
A Review of Progress on Targeting LDL Receptor-Dependent and -Independent Pathways for the Treatment of Hypercholesterolemia, a Major Risk Factor of ASCVD

Abstract
1. Introduction

{kind=link}
{kind=link}
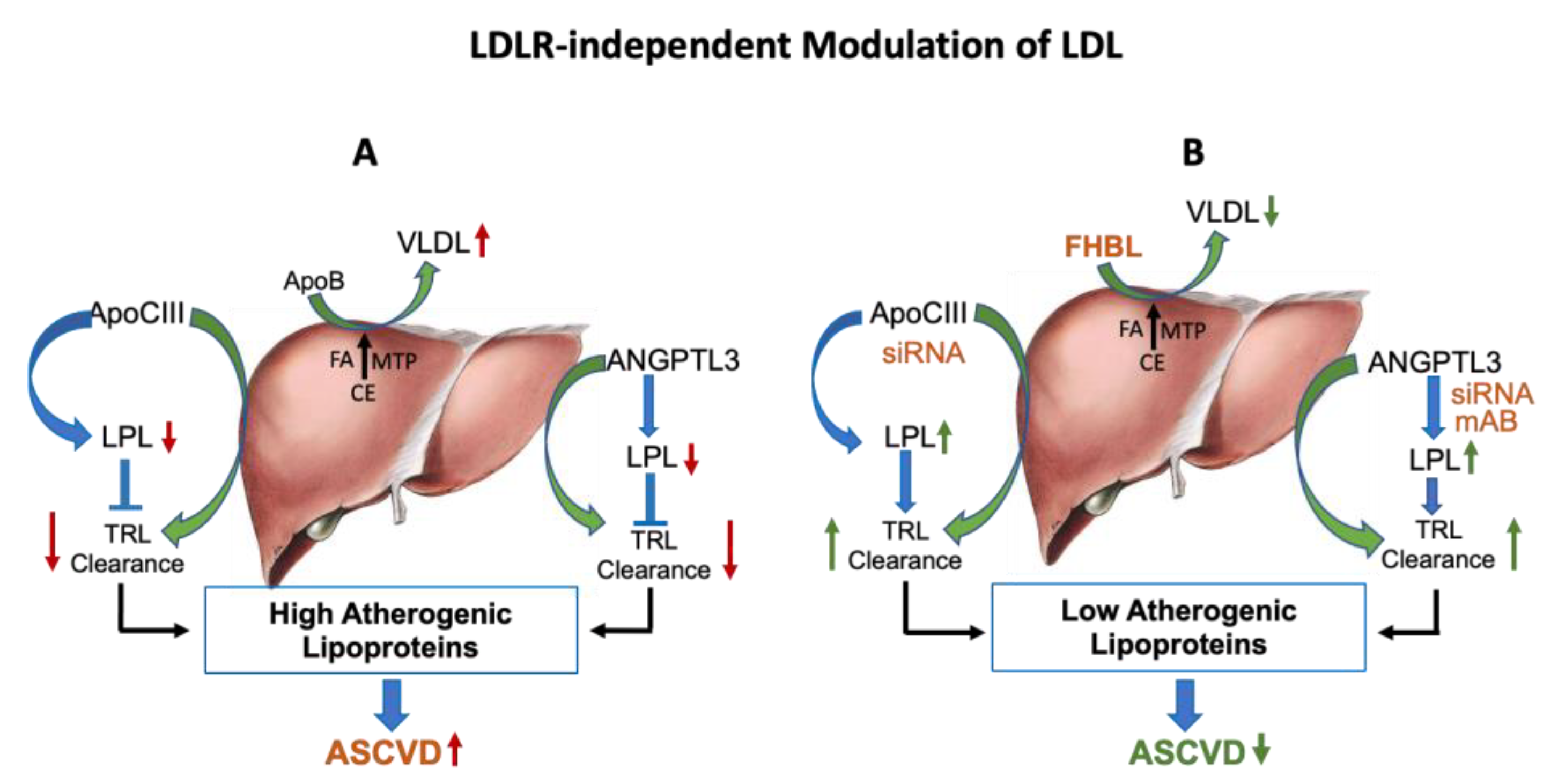{kind=link}
{kind=link}
{kind=link}
| Drug Name | Drug Class | Primary Endpoint | Mechanism | Refs. |
|---|---|---|---|---|
| Statins | LDL lowering | LDL | HMG-CoA Inhibitor | [27,28] |
| Cholestyramine | Bile acid sequestrants | LDL | Bile acid binding | [32,33] |
| Apheresis | LDL | Apheresis | [78] | |
| Niacin | LDL lowering | TG, LDL HDL | DGAT2 Inhibitor | [34,35] |
| Ezetimibe | LDL Lowering | LDL | NPC1L1 inhibitor | [29,30] |
| Lomitapide | LDL lowering | VLDL, LDL | MTP Inhibitor | [46] |
| Nexletol | LDL lowering | LDL | ACLY Inhibitor | [42,44] |
| Mipomersen | ASO | LDL | ApoB silencing | [46,47] |
| Volanesorsen | siRNA | TG | ApoCIII silencing | [58,59] |
| ARO-ApoCIII | siRNA | TG | ApoCIII silencing | [59] |
| Vupanorsen | siRNA | LDL, TG | ANGPTL3 silencing | [21] |
| IONIS-ANGPTL3 | ASO | LDL, TG | ANGPTL3 silencing | [59] |
| ARO-ANG3 | siRNA | LDL, TG | ANGPTL3 silencing | [59] |
| Evinacumab | mAb | LDL, TG | ANGPTL3 silencing | [51] |
| Inclisiran | siRNA | LDL | PCSK9 silencing | [72,73] |
| Evolocumab | mAb | LDL | PCSK9 silencing | [74,75] |
| Alirocumab | mAb | LDL | PCSK9 silencing | [76,77] |
2. LDLR Pathway Controlling LDL Level
2.1. LDLR-Independent Pathway Affecting LDL Level
2.2. LDLR Activity and Circulating LDL
2.3. LDLR Activity Determines Circulating LDL
2.3.1. LDLR Activity and LDL Association in Mice and Rats
2.3.2. Regulation of LDL in LDLR-Deficient Hamsters
2.3.3. LDLR-Deficient Rabbits as a Model of HoFH
2.3.4. LDLR Loss-of-Function Studies in Pig
3. LDLR Degradation Pathway Regulates LDL
3.1. PCSK9 Regulation of LDLR and LDL
3.2. IDOL Regulation of LDLR and LDL
4. Management of Hypercholesterolemia via ApoC-III and ANGPTL3 Inactivation
5. LDLR Gain-of-Function Mutations
6. Gene Therapy and Genome/Base Editing Approaches to Manage Refractory Hypercholesterolemia
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AAV | adeno-associated virus |
| ABE | adenosine base editors |
| ACLY | ATP citrate lyase |
| ANGPTL3 | angiopoietin-like 3 |
| Apo | apolipoprotein |
| ASO | antisense oligo |
| ASCVD | atherosclerotic cardiovascular disease |
| CAD | coronary artery disease |
| CBE | cytosine base editors |
| CHD | coronary heart disease |
| CETP | cholesteryl ester transfer protein |
| CRISPR | clustered regularly interspaced short palindromic repeats |
| FHBL | familial hypobetalipoproteinemia |
| HCHF | high cholesterol high fat |
| HeFH | heterozygous familial hypercholesterolemia |
| HMG-CoA | β-Hydroxy β-methylglutaryl-CoA |
| HoFH | homozygous familial hypercholesterolemia |
| HOMA | homeostatic model assessment |
| IDOL | inducible degrader of LDLR |
| KO | knockout |
| LDLR | low-density lipoprotein receptor |
| LCAT | lecithin:cholesterol acyltransferase |
| LNP | lipid nanoparticles |
| Lp(a) | lipoprotein(a) |
| LPL | lipoprotein lipase |
| mAb | monoclonal antibody |
| NPC1L1 | Niemann–Pick C1-Like1 |
| oxLDL | oxidized LDL |
| PCSK9 | proprotein convertase subtilisin/kexin type 9 |
| RH | refractory hypercholesterolemia |
| TRL | triglyceride-rich lipoproteins |
| UTR | Untranslated region |
| WHHL | Watanabe heritable hyperlipidemic |
| WT | wild type |
References
- Goldstein, J.L.; Brown, M.S. The LDL receptor defect in familial hypercholesterolemia: Implications for pathogenesis and therapy. Med. Clin. N. Am. 1982, 66, 335–362. [Google Scholar] [CrossRef]
