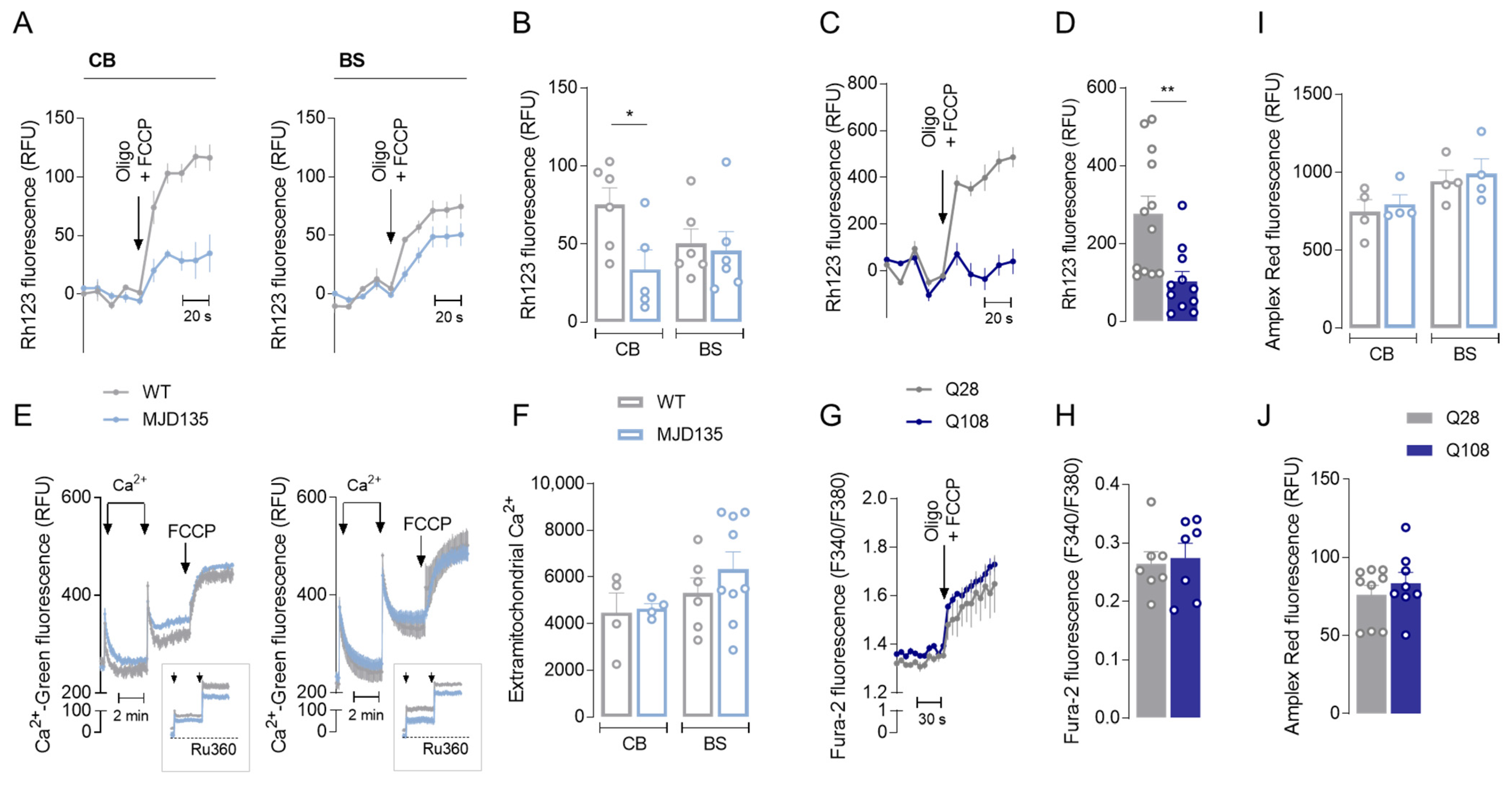
3.3. MJD135 Mouse Cerebellum and PC6-3 Q108 Cells Display Decreased Mitochondrial Membrane Potential (ΔΨm)
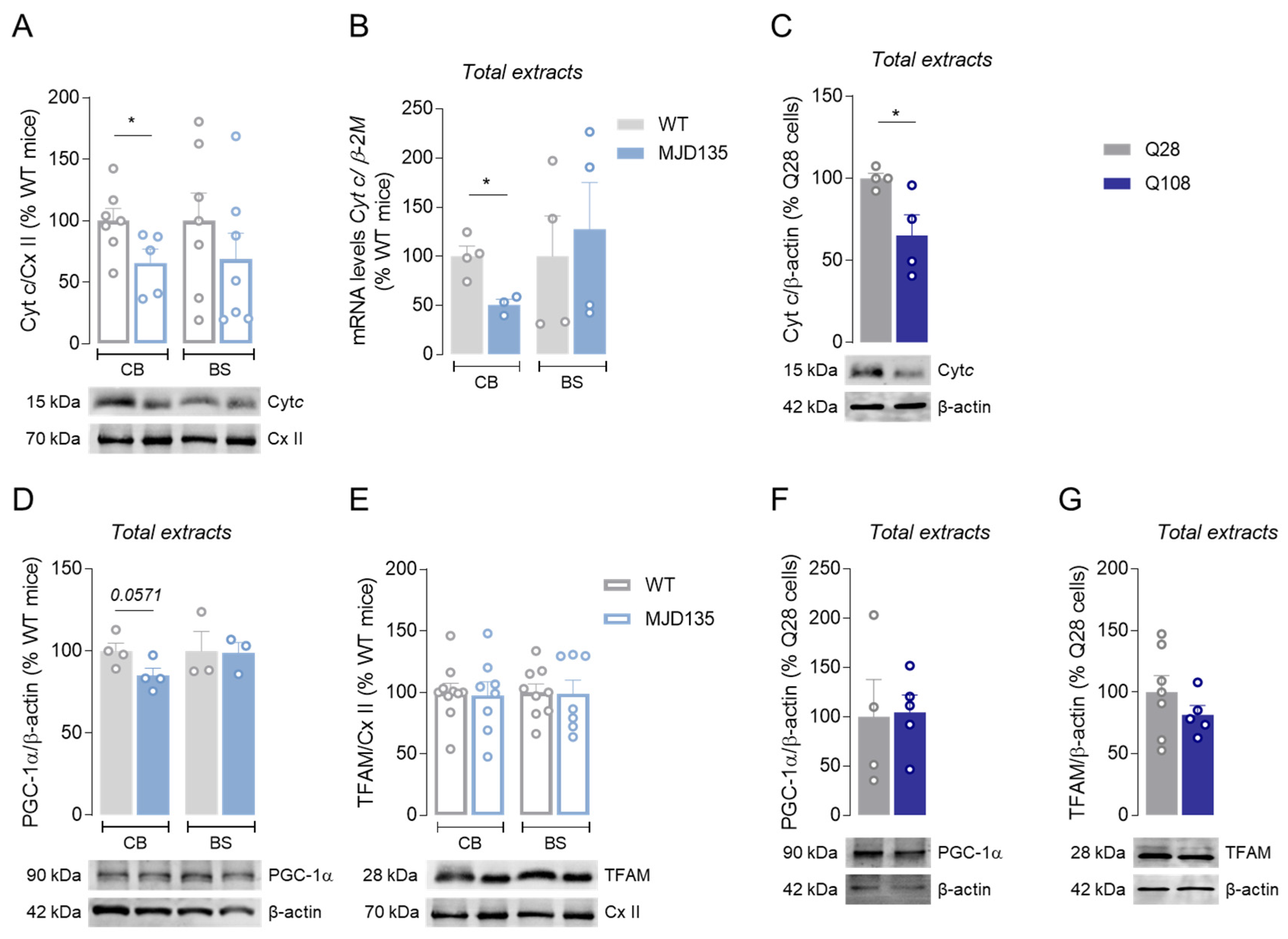
3.4. Decreased Cytochrome c in Cerebellum of MJD135 Mice and PC6-3 Q108 Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sequeiros, J.; Coutinho, P. Epidemiology and clinical aspects of Machado-Joseph disease. Adv. Neurol. 1993, 61, 139–153. [Google Scholar]
- Ruano, L.; Melo, C.; Silva, M.C.; Coutinho, P. The Global Epidemiology of Hereditary Ataxia and Spastic Paraplegia: A Systematic Review of Prevalence Studies. Neuroepidemiology 2014, 42, 174–183. [Google Scholar] [CrossRef]
- Bettencourt, C.; Santos, C.; Kay, T.; Vasconcelos, J.; Lima, M. Analysis of segregation patterns in Machado–Joseph disease pedigrees. J. Hum. Genet. 2008, 53, 920–923. [Google Scholar] [CrossRef]
- Matos, C.A.; Pereira de Almeida, L.; Nóbrega, C. Machado–Joseph disease/spinocerebellar ataxia type 3: Lessons from disease pathogenesis and clues into therapy. J. Neurochem. 2019, 148, 8–28. [Google Scholar] [CrossRef]
- Chen, Y.-S.; Hong, Z.-X.; Lin, S.-Z.; Harn, H.-J. Identifying Therapeutic Targets for Spinocerebellar Ataxia Type 3/Machado–Joseph Disease through Integration of Pathological Biomarkers and Therapeutic Strategies. Int. J. Mol. Sci. 2020, 21, 3063. [Google Scholar] [CrossRef]
- Naia, L.; Ferreira, I.L.; Ferreiro, E.; Rego, A.C. Mitochondrial Ca2+ handling in Huntington’s and Alzheimer’s diseases-Role of ER-mitochondria crosstalk. Biochem. Biophys. Res. Commun. 2017, 483, 1069–1077. [Google Scholar] [CrossRef]
- Laço, M.N.; Oliveira, C.R.; Paulson, H.L.; Rego, A.C. Compromised mitochondrial complex II in models of Machado–Joseph disease. Biochim. Biophys. Acta 2012, 1822, 139–149. [Google Scholar] [CrossRef]
- Harmuth, T.; Prell-Schicker, C.; Weber, J.J.; Gellerich, F.; Funke, C.; Drießen, S.; Magg, J.C.D.; Krebiehl, G.; Wolburg, H.; Hayer, S.N.; et al. Mitochondrial Morphology, Function and Homeostasis Are Impaired by Expression of an N-terminal Calpain Cleavage Fragment of Ataxin-3. Front. Mol. Neurosci. 2018, 11, 368. [Google Scholar] [CrossRef]
- Noronha, C.; Perfeito, R.; Laço, M.; Wüllner, U.; Rego, A.C. Expanded and Wild-type Ataxin-3 Modify the Redox Status of SH-SY5Y Cells Overexpressing α-Synuclein. Neurochem. Res. 2017, 42, 1430–1437. [Google Scholar] [CrossRef]
