
NAD+ Acts as a Protective Factor in Cellular Stress Response to DNA Alkylating Agents

 , , and
, , and 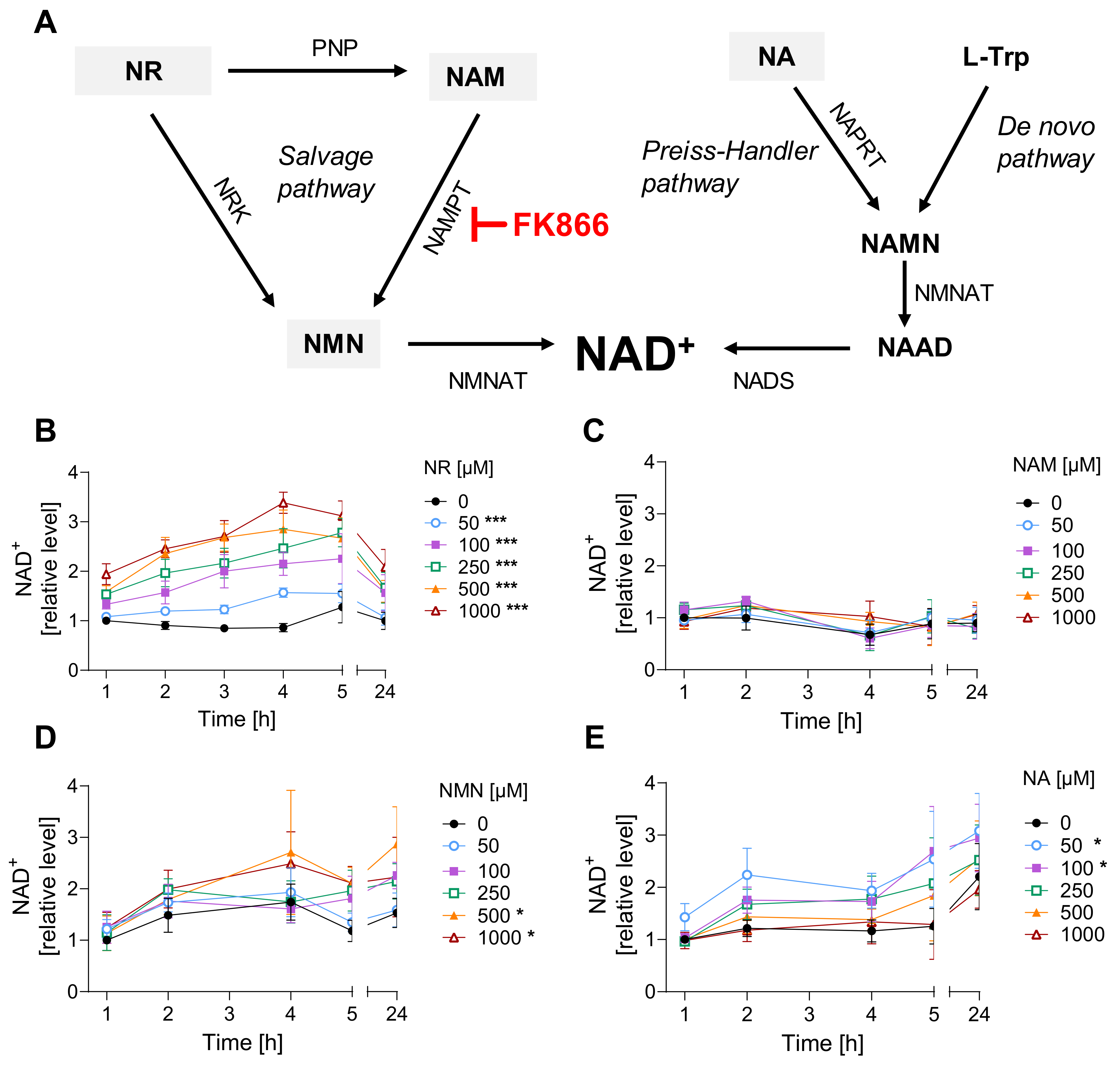{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Treatment with Chemicals
2.3. Cellular NAD+ Levels
2.4. Cellular ATP Levels
2.5. Annexin V/propidium Iodide Staining
2.6. AlamarBlue Viability Assay
2.7. Clonogenic Survival Assay
2.8. DNA Damage
2.9. PAR Formation
2.10. Statistical Analysis
3. Results
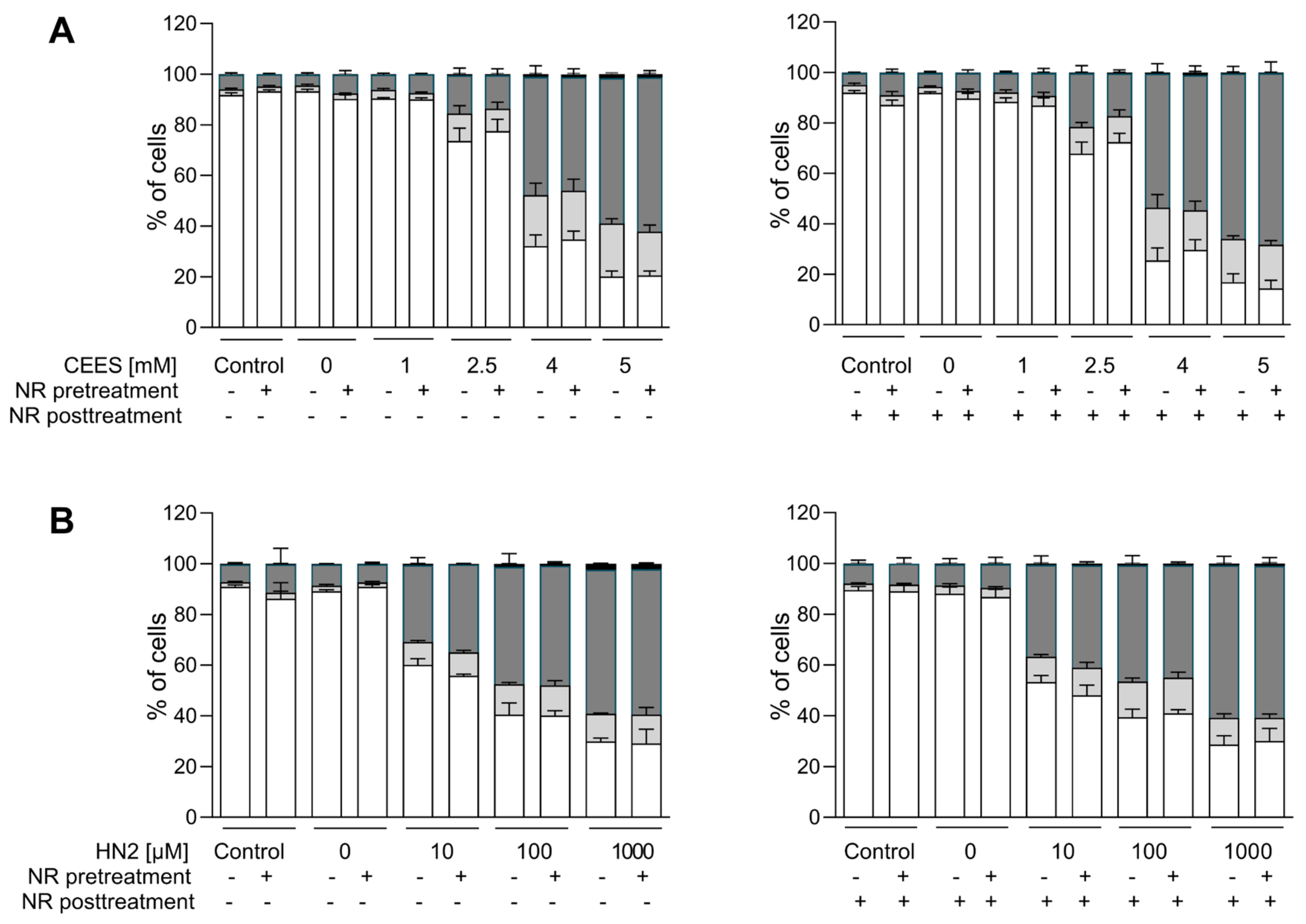
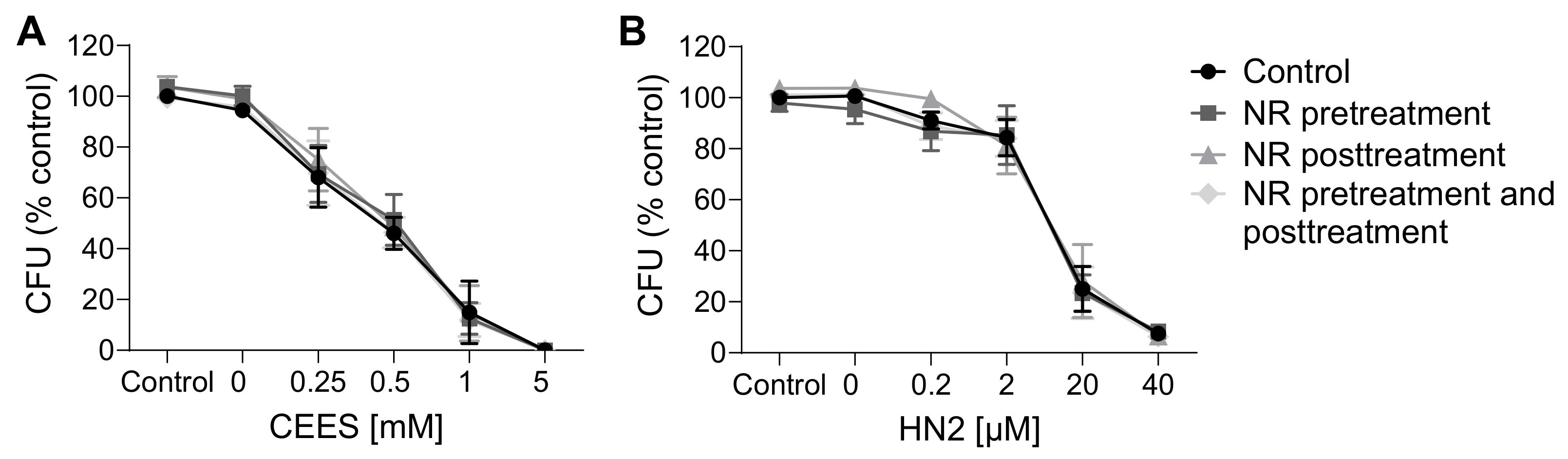
3.1. NR Does Not Affect Cellular Response to Genotoxic Stress in HaCaT
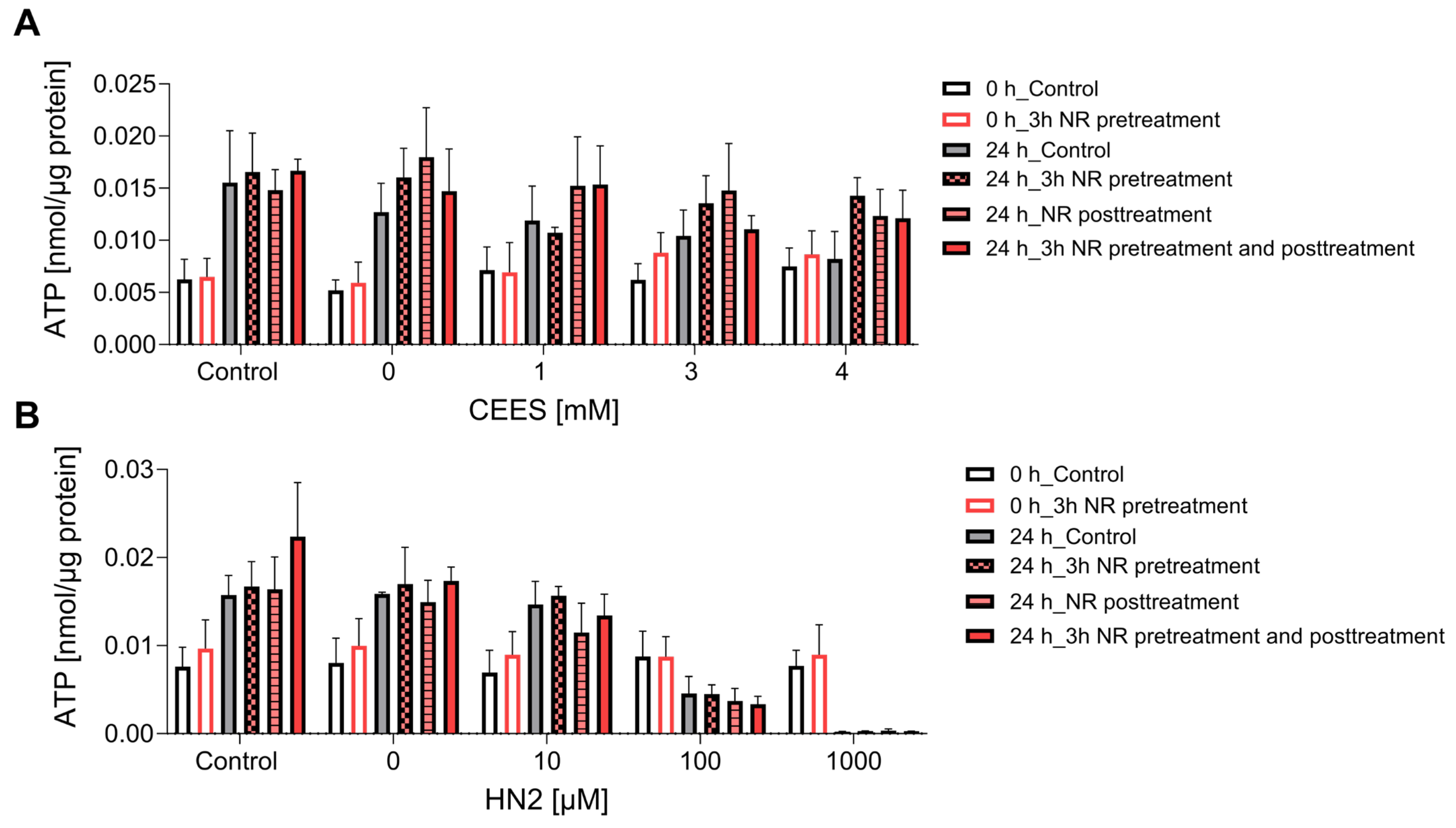
3.2. NR Does Not Affect Short- or Long-Term Cytotoxicity in HaCaT Cells
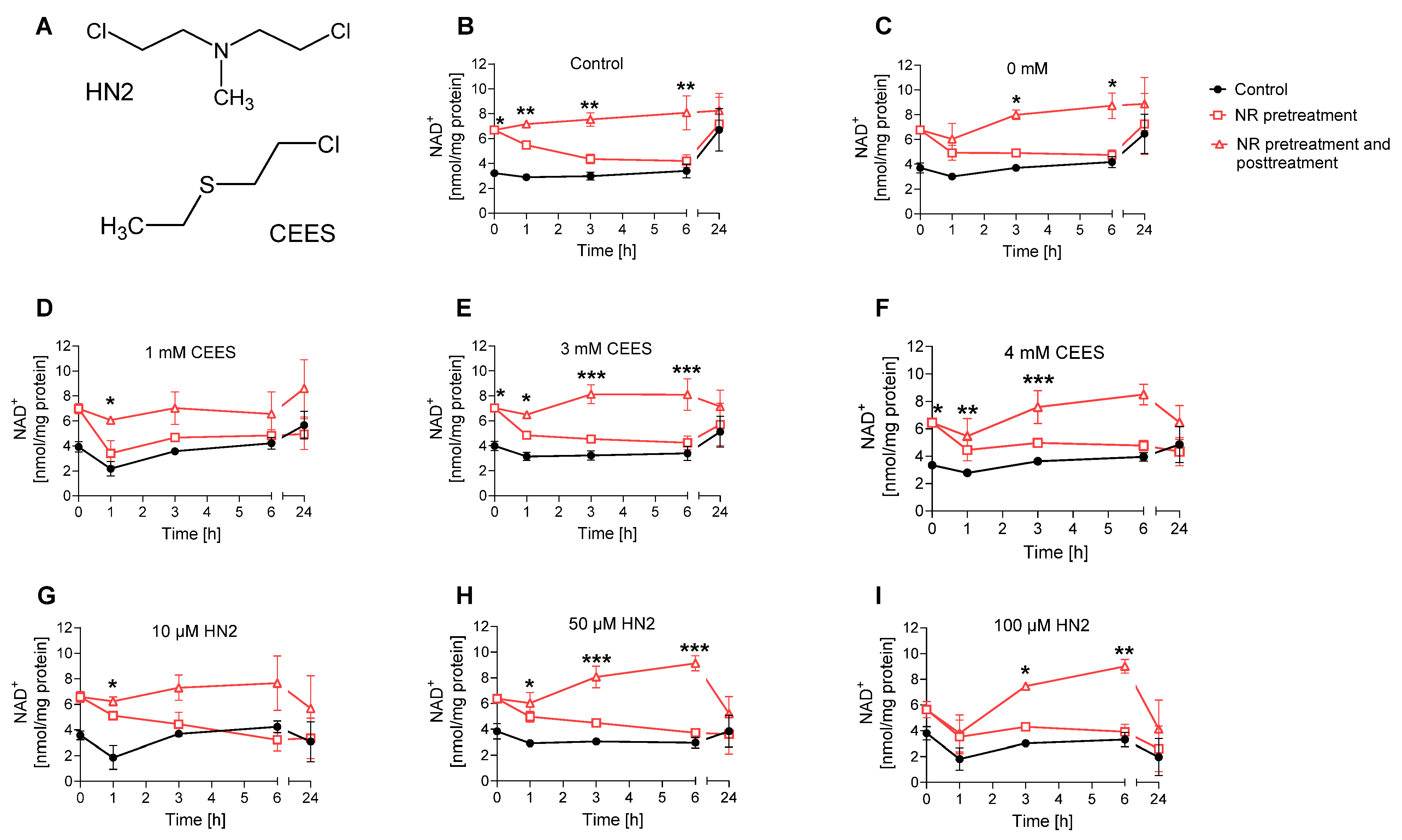
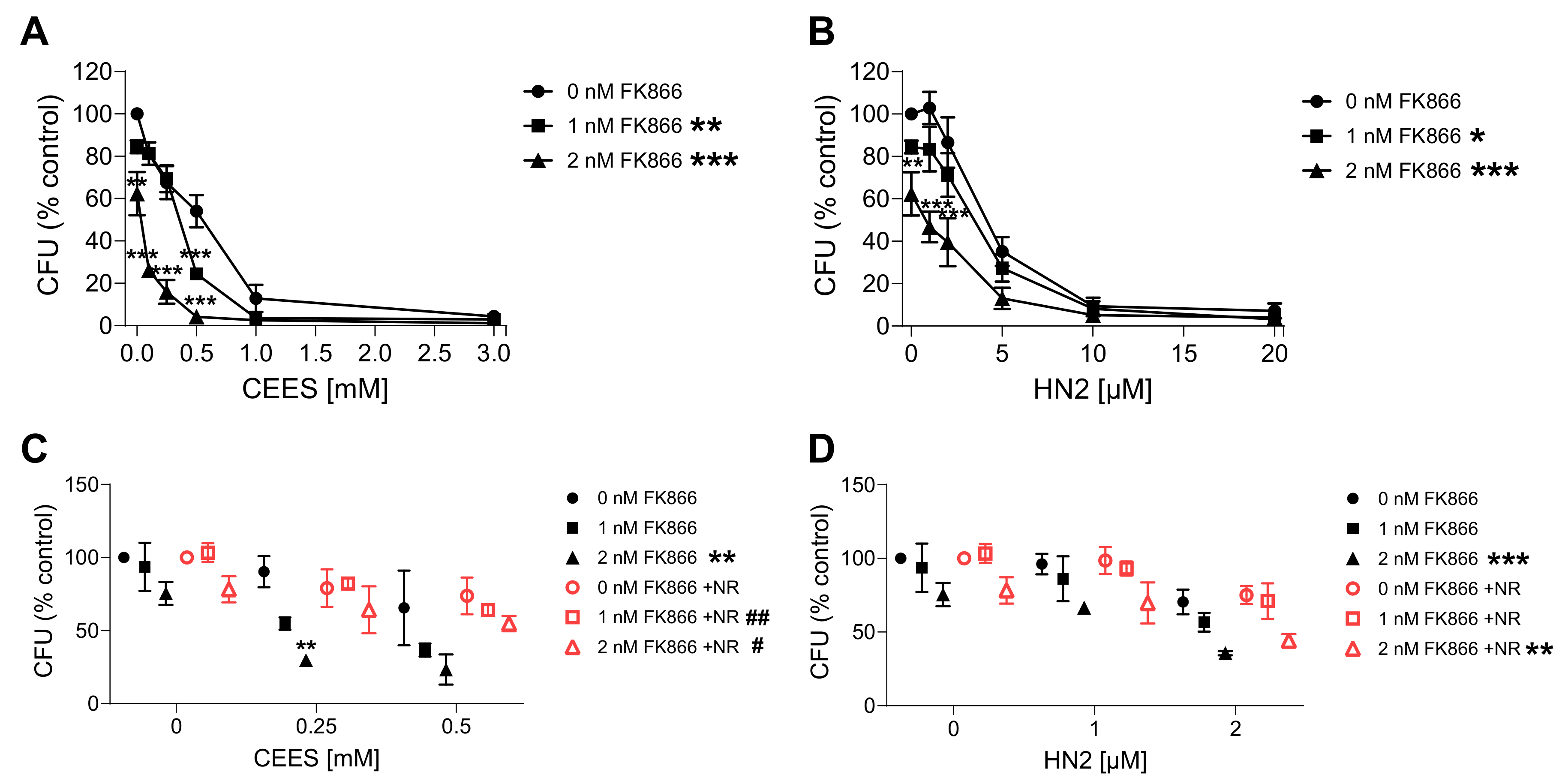
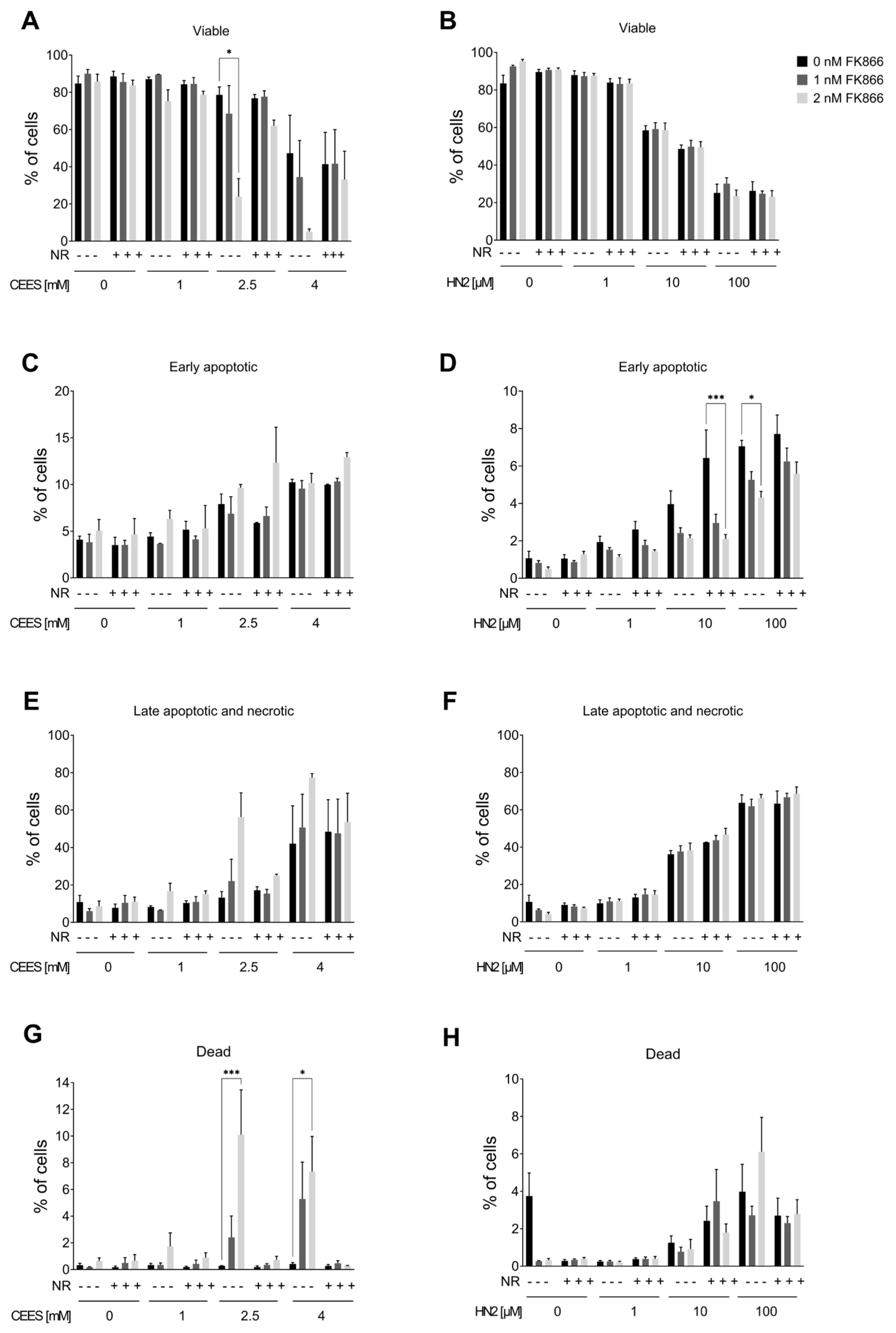
3.3. NAD+ Depletion Sensitizes Cells to Genotoxic Stress in a Compound-Specific Manner
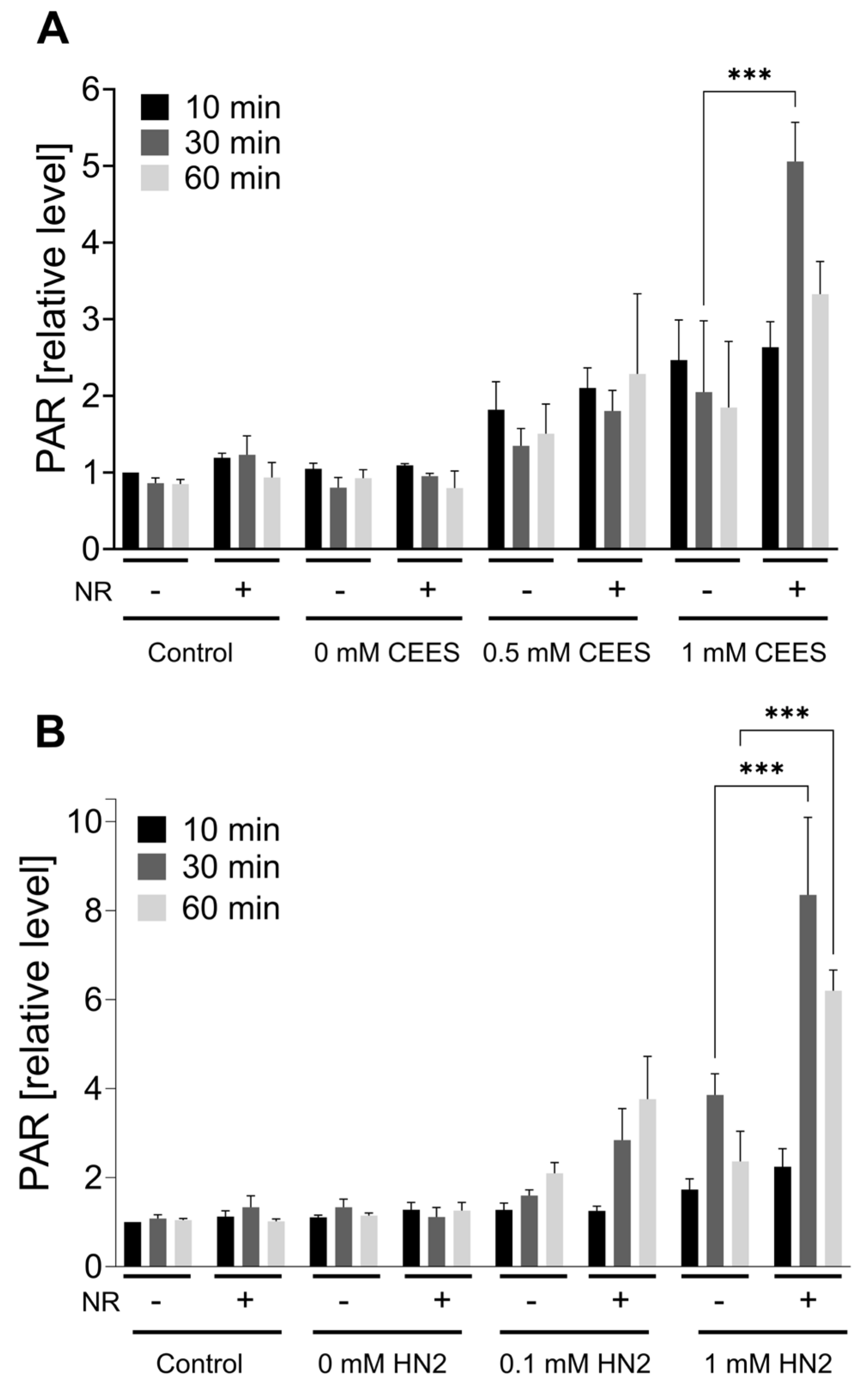
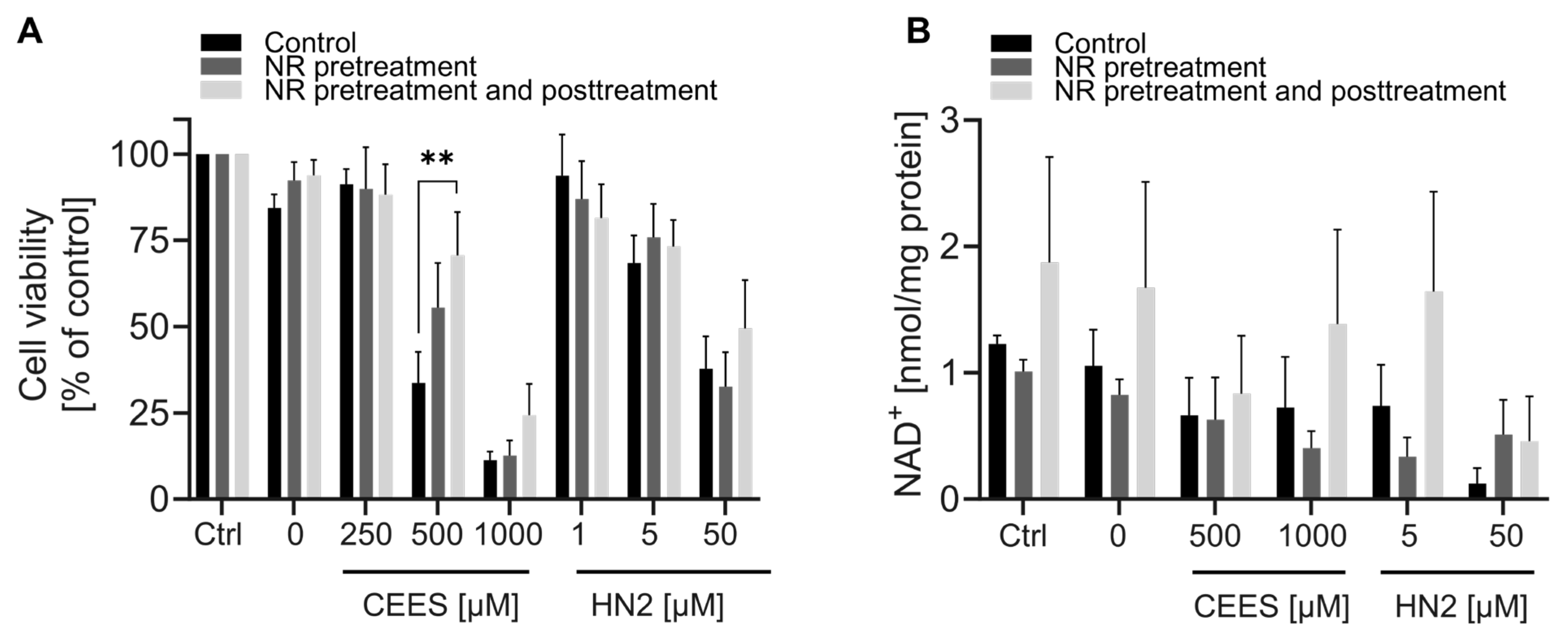
3.4. NR Attenuates THP-1 Susceptibility to Mustards
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mangerich, A.; Esser, C. Chemical warfare in the First World War: Reflections 100 years later. Arch. Toxicol. 2014, 88, 1909–1911. [Google Scholar] [CrossRef] [PubMed]
- Panahi, Y.; Abdolghaffari, A.H.; Sahebkar, A. A review on symptoms, treatments protocols, and proteomic profile in sulfur mustard-exposed victims. J. Cell. Biochem. 2018, 119, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, S.; Abdollahi, M. Novel treatment opportunities for sulfur mustard-related cancers: Genetic and epigenetic perspectives. Arch. Toxicol. 2017, 91, 3717–3735. [Google Scholar] [CrossRef] [PubMed]
- Etemad, L.; Moshiri, M.; Balali-Mood, M. Advances in treatment of acute sulfur mustard poisoning—A critical review. Crit. Rev. Toxicol. 2019, 49, 191–214. [Google Scholar] [CrossRef] [PubMed]
- Neidle, S.; Thurston, D.E. Chemical approaches to the discovery and development of cancer therapies. Nat. Rev. Cancer 2005, 5, 285–296. [Google Scholar] [CrossRef]
- Ludlum, D.B.; Austin-Ritchie, P.; Hagopian, M.; Niu, T.-Q.; Yu, D. Detection of sulfur mustard-induced DNA modifications. Chem. Biol. Interact. 1994, 91, 39–49. [Google Scholar] [CrossRef]