- Santos, P.C.; Pereira, A.C. Type of LDLR mutation and the pharmacogenetics of familial hypercholesterolemia treatment. Pharmacogenomics 2015, 16, 1743–1750. [Google Scholar] [CrossRef]
- Sharifi, M.; Futema, M.; Nair, D.; Humphries, S.E. Genetic Architecture of Familial Hypercholesterolaemia. Curr. Cardiol. Rep. 2017, 19, 44. [Google Scholar] [CrossRef]
- Nohara, A.; Tada, H.; Ogura, M.; Okazaki, S.; Ono, K.; Shimano, H.; Daida, H.; Dobashi, K.; Hayashi, T.; Hori, M.; et al. Homozygous Familial Hypercholesterolemia. J. Atheroscler. Thromb. 2021, 28, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Pisciotta, L.; Priore Oliva, C.; Cefalù, A.B.; Noto, D.; Bellocchio, A.; Fresa, R.; Cantafora, A.; Patel, D.; Averna, M.; Tarugi, P.; et al. Additive effect of mutations in LDLR and PCSK9 genes on the phenotype of familial hypercholesterolemia. Atherosclerosis 2006, 186, 433–440. [Google Scholar] [CrossRef]
- Dyrbuś, K.; Gąsior, M.; Desperak, P.; Trzeciak, P.; Nowak, J.; Penson, P.E.; Osadnik, T.; Banach, M. Risk-factors associated with extremely high cardiovascular risk of mid- and long-term mortality following myocardial infarction: Analysis of the Hyperlipidaemia Therapy in tERtiary Cardiological cEnTer (TERCET) registry. Atherosclerosis 2021, 333, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Ference, B.A.; Ginsberg, H.N.; Graham, I.; Ray, K.K.; Packard, C.J.; Bruckert, E.; Hegele, R.A.; Krauss, R.M.; Raal, F.J.; Schunkert, H.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2017, 38, 2459–2472. [Google Scholar] [CrossRef]
- Pinkosky, S.L.; Filippov, S.; Srivastava, R.A.; Hanselman, J.C.; Bradshaw, C.D.; Hurley, T.R.; Cramer, C.T.; Spahr, M.A.; Brant, A.F.; Houghton, J.L.; et al. AMP-activated protein kinase and ATP-citrate lyase are two distinct molecular targets for ETC-1002, a novel small molecule regulator of lipid and carbohydrate metabolism. J. Lipid Res. 2013, 54, 134–151. [Google Scholar] [CrossRef]
- Srivastava, R.A.K.; Hurley, T.R.; Oniciu, D.; Adeli, K.; Newton, R.S. Discovery of analogues of non-β oxidizable long-chain dicarboxylic fatty acids as dual inhibitors of fatty acids and cholesterol synthesis: Efficacy of lead compound in hyperlipidemic hamsters reveals novel mechanism. Nutr. Metab. Cardiovasc. Dis. 2021, 31, 2490–2506. [Google Scholar] [CrossRef]
- Srivastava, R.A.; Srivastava, N.; Averna, M.; Cefalu, A.B.; Schonfeld, G. Molecular bases of low production rates of apolipoprotein B-100 and truncated apoB-82 in a mutant HepG2 cell line generated by targeted modification of the apolipoprotein B gene. J. Lipid Res. 1999, 40, 901–912. [Google Scholar] [CrossRef]
- Plump, A.S.; Masucci-Magoulas, L.; Bruce, C.; Bisgaier, C.L.; Breslow, J.L.; Tall, A.R. Increased atherosclerosis in ApoE and LDL receptor gene knock-out mice as a result of human cholesteryl ester transfer protein transgene expression. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1105–1110. [Google Scholar] [CrossRef]
- Davidson, M.; Liu, S.X.; Barter, P.; Brinton, E.A.; Cannon, C.P.; Gotto, A.M., Jr.; Leary, E.T.; Shah, S.; Stepanavage, M.; Mitchel, Y.; et al. Measurement of LDL-C after treatment with the CETP inhibitor anacetrapib. J. Lipid Res. 2013, 54, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, S.; Brown, M.S.; Goldstein, J.L.; Gerard, R.D.; Hammer, R.E.; Herz, J. Hypercholesterolemia in low density lipoprotein receptor knockout mice and its reversal by adenovirus-mediated gene delivery. J. Clin. Investig. 1993, 92, 883–893. [Google Scholar] [CrossRef] [PubMed]