- Yu, Y.-C.; Kuo, C.-L.; Cheng, W.-L.; Liu, C.-S.; Hsieh, M. Decreased antioxidant enzyme activity and increased mitochondrial DNA damage in cellular models of Machado-Joseph disease. J. Neurosci. Res. 2009, 87, 1884–1891. [Google Scholar] [CrossRef]
- Kazachkova, N.; Raposo, M.; Montiel, R.; Cymbron, T.; Bettencourt, C.; Silva-Fernandes, A.; Duarte-Silva, S.; Maciel, P.; Lima, M. Patterns of Mitochondrial DNA Damage in Blood and Brain Tissues of a Transgenic Mouse Model of Machado-Joseph Disease. Neurodegener. Dis. 2013, 11, 206–214. [Google Scholar] [CrossRef]
- Ramos, A.; Kazachkova, N.; Silva, F.; Maciel, P.; Silva-Fernandes, A.; Duarte-Silva, S.; Santos, C.; Lima, M. Differential mtDNA Damage Patterns in a Transgenic Mouse Model of Machado–Joseph Disease (MJD/SCA3). J. Mol. Neurosci. 2015, 55, 449–453. [Google Scholar] [CrossRef]
- Kristensen, L.V.; Oppermann, F.S.; Rauen, M.J.; Fog, K.; Schmidt, T.; Schmidt, J.; Harmuth, T.; Hartmann-Petersen, R.; Thirstrup, K. Mass spectrometry analyses of normal and polyglutamine expanded ataxin-3 reveal novel interaction partners involved in mitochondrial function. Neurochem. Int. 2018, 112, 5–17. [Google Scholar] [CrossRef]
- Chang, J.-C.; Wu, S.-L.; Hoel, F.; Cheng, Y.-S.; Liu, K.-H.; Hsieh, M.; Hoel, A.; Tronstad, K.J.; Yan, K.-C.; Hsieh, C.-L.; et al. Far-infrared radiation protects viability in a cell model of Spinocerebellar Ataxia by preventing polyQ protein accumulation and improving mitochondrial function. Sci. Rep. 2016, 6, 30436. [Google Scholar] [CrossRef] [PubMed]
- Duarte-Silva, S.; Neves-Carvalho, A.; Soares-Cunha, C.; Silva, J.M.; Teixeira-Castro, A.; Vieira, R.; Silva-Fernandes, A.; Maciel, P. Neuroprotective Effects of Creatine in the CMVMJD135 Mouse Model of Spinocerebellar Ataxia Type 3. Mov. Disord. 2018, 33, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Wiatr, K.; Marczak, Ł.; Pérot, J.-B.; Brouillet, E.; Flament, J.; Figiel, M. Broad Influence of Mutant Ataxin-3 on the Proteome of the Adult Brain, Young Neurons, and Axons Reveals Central Molecular Processes and Biomarkers in SCA3/MJD Using Knock-In Mouse Model. Front. Mol. Neurosci. 2021, 14, 658339. [Google Scholar] [CrossRef]