- Zubel, T.; Hochgesand, S.; John, H.; Steinritz, D.; Schmidt, A.; Bürkle, A.; Mangerich, A. A mass spectrometric platform for the quantitation of sulfur mustard-induced nucleic acid adducts as mechanistically relevant biomarkers of exposure. Arch. Toxicol. 2019, 93, 61–79. [Google Scholar] [CrossRef]
- Kehe, K.; Balszuweit, F.; Steinritz, D.; Thiermann, H. Molecular toxicology of sulfur mustard-induced cutaneous inflammation and blistering. Toxicology 2009, 263, 12–19. [Google Scholar] [CrossRef]
- Powell, K.L.; Boulware, S.; Thames, H.; Vasquez, K.M.; MacLeod, M.C. 2,6-Dithiopurine blocks toxicity and mutagenesis in human skin cells exposed to sulfur mustard analogues, 2-chloroethyl ethyl sulfide and 2-chloroethyl methyl sulfide. Chem. Res. Toxicol. 2010, 23, 497–503. [Google Scholar] [CrossRef]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Papirmeister, B.; Gross, C.L.; Meier, H.L.; Petrali, J.P.; Johnson, J.B. Molecular basis for mustard-induced vesication. Fundam. Appl. Toxicol. 1985, 5, S134–S149. [Google Scholar] [CrossRef] [PubMed]
- Bhute, V.J.; Palecek, S.P. Metabolic responses induced by DNA damage and poly (ADP-ribose) polymerase (PARP) inhibition in MCF-7 cells. Metabolomics Off. J. Metabolomic Soc. 2015, 11, 1779–1791. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Hogan-Cann, A.D.; Kamboj, A.; Roy Chowdhury, S.K.; Aghanoori, M.-R.; Fernyhough, P.; Anderson, C.M. Poly(ADP-ribose) polymerase-1 inhibits mitochondrial respiration by suppressing PGC-1α activity in neurons. Neuropharmacology 2019, 160, 107755. [Google Scholar] [CrossRef]
- Tateishi, K.; Higuchi, F.; Miller, J.J.; Koerner, M.V.A.; Lelic, N.; Shankar, G.M.; Tanaka, S.; Fisher, D.E.; Batchelor, T.T.; Iafrate, A.J.; et al. The Alkylating Chemotherapeutic Temozolomide Induces Metabolic Stress in IDH1-Mutant Cancers and Potentiates NAD(+) Depletion-Mediated Cytotoxicity. Cancer Res. 2017, 77, 4102–4115. [Google Scholar] [CrossRef] [PubMed]
- Kehe, K.; Raithel, K.; Kreppel, H.; Jochum, M.; Worek, F.; Thiermann, H. Inhibition of poly(ADP-ribose) polymerase (PARP) influences the mode of sulfur mustard (SM)-induced cell death in HaCaT cells. Arch. Toxicol. 2008, 82, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Beigi Harchegani, A.; Khor, A.; Tahmasbpour, E.; Ghatrehsamani, M.; Bakhtiari Kaboutaraki, H.; Shahriary, A. Role of oxidative stress and antioxidant therapy in acute and chronic phases of sulfur mustard injuries: A review. Cutan. Ocul. Toxicol. 2019, 38, 9–17. [Google Scholar] [CrossRef]