- Benito-Vicente, A.; Uribe, K.B.; Jebari, S.; Galicia-Garcia, U.; Ostolaza, H.; Martin, C. Familial Hypercholesterolemia: The Most Frequent Cholesterol Metabolism Disorder Caused Disease. Int. J. Mol. Sci. 2018, 19, 3426. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S. Role of VLDL receptor in atherogenesis. Curr. Opin. Lipidol. 2021, 32, 219–225. [Google Scholar] [CrossRef]
- Abifadel, M.; Varret, M.; Rabès, J.P.; Allard, D.; Ouguerram, K.; Devillers, M.; Cruaud, C.; Benjannet, S.; Wickham, L.; Erlich, D.; et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 2003, 34, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Lagace, T.A.; Curtis, D.E.; Garuti, R.; McNutt, M.C.; Park, S.W.; Prather, H.B.; Anderson, N.N.; Ho, Y.K.; Hammer, R.E.; Horton, J.D. Secreted PCSK9 decreases the number of LDL receptors in hepatocytes and in livers of parabiotic mice. J. Clin. Investig. 2006, 116, 2995–3005. [Google Scholar] [CrossRef] [PubMed]
- Dong, B.; Wu, M.; Cao, A.; Li, H.; Liu, J. Suppression of Idol expression is an additional mechanism underlying statin-induced up-regulation of hepatic LDL receptor expression. Int. J. Mol. Med. 2011, 27, 103–110. [Google Scholar] [CrossRef]
- Schmitz, J.; Gouni-Berthold, I. APOC-III Antisense Oligonucleotides: A New Option for the Treatment of Hypertriglyceridemia. Curr. Med. Chem. 2018, 25, 1567–1576. [Google Scholar] [CrossRef]
- Lightbourne, M.; Startzell, M.; Bruce, K.D.; Brite, B.; Muniyappa, R.; Skarulis, M.; Shamburek, R.; Gharib, A.M.; Ouwerkerk, R.; Walter, M.; et al. Volanesorsen, an antisense oligonucleotide to apolipoprotein C-III, increases lipoprotein lipase activity and lowers triglycerides in partial lipodystrophy. J. Clin. Lipidol. 2022, 16, 850–862. [Google Scholar] [CrossRef]
- Gaudet, D.; Karwatowska-Prokopczuk, E.; Baum, S.J.; Hurh, E.; Kingsbury, J.; Bartlett, V.J.; Figueroa, A.L.; Piscitelli, P.; Singleton, W.; Witztum, J.L.; et al. Vupanorsen, an N-acetyl galactosamine-conjugated antisense drug to ANGPTL3 mRNA, lowers triglycerides and atherogenic lipoproteins in patients with diabetes, hepatic steatosis, and hypertriglyceridaemia. Eur. Heart J. 2020, 41, 3936–3945. [Google Scholar] [CrossRef] [PubMed]
- Dewey, F.E.; Gusarova, V.; Dunbar, R.L.; O’Dushlaine, C.; Schurmann, C.; Gottesman, O.; McCarthy, S.; Van Hout, C.V.; Bruse, S.; Dansky, H.M.; et al. Genetic and Pharmacologic Inactivation of ANGPTL3 and Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 211–221. [Google Scholar] [CrossRef]
- Davis, H.R., Jr.; Lowe, R.S.; Neff, D.R. Effects of ezetimibe on atherosclerosis in preclinical models. Atherosclerosis 2011, 215, 266–278. [Google Scholar] [CrossRef]
- Hayashi, H.; Kawamura, M. Lowering LDL cholesterol, but not raising LDL receptor activity, by ezetimibe. J. Clin. Lipidol. 2013, 7, 632–636. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Calvo, M.; Lisnock, J.; Bull, H.G.; Hawes, B.E.; Burnett, D.A.; Braun, M.P.; Crona, J.H.; Davis, H.R., Jr.; Dean, D.C.; Detmers, P.A.; et al. The target of ezetimibe is Niemann-Pick C1-Like 1 (NPC1L1). Proc. Natl. Acad. Sci. USA 2005, 102, 8132–8137. [Google Scholar] [CrossRef]
- LaRosa, J.C.; He, J.; Vupputuri, S. Effect of statins on risk of coronary disease: A meta-analysis of randomized controlled trials. JAMA 1999, 282, 2340–2346. [Google Scholar] [CrossRef]
- Murdoch, D.; Scott, L.J. Ezetimibe/Simvastatin: A review of its use in the management of hypercholesterolemia. Am. J. Cardiovasc. Drugs 2004, 4, 405–422. [Google Scholar] [CrossRef]