- Harmuth, T.; Weber, J.J.; Zimmer, A.J.; Sowa, A.S.; Schmidt, J.; Fitzgerald, J.C.; Schöls, L.; Riess, O.; Hübener-Schmid, J. Mitochondrial Dysfunction in Spinocerebellar Ataxia Type 3 Is Linked to VDAC1 Deubiquitination. Int. J. Mol. Sci. 2022, 23, 5933. [Google Scholar] [CrossRef]
- Pittman, R.N.; Wang, S.; DiBenedetto, A.; Mills, J. A system for characterizing cellular and molecular events in programmed neuronal cell death. J. Neurosci. 1993, 13, 3669–3680. [Google Scholar] [CrossRef] [PubMed]
- Silva-Fernandes, A.; Duarte-Silva, S.; Neves-Carvalho, A.; Amorim, M.; Soares-Cunha, C.; Oliveira, P.; Thirstrup, K.; Teixeira-Castro, A.; Maciel, P. Chronic Treatment with 17-DMAG Improves Balance and Coordination in A New Mouse Model of Machado-Joseph Disease. Neurotherapeutics 2014, 11, 433–449. [Google Scholar] [CrossRef]
- Silva-Fernandes, A.; Costa, M.D.C.; Duarte-Silva, S.; Oliveira, P.; Botelho, C.M.; Martins, L.; Mariz, J.A.; Ferreira, T.; Ribeiro, F.; Correia-Neves, M.; et al. Motor uncoordination and neuropathology in a transgenic mouse model of Machado–Joseph disease lacking intranuclear inclusions and ataxin-3 cleavage products. Neurobiol. Dis. 2010, 40, 163–176. [Google Scholar] [CrossRef]
- Ferreira, I.L.; Carmo, C.; Naia, L.; Mota, S.I.; Cristina Rego, A. Assessing Mitochondrial Function in In Vitro and Ex Vivo Models of Huntington’s Disease. In Huntington’s Disease; Precious, S.V., Rosser, A.E., Dunnett, S.B., Eds.; Springer: New York, NY, USA, 2018; pp. 415–442. [Google Scholar]
- TeSlaa, T.; Teitell, M.A. Techniques to Monitor Glycolysis. Methods Enzymol. 2014, 542, 91–114. [Google Scholar] [CrossRef] [PubMed]
- Stocchi, V.; Cucchiarini, L.; Magnani, M.; Chiarantini, L.; Palma, P.; Crescentini, G. Simultaneous extraction and reverse-phase high-performance liquid chromatographic determination of adenine and pyridine nucleotides in human red blood cells. Anal. Biochem. 1985, 146, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Pellman, J.J.; Hamilton, J.; Brustovetsky, T.; Brustovetsky, N. Ca2+ handling in isolated brain mitochondria and cultured neurons derived from the YAC128 mouse model of Huntington’s disease. J. Neurochem. 2015, 134, 652–667. [Google Scholar] [CrossRef] [PubMed]