- Stenger, B.; Popp, T.; John, H.; Siegert, M.; Tsoutsoulopoulos, A.; Schmidt, A.; Mückter, H.; Gudermann, T.; Thiermann, H.; Steinritz, D. N-Acetyl-l-cysteine inhibits sulfur mustard-induced and TRPA1-dependent calcium influx. Arch. Toxicol. 2017, 91, 2179–2189. [Google Scholar] [CrossRef]
- Borna, H.; Hosseini Qale Noe, S.H.; Harchegani, A.B.; Talatappe, N.R.; Ghatrehsamani, M.; Ghanei, M.; Shahriary, A. A review on proteomics analysis to reveal biological pathways and predictive proteins in sulfur mustard exposed patients: Roles of inflammation and oxidative stress. Inhal. Toxicol. 2019, 31, 3–11. [Google Scholar] [CrossRef]
- Ruszkiewicz, J.A.; Burkle, A.; Mangerich, A. NAD(+) in sulfur mustard toxicity. Toxicol. Lett. 2020, 324, 95–103. [Google Scholar] [CrossRef]
- Covarrubias, A.J.; Perrone, R.; Grozio, A.; Verdin, E. NAD(+) metabolism and its roles in cellular processes during ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 119–141. [Google Scholar] [CrossRef]
- Katsyuba, E.; Romani, M.; Hofer, D.; Auwerx, J. NAD+ homeostasis in health and disease. Nat. Metab. 2020, 2, 9–31. [Google Scholar] [CrossRef] [PubMed]
- Mateuszuk, Ł.; Campagna, R.; Kutryb-Zając, B.; Kuś, K.; Słominska, E.M.; Smolenski, R.T.; Chlopicki, S. Reversal of endothelial dysfunction by nicotinamide mononucleotide via extracellular conversion to nicotinamide riboside. Biochem. Pharmacol. 2020, 178, 114019. [Google Scholar] [CrossRef] [PubMed]
- Neelakantan, H.; Vance, V.; Wetzel, M.D.; Wang, H.-Y.L.; McHardy, S.F.; Finnerty, C.C.; Hommel, J.D.; Watowich, S.J. Selective and membrane-permeable small molecule inhibitors of nicotinamide N-methyltransferase reverse high fat diet-induced obesity in mice. Biochem. Pharmacol. 2018, 147, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Conze, D.; Brenner, C.; Kruger, C.L. Safety and Metabolism of Long-term Administration of NIAGEN (Nicotinamide Riboside Chloride) in a Randomized, Double-Blind, Placebo-controlled Clinical Trial of Healthy Overweight Adults. Sci. Rep. 2019, 9, 9772. [Google Scholar] [CrossRef] [PubMed]