- Kastelein, J.J.; Akdim, F.; Stroes, E.S.; Zwinderman, A.H.; Bots, M.L.; Stalenhoef, A.F.; Visseren, F.L.; Sijbrands, E.J.; Trip, M.D.; Stein, E.A.; et al. Simvastatin with or without ezetimibe in familial hypercholesterolemia. N. Engl. J. Med. 2008, 358, 1431–1443. [Google Scholar] [CrossRef]
- Yamamoto, A.; Harada-Shiba, M.; Endo, M.; Kusakabe, N.; Tanioka, T.; Kato, H.; Shoji, T. The effect of ezetimibe on serum lipids and lipoproteins in patients with homozygous familial hypercholesterolemia undergoing LDL-apheresis therapy. Atherosclerosis 2006, 186, 126–131. [Google Scholar] [CrossRef]
- Yang, Y.J.; Lee, S.H.; Kim, B.S.; Cho, Y.K.; Cho, H.J.; Cho, K.I.; Kim, S.Y.; Ryu, J.K.; Cho, J.M.; Park, J.I.; et al. Combination Therapy of Rosuvastatin and Ezetimibe in Patients with High Cardiovascular Risk. Clin. Ther. 2017, 39, 107–117. [Google Scholar] [CrossRef]
- Islam, M.S.; Sharif, A.; Kwan, N.; Tam, K.C. Bile Acid Sequestrants for Hypercholesterolemia Treatment Using Sustainable Biopolymers: Recent Advances and Future Perspectives. Mol. Pharm. 2022, 19, 1248–1272. [Google Scholar] [CrossRef] [PubMed]
- Hashim, S.A.; Vanitallie, T.B. Cholestyramine Resin Therapy for Hypercholesteremia: Clinical and Metabolic Studies. JAMA 1965, 192, 289–293. [Google Scholar] [CrossRef] [PubMed]
- LaRosa, J. Review of clinical studies of bile acid sequestrants for lowering plasma lipid levels. Cardiology 1989, 76 (Suppl. S1), 55–61; discussion 61–64. [Google Scholar] [CrossRef]
- Drexel, H. Nicotinic acid in the treatment of hyperlipidaemia. Fundam. Clin. Pharmacol. 2007, 21 (Suppl. S2), 5–6. [Google Scholar] [CrossRef]
- Kamanna, V.S.; Kashyap, M.L. Mechanism of action of niacin. Am. J. Cardiol. 2008, 101, 20b–26b. [Google Scholar] [CrossRef]
- Yim, B.T.; Chong, P.H. Niacin-ER and lovastatin treatment of hypercholesterolemia and mixed dyslipidemia. Ann. Pharmacother. 2003, 37, 106–115. [Google Scholar] [CrossRef]
- Sanford, M.; Curran, M.P. Niacin extended-release/simvastatin. Drugs 2008, 68, 2373–2386. [Google Scholar] [CrossRef]
- Stefanutti, C. Lomitapide—A Microsomal Triglyceride Transfer Protein Inhibitor for Homozygous Familial Hypercholesterolemia. Curr. Atheroscler. Rep. 2020, 22, 38. [Google Scholar] [CrossRef]
- Parham, J.S.; Goldberg, A.C. Mipomersen and its use in familial hypercholesterolemia. Expert Opin. Pharmacother. 2019, 20, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Chambergo-Michilot, D.; Alur, A.; Kulkarni, S.; Agarwala, A. Mipomersen in Familial Hypercholesterolemia: An Update on Health-Related Quality of Life and Patient-Reported Outcomes. Vasc. Health Risk Manag. 2022, 18, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Stefanutti, C.; Thompson, G.R. Lipoprotein apheresis in the management of familial hypercholesterolaemia: Historical perspective and recent advances. Curr. Atheroscler. Rep. 2015, 17, 465. [Google Scholar] [CrossRef]
- Ray, K.K.; Bays, H.E.; Catapano, A.L.; Lalwani, N.D.; Bloedon, L.T.; Sterling, L.R.; Robinson, P.L.; Ballantyne, C.M. Safety and Efficacy of Bempedoic Acid to Reduce LDL Cholesterol. N. Engl. J. Med. 2019, 380, 1022–1032. [Google Scholar] [CrossRef]
- Aguilar-Salinas, C.A.; Gómez-Díaz, R.A.; Corral, P. New Therapies for Primary Hyperlipidemia. J. Clin. Endocrinol. Metab. 2022, 107, 1216–1224. [Google Scholar] [CrossRef]
- Laufs, U.; Banach, M.; Mancini, G.B.J.; Gaudet, D.; Bloedon, L.T.; Sterling, L.R.; Kelly, S.; Stroes, E.S.G. Efficacy and Safety of Bempedoic Acid in Patients With Hypercholesterolemia and Statin Intolerance. J. Am. Heart Assoc. 2019, 8, e011662. [Google Scholar] [CrossRef] [PubMed]