- De Munter, S.; Verheijden, S.; Vanderstuyft, E.; Malheiro, A.R.; Brites, P.; Gall, D.; Schiffmann, S.N.; Baes, M. Early-onset Purkinje cell dysfunction underlies cerebellar ataxia in peroxisomal multifunctional protein-2 deficiency. Neurobiol. Dis. 2016, 94, 157–168. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Panov, A.V.; Burke, J.R.; Strittmatter, W.J.; Greenamyre, J.T. In vitro effects of polyglutamine tracts on Ca2+-dependent depolarization of rat and human mitochondria: Relevance to Huntington’s disease. Arch. Biochem. Biophys. 2003, 410, 1–6. [Google Scholar] [CrossRef]
- Jastroch, M.; Divakaruni, A.S.; Mookerjee, S.; Treberg, J.R.; Brand, M.D. Mitochondrial proton and electron leaks. Essays Biochem. 2010, 47, 53–67. [Google Scholar] [CrossRef]
- Lengacher, S.; Magistretti, P.J.; Pellerin, L. Quantitative RT-PCR Analysis of Uncoupling Protein Isoforms in Mouse Brain Cortex: Methodological Optimization and Comparison of Expression with Brown Adipose Tissue and Skeletal Muscle. J. Cereb. Blood Flow Metab. 2004, 24, 780–788. [Google Scholar] [CrossRef]
- Trottier, Y.; Cancel, G.; An-Gourfinkel, I.; Lutz, Y.; Weber, C.; Brice, A.; Hirsch, E.; Mandel, J.L. Heterogeneous intracellular localization and expression of ataxin-3. Neurobiol. Dis. 1998, 5, 335–347. [Google Scholar] [CrossRef]
- Hroudová, J.; Fišar, Z. Control mechanisms in mitochondrial oxidative phosphorylation. Neural. Regen. Res. 2013, 8, 363–375. [Google Scholar] [CrossRef]
- Rich, P.R.; Maréchal, A. The mitochondrial respiratory chain. Essays Biochem. 2010, 47, 1–23. [Google Scholar] [PubMed]
- Lopes, C.; Ferreira, I.L.; Maranga, C.; Beatriz, M.; Mota, S.I.; Sereno, J.; Castelhano, J.; Abrunhosa, A.; Oliveira, F.; De Rosa, M.; et al. Mitochondrial and redox modifications in early stages of Huntington’s disease. Redox Biol. 2022, 56, 102424. [Google Scholar] [CrossRef] [PubMed]
- Austin, S.; St-Pierre, J. PGC1α and mitochondrial metabolism–emerging concepts and relevance in ageing and neurodegenerative disorders. J. Cell Sci. 2012, 125(Pt21), 4963–4971. [Google Scholar] [CrossRef] [PubMed]
- Mudò, G.; Mäkelä, J.; Di Liberto, V.; Tselykh, T.V.; Olivieri, M.; Piepponen, P.; Eriksson, O.; Mälkiä, A.; Bonomo, A.; Kairisalo, M.; et al. Transgenic expression and activation of PGC-1α protect dopaminergic neurons in the MPTP mouse model of Parkinson’s disease. Cell. Mol. Life Sci. 2011, 69, 1153–1165. [Google Scholar] [CrossRef]