- Trammell, S.A.; Schmidt, M.S.; Weidemann, B.J.; Redpath, P.; Jaksch, F.; Dellinger, R.W.; Li, Z.; Abel, E.D.; Migaud, M.E.; Brenner, C. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat. Commun. 2016, 7, 12948. [Google Scholar] [CrossRef]
- Hasmann, M.; Schemainda, I. FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis. Cancer Res. 2003, 63, 7436–7442. [Google Scholar]
- Wang, Q.Q.; Begum, R.A.; Day, V.W.; Bowman-James, K. Sulfur, oxygen, and nitrogen mustards: Stability and reactivity. Org. Biomol. Chem. 2012, 10, 8786–8793. [Google Scholar] [CrossRef]
- Mangerich, A.; Debiak, M.; Birtel, M.; Ponath, V.; Balszuweit, F.; Lex, K.; Martello, R.; Burckhardt-Boer, W.; Strobelt, R.; Siegert, M.; et al. Sulfur and nitrogen mustards induce characteristic poly(ADP-ribosyl)ation responses in HaCaT keratinocytes with distinctive cellular consequences. Toxicol. Lett. 2016, 244, 56–71. [Google Scholar] [CrossRef][Green Version]
- Boukamp, P.; Petrussevska, R.T.; Breitkreutz, D.; Hornung, J.; Markham, A.; Fusenig, N.E. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 1988, 106, 761–771. [Google Scholar] [CrossRef]
- Tsuchiya, S.; Yamabe, M.; Yamaguchi, Y.; Kobayashi, Y.; Konno, T.; Tada, K. Establishment and characterization of a human acute monocytic leukemia cell line (THP-1). Int. J. Cancer 1980, 26, 171–176. [Google Scholar] [CrossRef]
- Debiak, M.; Lex, K.; Ponath, V.; Burckhardt-Boer, W.; Thiermann, H.; Steinritz, D.; Schmidt, A.; Mangerich, A.; Burkle, A. Immunochemical analysis of poly(ADP-ribosyl)ation in HaCaT keratinocytes induced by the mono-alkylating agent 2-chloroethyl ethyl sulfide (CEES): Impact of experimental conditions. Toxicol. Lett. 2016, 244, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, E.L.; Jacobson, M.K. Pyridine nucleotide levels as a function of growth in normal and transformed 3T3 cells. Arch. Biochem. Biophys. 1976, 175, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Geissmann, Q. OpenCFU, a new free and open-source software to count cell colonies and other circular objects. PLoS ONE 2013, 8, e54072. [Google Scholar] [CrossRef] [PubMed]
- Debiak, M.; Panas, A.; Steinritz, D.; Kehe, K.; Burkle, A. High-throughput analysis of DNA interstrand crosslinks in human peripheral blood mononuclear cells by automated reverse FADU assay. Toxicology 2011, 280, 53–60. [Google Scholar] [CrossRef]