- Kong, S.X.; Crawford, S.Y.; Gandhi, S.K.; Seeger, J.D.; Schumock, G.T.; Lam, N.P.; Stubbings, J.; Schoen, M.D. Efficacy of 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitors in the treatment of patients with hypercholesterolemia: A meta-analysis of clinical trials. Clin. Ther. 1997, 19, 778–797. [Google Scholar] [CrossRef]
- Rader, D.J.; Kastelein, J.J. Lomitapide and mipomersen: Two first-in-class drugs for reducing low-density lipoprotein cholesterol in patients with homozygous familial hypercholesterolemia. Circulation 2014, 129, 1022–1032. [Google Scholar] [CrossRef]
- Ricotta, D.N.; Frishman, W. Mipomersen: A safe and effective antisense therapy adjunct to statins in patients with hypercholesterolemia. Cardiol. Rev. 2012, 20, 90–95. [Google Scholar] [CrossRef]
- Fu, Y.S.; Wang, J.H.; Lee, C.J.; Hsu, B.G. Positive correlation of the serum angiopoietin-like protein 3 levels with the aortic augmentation index in patients with coronary artery disease. Ther. Clin. Risk Manag. 2018, 14, 231–236. [Google Scholar] [CrossRef]
- Jin, M.; Meng, F.; Yang, W.; Liang, L.; Wang, H.; Fu, Z. Efficacy and Safety of Evinacumab for the Treatment of Hypercholesterolemia: A Meta-Analysis. J. Cardiovasc. Pharmacol. 2021, 78, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhang, B.; Yang, J. Evinacumab for the treatment of homozygous familial hypercholesterolemia. Expert Rev. Clin. Pharmacol. 2022, 15, 139–145. [Google Scholar] [CrossRef]
- Raal, F.J.; Rosenson, R.S.; Reeskamp, L.F.; Hovingh, G.K.; Kastelein, J.J.P.; Rubba, P.; Ali, S.; Banerjee, P.; Chan, K.C.; Gipe, D.A.; et al. Evinacumab for Homozygous Familial Hypercholesterolemia. N. Engl. J. Med. 2020, 383, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, D.; Gipe, D.A.; Pordy, R.; Ahmad, Z.; Cuchel, M.; Shah, P.K.; Chyu, K.Y.; Sasiela, W.J.; Chan, K.C.; Brisson, D.; et al. ANGPTL3 Inhibition in Homozygous Familial Hypercholesterolemia. N. Engl. J. Med. 2017, 377, 296–297. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, F.; Mansfield, B.S.; Raal, F.J. ANGPTL3 as a Drug Target in Hyperlipidemia and Atherosclerosis. Curr. Atheroscler. Rep. 2022, 24, 959–967. [Google Scholar] [CrossRef]
- Khoury, E.; Lauzière, A.; Raal, F.J.; Mancini, J.; Gaudet, D. Atherosclerotic plaque regression in homozygous familial hypercholesterolaemia: A case report of a long-term lipid-lowering therapy involving LDL-receptor-independent mechanisms. Eur. Heart J. Case Rep. 2023, 7, ytad029. [Google Scholar] [CrossRef] [PubMed]
- Stitziel, N.O.; Khera, A.V.; Wang, X.; Bierhals, A.J.; Vourakis, A.C.; Sperry, A.E.; Natarajan, P.; Klarin, D.; Emdin, C.A.; Zekavat, S.M.; et al. ANGPTL3 Deficiency and Protection Against Coronary Artery Disease. J. Am. Coll. Cardiol. 2017, 69, 2054–2063. [Google Scholar] [CrossRef]
- Kersten, S. ANGPTL3 as therapeutic target. Curr. Opin. Lipidol. 2021, 32, 335–341. [Google Scholar] [CrossRef]
- Gouni-Berthold, I. The role of antisense oligonucleotide therapy against apolipoprotein-CIII in hypertriglyceridemia. Atheroscler. Suppl. 2017, 30, 19–27. [Google Scholar] [CrossRef]
- Calcaterra, I.; Lupoli, R.; Di Minno, A.; Di Minno, M.N.D. Volanesorsen to treat severe hypertriglyceridaemia: A pooled analysis of randomized controlled trials. Eur. J. Clin. Investig. 2022, 52, e13841. [Google Scholar] [CrossRef]
- Akoumianakis, I.; Zvintzou, E.; Kypreos, K.; Filippatos, T.D. ANGPTL3 and Apolipoprotein C-III as Novel Lipid-Lowering Targets. Curr. Atheroscler. Rep. 2021, 23, 20. [Google Scholar] [CrossRef]
- Shimizugawa, T.; Ono, M.; Shimamura, M.; Yoshida, K.; Ando, Y.; Koishi, R.; Ueda, K.; Inaba, T.; Minekura, H.; Kohama, T.; et al. ANGPTL3 decreases very low density lipoprotein triglyceride clearance by inhibition of lipoprotein lipase. J. Biol. Chem. 2002, 277, 33742–33748. [Google Scholar] [CrossRef]