- Mäkelä, J.; Mudò, G.; Pham, D.D.; Di Liberto, V.; Eriksson, O.; Louhivuori, L.; Bruelle, C.; Soliymani, R.; Baumann, M.; Korhonen, L.; et al. Peroxisome proliferator-activated receptor-γ coactivator-1α mediates neuroprotection against excitotoxic brain injury in transgenic mice: Role of mitochondria and X-linked inhibitor of apoptosis protein. Eur. J. Neurosci. 2016, 43, 626–639. [Google Scholar] [CrossRef]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jäger, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of Reactive Oxygen Species and Neurodegeneration by the PGC-1 Transcriptional Coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef]
- Kaufman, B.A.; Durisic, N.; Mativetsky, J.M.; Costantino, S.; Hancock, M.A.; Grutter, P.; Shoubridge, E.A. The Mitochondrial Transcription Factor TFAM Coordinates the Assembly of Multiple DNA Molecules into Nucleoid-like Structures. Mol. Biol. Cell 2007, 18, 3225–3236. [Google Scholar] [CrossRef]
- Cui, L.; Jeong, H.; Borovecki, F.; Parkhurst, C.N.; Tanese, N.; Krainc, D. Transcriptional Repression of PGC-1alpha by Mutant Huntingtin Leads to Mitochondrial Dysfunction and Neurodegeneration. Cell 2006, 127, 59–69. [Google Scholar] [CrossRef]
- Kim, J.; Moody, J.P.; Edgerly, C.K.; Bordiuk, O.L.; Cormier, K.; Smith, K.; Flint Beal, M.; Ferrante, R.J. Mitochondrial loss, dysfunction and altered dynamics in Huntington’s disease. Hum. Mol. Genet. 2010, 19, 3919–3935. [Google Scholar] [CrossRef]
- Nicholls, D.G.; Ward, M.W. Mitochondrial membrane potential and neuronal glutamate excitotoxicity: Mortality and millivolts. Trends Neurosci. 2000, 23, 166–174. [Google Scholar] [CrossRef]
- Perry, S.W.; Norman, J.P.; Barbieri, J.; Brown, E.B.; Gelbard, H.A. Mitochondrial membrane potential probes and the proton gradient: A practical usage guide. BioTechniques 2011, 50, 98–115. [Google Scholar] [CrossRef] [PubMed]
- Jeub, M.; Herbst, M.; Spauschus, A.; Fleischer, H.; Klockgether, T.; Wuellner, U.; Evert, B.O. Potassium channel dysfunction and depolarized resting membrane potential in a cell model of SCA3. Exp. Neurol. 2006, 201, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Tang, T.-S.; Tu, H.; Nelson, O.; Pook, M.; Hammer, R.; Nukina, N.; Bezprozvanny, I. Deranged Calcium Signaling and Neurodegeneration in Spinocerebellar Ataxia Type 3. J. Neurosci. 2008, 28, 12713–12724. [Google Scholar] [CrossRef]