- Moreno-Villanueva, M.; Pfeiffer, R.; Sindlinger, T.; Leake, A.; Muller, M.; Kirkwood, T.B.; Burkle, A. A modified and automated version of the ‘Fluorimetric Detection of Alkaline DNA Unwinding’ method to quantify formation and repair of DNA strand breaks. BMC Biotechnol. 2009, 9, 39. [Google Scholar] [CrossRef]
- Kawamitsu, H.; Hoshino, H.; Okada, H.; Miwa, M.; Momoi, H.; Sugimura, T. Monoclonal antibodies to poly(adenosine diphosphate ribose) recognize different structures. Biochemistry 1984, 23, 3771–3777. [Google Scholar] [CrossRef]
- Rank, L.; Veith, S.; Gwosch, E.C.; Demgenski, J.; Ganz, M.; Jongmans, M.C.; Vogel, C.; Fischbach, A.; Buerger, S.; Fischer, J.M.; et al. Analyzing structure-function relationships of artificial and cancer-associated PARP1 variants by reconstituting TALEN-generated HeLa PARP1 knock-out cells. Nucleic Acids Res. 2016, 44, 10386–10405. [Google Scholar] [CrossRef]
- Martens, C.R.; Denman, B.A.; Mazzo, M.R.; Armstrong, M.L.; Reisdorph, N.; McQueen, M.B.; Chonchol, M.; Seals, D.R. Chronic nicotinamide riboside supplementation is well-tolerated and elevates NAD+ in healthy middle-aged and older adults. Nat. Commun. 2018, 9, 1286. [Google Scholar] [CrossRef]
- Cohen, M.S. Interplay between compartmentalized NAD(+) synthesis and consumption: A focus on the PARP family. Genes. Dev. 2020, 34, 254–262. [Google Scholar] [CrossRef]
- Weidele, K.; Kunzmann, A.; Schmitz, M.; Beneke, S.; Burkle, A. Ex vivo supplementation with nicotinic acid enhances cellular poly(ADP-ribosyl)ation and improves cell viability in human peripheral blood mononuclear cells. Biochem. Pharmacol. 2010, 80, 1103–1112. [Google Scholar] [CrossRef]
- Boyonoski, A.C.; Spronck, J.C.; Jacobs, R.M.; Shah, G.M.; Poirier, G.G.; Kirkland, J.B. Pharmacological intakes of niacin increase bone marrow poly(ADP-ribose) and the latency of ethylnitrosourea-induced carcinogenesis in rats. J. Nutr. 2002, 132, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Hinshaw, D.B.; Lodhi, I.J.; Hurley, L.L.; Atkins, K.B.; Dabrowska, M.I. Activation of poly [ADP-Ribose] polymerase in endothelial cells and keratinocytes: Role in an in vitro model of sulfur mustard-mediated vesication. Toxicol. Appl. Pharmacol. 