- Pinkosky, S.L.; Newton, R.S.; Day, E.A.; Ford, R.J.; Lhotak, S.; Austin, R.C.; Birch, C.M.; Smith, B.K.; Filippov, S.; Groot, P.H.E.; et al. Liver-specific ATP-citrate lyase inhibition by bempedoic acid decreases LDL-C and attenuates atherosclerosis. Nat. Commun. 2016, 7, 13457. [Google Scholar] [CrossRef] [PubMed]
- Ballantyne, C.M.; Laufs, U.; Ray, K.K.; Leiter, L.A.; Bays, H.E.; Goldberg, A.C.; Stroes, E.S.; MacDougall, D.; Zhao, X.; Catapano, A.L. Bempedoic acid plus ezetimibe fixed-dose combination in patients with hypercholesterolemia and high CVD risk treated with maximally tolerated statin therapy. Eur. J. Prev. Cardiol. 2020, 27, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, E.; Johns, T. Bempedoic Acid (Nexletol) for the Treatment of Hyperlipidemia and Familial Hypercholesterolemia. Am. Fam. Physician 2021, 103, 377–378. [Google Scholar]
- LaRosa, J.C.; Grundy, S.M.; Waters, D.D.; Shear, C.; Barter, P.; Fruchart, J.C.; Gotto, A.M.; Greten, H.; Kastelein, J.J.; Shepherd, J.; et al. Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N. Engl. J. Med. 2005, 352, 1425–1435. [Google Scholar] [CrossRef]
- Gonzalez, M.A.; Selwyn, A.P. Endothelial function, inflammation, and prognosis in cardiovascular disease. Am. J. Med. 2003, 115 (Suppl. S8A), 99s–106s. [Google Scholar] [CrossRef]
- Ceriello, A.; Quagliaro, L.; Piconi, L.; Assaloni, R.; Da Ros, R.; Maier, A.; Esposito, K.; Giugliano, D. Effect of postprandial hypertriglyceridemia and hyperglycemia on circulating adhesion molecules and oxidative stress generation and the possible role of simvastatin treatment. Diabetes 2004, 53, 701–710. [Google Scholar] [CrossRef]
- Rhoads, J.P.; Major, A.S. How Oxidized Low-Density Lipoprotein Activates Inflammatory Responses. Crit. Rev. Immunol. 2018, 38, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Liu, J. Association between circulating oxidized low-density lipoprotein and atherosclerotic cardiovascular disease. Chronic Dis. Transl. Med. 2017, 3, 89–94. [Google Scholar] [CrossRef]
- Srivastava, R.A.K. Dysfunctional HDL in diabetes mellitus and its role in the pathogenesis of cardiovascular disease. Mol. Cell. Biochem. 2018, 440, 167–187. [Google Scholar] [CrossRef]
- Pichler, G.; Amigo, N.; Tellez-Plaza, M.; Pardo-Cea, M.A.; Dominguez-Lucas, A.; Marrachelli, V.G.; Monleon, D.; Martin-Escudero, J.C.; Ascaso, J.F.; Chaves, F.J.; et al. LDL particle size and composition and incident cardiovascular disease in a South-European population: The Hortega-Liposcale Follow-up Study. Int. J. Cardiol. 2018, 264, 172–178. [Google Scholar] [CrossRef]
- Li, C.; Chen, Q.; Zhang, M.; Liu, Y.; Chu, Y.; Meng, F.; Wang, J.; Tang, J.; Luo, J.; Niu, X.; et al. The correlation between lipoprotein(a) and coronary atherosclerotic lesion is stronger than LDL-C, when LDL-C is less than 104 mg/dL. BMC Cardiovasc. Disord. 2021, 21, 41. [Google Scholar] [CrossRef]
- Ray, K.K.; Wright, R.S.; Kallend, D.; Koenig, W.; Leiter, L.A.; Raal, F.J.; Bisch, J.A.; Richardson, T.; Jaros, M.; Wijngaard, P.L.J.; et al. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. N. Engl. J. Med. 2020, 382, 1507–1519. [Google Scholar] [CrossRef] [PubMed]
- Gareri, C.; Polimeni, A.; Giordano, S.; Tammè, L.; Curcio, A.; Indolfi, C. Antisense Oligonucleotides and Small Interfering RNA for the Treatment of Dyslipidemias. J. Clin. Med. 2022, 11, 3884. [Google Scholar] [CrossRef] [PubMed]