- Chou, A.-H.; Chen, C.-Y.; Chen, S.-Y.; Chen, W.-J.; Chen, Y.-L.; Weng, Y.-S.; Wang, H.-L. Polyglutamine-expanded ataxin-7 causes cerebellar dysfunction by inducing transcriptional dysregulation. Neurochem. Int. 2010, 56, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.-Y.; Jhang, Y.-L.; Cheng, P.-H.; Chang, Y.-F.; Mao, S.-H.; Yang, H.-I.; Lin, C.-W.; Chen, C.-M.; Yang, S.-H. The Truncated C-terminal Fragment of Mutant ATXN3 Disrupts Mitochondria Dynamics in Spinocerebellar Ataxia Type 3 Models. Front. Mol. Neurosci. 2017, 10, 196. [Google Scholar] [CrossRef] [PubMed]
- Naia, L.; Ly, P.; Mota, S.I.; Lopes, C.; Maranga, C.; Coelho, P.; Gershoni-Emek, N.; Ankarcrona, M.; Geva, M.; Hayden, M.R.; et al. The Sigma-1 Receptor Mediates Pridopidine Rescue of Mitochondrial Function in Huntington Disease Models. Neurotherapeutics 2021, 18, 1017–1038. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almeida, F.; Ferreira, I.L.; Naia, L.; Marinho, D.; Vilaça-Ferreira, A.C.; Costa, M.D.; Duarte-Silva, S.; Maciel, P.; Rego, A.C. Mitochondrial Dysfunction and Decreased Cytochrome c in Cell and Animal Models of Machado–Joseph Disease. Cells 2023, 12, 2397. https://doi.org/10.3390/cells12192397
Almeida F, Ferreira IL, Naia L, Marinho D, Vilaça-Ferreira AC, Costa MD, Duarte-Silva S, Maciel P, Rego AC. Mitochondrial Dysfunction and Decreased Cytochrome c in Cell and Animal Models of Machado–Joseph Disease. Cells. 2023; 12(19):2397. https://doi.org/10.3390/cells12192397
Chicago/Turabian StyleAlmeida, Filipa, Ildete L. Ferreira, Luana Naia, Daniela Marinho, Ana Catarina Vilaça-Ferreira, Marta D. Costa, Sara Duarte-Silva, Patrícia Maciel, and A. Cristina Rego. 2023. "Mitochondrial Dysfunction and Decreased Cytochrome c in Cell and Animal Models of Machado–Joseph Disease" Cells 12, no. 19: 2397. https://doi.org/10.3390/cells12192397
APA StyleAlmeida, F., Ferreira, I. L., Naia, L., Marinho, D., Vilaça-Ferreira, A. C., Costa, M. D., Duarte-Silva, S., Maciel, P., & Rego, A. C. (2023). Mitochondrial Dysfunction and Decreased Cytochrome c in Cell and Animal Models of Machado–Joseph Disease. Cells, 12(19), 2397. https://doi.org/10.3390/cells12192397