1999, 156, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Martens, M.E.; Smith, W.J. The role of NAD+ depletion in the mechanism of sulfur mustard-induced metabolic injury. Cutan. Ocul. Toxicol. 2008, 27, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Mol, M.A.; van de Ruit, A.M.; Kluivers, A.W. NAD+ levels and glucose uptake of cultured human epidermal cells exposed to sulfur mustard. Toxicol. Appl. Pharmacol. 1989, 98, 159–165. [Google Scholar] [CrossRef]
- Thompson, B.C.; Halliday, G.M.; Damian, D.L. Nicotinamide enhances repair of arsenic and ultraviolet radiation-induced DNA damage in HaCaT keratinocytes and ex vivo human skin. PLoS ONE 2015, 10, e0117491. [Google Scholar] [CrossRef]
- Zhen, A.X.; Piao, M.J.; Kang, K.A.; Fernando, P.; Kang, H.K.; Koh, Y.S.; Yi, J.M.; Hyun, J.W. Niacinamide Protects Skin Cells from Oxidative Stress Induced by Particulate Matter. Biomol. Ther. 2019, 27, 562–569. [Google Scholar] [CrossRef]
- Grozio, A.; Mills, K.F.; Yoshino, J.; Bruzzone, S.; Sociali, G.; Tokizane, K.; Lei, H.C.; Cunningham, R.; Sasaki, Y.; Migaud, M.E.; et al. Slc12a8 is a nicotinamide mononucleotide transporter. Nat. Metab. 2019, 1, 47–57. [Google Scholar] [CrossRef]
- Schmidt, M.S.; Brenner, C. Absence of evidence that Slc12a8 encodes a nicotinamide mononucleotide transporter. Nat. Metab. 2019, 1, 660–661. [Google Scholar] [CrossRef]
- Ratajczak, J.; Joffraud, M.; Trammell, S.A.J.; Ras, R.; Canela, N.; Boutant, M.; Kulkarni, S.S.; Rodrigues, M.; Redpath, P.; Migaud, M.E.; et al. NRK1 controls nicotinamide mononucleotide and nicotinamide riboside metabolism in mammalian cells. Nat. Commun. 2016, 7, 13103. [Google Scholar] [CrossRef]
- Palmer, R.D.; Elnashar, M.M.; Vaccarezza, M. Precursor comparisons for the upregulation of nicotinamide adenine dinucleotide. Novel approaches for better aging. Aging Med. 2021, 4, 214–220. [Google Scholar] [CrossRef]
- Dellinger, R.W.; Santos, S.R.; Morris, M.; Evans, M.; Alminana, D.; Guarente, L.; Marcotulli, E. Repeat dose NRPT (nicotinamide riboside and pterostilbene) increases NAD(+) levels in humans safely and sustainably: A randomized, double-blind, placebo-controlled study. NPJ Aging Mech. Dis. 2017, 3, 17. [Google Scholar] [CrossRef] [PubMed]
- Poljšak, B.; Kovač, V.; Milisav, I. Current Uncertainties and Future Challenges Regarding NAD+ Boosting Strategies. Antioxidants 2022, 11, 1673. [Google Scholar] [CrossRef]