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A.; Kuder, J.F.; Wang, H.; Liu, T.; Wasserman, S.M.; et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N. Engl. J. Med. 2017, 376, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Koren, M.J.; Sabatine, M.S.; Giugliano, R.P.; Langslet, G.; Wiviott, S.D.; Ruzza, A.; Ma, Y.; Hamer, A.W.; Wasserman, S.M.; Raal, F.J. Long-Term Efficacy and Safety of Evolocumab in Patients With Hypercholesterolemia. J. Am. Coll. Cardiol. 2019, 74, 2132–2146. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.G.; Farnier, M.; Krempf, M.; Bergeron, J.; Luc, G.; Averna, M.; Stroes, E.S.; Langslet, G.; Raal, F.J.; El Shahawy, M.; et al. Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N. Engl. J. Med. 2015, 372, 1489–1499. [Google Scholar] [CrossRef]
- Gaudet, D.; López-Sendón, J.L.; Averna, M.; Bigot, G.; Banach, M.; Letierce, A.; Loy, M.; Samuel, R.; Manvelian, G.; Batsu, I.; et al. Safety and efficacy of alirocumab in a real-life setting: The ODYSSEY APPRISE study. Eur. J. Prev. Cardiol. 2022, 28, 1864–1872. [Google Scholar] [CrossRef]
- Pokrovsky, S.N.; Afanasieva, O.I.; Ezhov, M.V. Therapeutic Apheresis for Management of Lp(a) Hyperlipoproteinemia. Curr. Atheroscler. Rep. 2020, 22, 68. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. Binding and degradation of low density lipoproteins by cultured human fibroblasts. Comparison of cells from a normal subject and from a patient with homozygous familial hypercholesterolemia. J. Biol. Chem. 1974, 249, 5153–5162. [Google Scholar] [CrossRef]
- Mabuchi, H.; Haba, T.; Tatami, R.; Miyamoto, S.; Sakai, Y.; Wakasugi, T.; Watanabe, A.; Koizumi, J.; Takeda, R. Effect of an inhibitor of 3-hydroxy-3-methyglutaryl coenzyme A reductase on serum lipoproteins and ubiquinone-10-levels in patients with familial hypercholesterolemia. N. Engl. J. Med. 1981, 305, 478–482. [Google Scholar] [CrossRef]
- Sprecher, D.L.; Hoeg, J.M.; Schaefer, E.J.; Zech, L.A.; Gregg, R.E.; Lakatos, E.; Brewer, H.B., Jr. The association of LDL receptor activity, LDL cholesterol level, and clinical course in homozygous familial hypercholesterolemia. Metabolism 1985, 34, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Gaddi, A.; Arca, M.; Ciarrocchi, A.; Fazio, S.; D’Alò, G.; Tiozzo, R.; Descovich, G.C.; Calandra, S. Pravastatin in heterozygous familial hypercholesterolemia: Low-density lipoprotein (LDL) cholesterol-lowering effect and LDL receptor activity on skin fibroblastS. Metabolism 1991, 40, 1074–1078. [Google Scholar] [CrossRef] [PubMed]
- Pecoraro, V.; Moja, L.; Dall’Olmo, L.; Cappellini, G.; Garattini, S. Most appropriate animal models to study the efficacy of statins: A systematic review. Eur. J. Clin. Investig. 2014, 44, 848–871. [Google Scholar] [CrossRef]
- Reihnér, E.; Rudling, M.; Ståhlberg, D.; Berglund, L.; Ewerth, S.; Björkhem, I.; Einarsson, K.; Angelin, B. Influence of pravastatin, a specific inhibitor of HMG-CoA reductase, on hepatic metabolism of cholesterol. N. Engl. J. Med. 1990, 323, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Barale, C.; Melchionda, E.; Morotti, A.; Russo, I. PCSK9 Biology and Its Role in Atherothrombosis. Int. J. Mol. Sci. 2021, 22, 5880. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Huang, Y.; Hobbs, H.H.; Cohen, J.C. Molecular characterization of proprotein convertase subtilisin/kexin type 9-mediated degradation of the LDLR. J. Lipid Res. 2012, 53, 1932–1943. [Google Scholar] [CrossRef]
- Fasano, T.; Cefalù, A.B.; Di Leo, E.; Noto, D.; Pollaccia, D.; Bocchi, L.; Valenti, V.; Bonardi, R.; Guardamagna, O.; Averna, M.; et al. A novel loss of function mutation of PCSK9 gene in white subjects with low-plasma low-density lipoprotein cholesterol. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 677–681. [Google Scholar] [CrossRef]