- Kourtzidis, I.A.; Dolopikou, C.F.; Tsiftsis, A.N.; Margaritelis, N.V.; Theodorou, A.A.; Zervos, I.A.; Tsantarliotou, M.P.; Veskoukis, A.S.; Vrabas, I.S.; Paschalis, V.; et al. Nicotinamide riboside supplementation dysregulates redox and energy metabolism in rats: Implications for exercise performance. Exp. Physiol. 2018, 103, 1357–1366. [Google Scholar] [CrossRef] [PubMed]
- Nakahata, Y.; Sahar, S.; Astarita, G.; Kaluzova, M.; Sassone-Corsi, P. Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science 2009, 324, 654–657. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.; Bernstein, I.A.; Vaughan, F.L. Failure to observe a relationship between bis-(beta-chloroethyl)sulfide-induced NAD depletion and cytotoxicity in the rat keratinocyte culture. J. Toxicol. Environ. Health 1994, 42, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Mol, M.E.; de Vries, R.; Kluivers, A.W. Effects of nicotinamide on biochemical changes and microblistering induced by sulfur mustard in human skin organ cultures. Toxicol. Appl. Pharmacol. 1991, 107, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.J.; Gross, C.L.; Chan, P.; Meier, H.L. The use of human epidermal keratinocytes in culture as a model for studying the biochemical mechanisms of sulfur mustard toxicity. Cell Biol. Toxicol. 1990, 6, 285–291. [Google Scholar] [CrossRef]
- Sayer, N.M.; Whiting, R.; Green, A.C.; Anderson, K.; Jenner, J.; Lindsay, C.D. Direct binding of sulfur mustard and chloroethyl ethyl sulphide to human cell membrane-associated proteins; implications for sulfur mustard pathology. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2010, 878, 1426–1432. [Google Scholar] [CrossRef]
- Sabnam, S.; Pal, A. Relevance of Erk1/2-PI3K/Akt signaling pathway in CEES-induced oxidative stress regulates inflammation and apoptosis in keratinocytes. Cell Biol. Toxicol. 2019, 35, 541–564. [Google Scholar] [CrossRef]
- Sabnam, S.; Rizwan, H.; Pal, S.; Pal, A. CEES-induced ROS accumulation regulates mitochondrial complications and inflammatory response in keratinocytes. Chem. Biol. Interact. 2020, 321, 109031. [Google Scholar] [CrossRef]
- Tewari-Singh, N.; Gu, M.; Agarwal, C.; White, C.W.; Agarwal, R. Biological and molecular mechanisms of sulfur mustard analogue-induced toxicity in JB6 and HaCaT cells: Possible role of ataxia telangiectasia-mutated/ataxia telangiectasia-Rad3-related cell cycle checkpoint pathway. Chem. Res. Toxicol. 2010, 23, 1034–1044. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Brakedal, B.; Dolle, C.; Riemer, F.; Ma, Y.; Nido, G.S.; Skeie, G.O.; Craven, A.R.; Schwarzlmuller, T.; Brekke, N.; Diab, J.; et al. The NADPARK study: A randomized phase I trial of nicotinamide riboside supplementation in Parkinson’s disease. Cell Metab. 2022, 34, 396–407.e396. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Cao, B.; Naval-Sanchez, M.; Pham, T.; Sun, Y.B.Y.; Williams, B.; Heazlewood, S.Y.; Deshpande, N.; Li, J.; Kraus, F.; et al. Nicotinamide riboside attenuates age-associated metabolic and functional changes in hematopoietic stem cells. Nat. Commun. 2021, 12, 2665. [Google Scholar] [CrossRef] [PubMed]
- Reiten, O.K.; Wilvang, M.A.; Mitchell, S.J.; Hu, Z.; Fang, E.F. Preclinical and clinical evidence of NAD(+) precursors in health, disease, and ageing. Mech. Ageing Dev. 2021, 199, 111567. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruszkiewicz, J.; Papatheodorou, Y.; Jäck, N.; Melzig, J.; Eble, F.; Pirker, A.; Thomann, M.; Haberer, A.; Rothmiller, S.; Bürkle, A.; et al. NAD+ Acts as a Protective Factor in Cellular Stress Response to DNA Alkylating Agents. Cells 2023, 12, 2396. https://doi.org/10.3390/cells12192396
Ruszkiewicz J, Papatheodorou Y, Jäck N, Melzig J, Eble F, Pirker A, Thomann M, Haberer A, Rothmiller S, Bürkle A, et al. NAD+ Acts as a Protective Factor in Cellular Stress Response to DNA Alkylating Agents. Cells. 2023; 12(19):2396. https://doi.org/10.3390/cells12192396
Chicago/Turabian StyleRuszkiewicz, Joanna, Ylea Papatheodorou, Nathalie Jäck, Jasmin Melzig, Franziska Eble, Annika Pirker, Marius Thomann, Andreas Haberer, Simone Rothmiller, Alexander Bürkle, and et al. 2023. "NAD+ Acts as a Protective Factor in Cellular Stress Response to DNA Alkylating Agents" Cells 12, no. 19: 2396. https://doi.org/10.3390/cells12192396
APA StyleRuszkiewicz, J., Papatheodorou, Y., Jäck, N., Melzig, J., Eble, F., Pirker, A., Thomann, M., Haberer, A., Rothmiller, S., Bürkle, A., & Mangerich, A. (2023). NAD+ Acts as a Protective Factor in Cellular Stress Response to DNA Alkylating Agents. Cells, 12(19), 2396. https://doi.org/10.3390/cells12192396