- Denis, M.; Marcinkiewicz, J.; Zaid, A.; Gauthier, D.; Poirier, S.; Lazure, C.; Seidah, N.G.; Prat, A. Gene inactivation of proprotein convertase subtilisin/kexin type 9 reduces atherosclerosis in mice. Circulation 2012, 125, 894–901. [Google Scholar] [CrossRef]
- Shan, Z.N.; Tian, R.; Zhang, M.; Gui, Z.H.; Wu, J.; Ding, M.; Zhou, X.F.; He, J. miR128-1 inhibits the growth of glioblastoma multiforme and glioma stem-like cells via targeting BMI1 and E2F3. Oncotarget 2016, 7, 78813–78826. [Google Scholar] [CrossRef]
- Zhang, S.; Li, D.; Jiao, G.J.; Wang, H.L.; Yan, T.B. miR-185 suppresses progression of Ewing’s sarcoma via inhibiting the PI3K/AKT and Wnt/β-catenin pathways. Onco Targets Ther. 2018, 11, 7967–7977. [Google Scholar] [CrossRef]
- Dostal, Z.; Modriansky, M. The effect of quercetin on microRNA expression: A critical review. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech Repub. 2019, 163, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Goedeke, L.; Rotllan, N.; Canfrán-Duque, A.; Aranda, J.F.; Ramírez, C.M.; Araldi, E.; Lin, C.S.; Anderson, N.N.; Wagschal, A.; de Cabo, R.; et al. MicroRNA-148a regulates LDL receptor and ABCA1 expression to control circulating lipoprotein levels. Nat. Med. 2015, 21, 1280–1289. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Zhang, J.; Du, Y.; Jia, X.; Yang, F.; Si, S.; Wang, L.; Hong, B. microRNA-185 modulates low density lipoprotein receptor expression as a key posttranscriptional regulator. Atherosclerosis 2015, 243, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Bell, D.A.; Hooper, A.J.; Burnett, J.R. Mipomersen, an antisense apolipoprotein B synthesis inhibitor. Expert Opin. Investig. Drugs 2011, 20, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Graham, M.J.; Lee, R.G.; Bell, T.A., 3rd; Fu, W.; Mullick, A.E.; Alexander, V.J.; Singleton, W.; Viney, N.; Geary, R.; Su, J.; et al. Antisense oligonucleotide inhibition of apolipoprotein C-III reduces plasma triglycerides in rodents, nonhuman primates, and humans. Circ. Res. 2013, 112, 1479–1490. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kim, N.H. New Therapeutic Approaches to the Treatment of Dyslipidemia 1: ApoC-III and ANGPTL3. J. Lipid Atheroscler. 2023, 12, 23–36. [Google Scholar] [CrossRef]
- Taghibiglou, C.; Rudy, D.; Van Iderstine, S.C.; Aiton, A.; Cavallo, D.; Cheung, R.; Adeli, K. Intracellular mechanisms regulating apoB-containing lipoprotein assembly and secretion in primary hamster hepatocytes. J. Lipid Res. 2000, 41, 499–513. [Google Scholar] [CrossRef]
- Srivastava, R.A.; Toth, L.; Srivastava, N.; Hinsdale, M.E.; Maeda, N.; Cefalu, A.B.; Averna, M.; Schonfeld, G. Regulation of the apolipoprotein B in heterozygous hypobetalipoproteinemic knock-out mice expressing truncated apoB, B81. Low production and enhanced clearance of apoB cause low levels of apoB. Mol. Cell. Biochem. 1999, 202, 37–46. [Google Scholar] [CrossRef]
- Srivastava, R.A.; Srivastava, N. High density lipoprotein, apolipoprotein A-I, and coronary artery disease. Mol. Cell. Biochem. 2000, 209, 131–144. [Google Scholar] [CrossRef]
- Srivastava, N.; Noto, D.; Averna, M.; Pulai, J.; Srivastava, R.A.; Cole, T.G.; Latour, M.A.; Patterson, B.W.; Schonfeld, G. A new apolipoprotein B truncation (apo B-43.7) in familial hypobetalipoproteinemia: Genetic and metabolic studies. Metabolism 1996, 45, 1296–1304. [Google Scholar] [CrossRef]
- Schonfeld, G.; Patterson, B.W.; Yablonskiy, D.A.; Tanoli, T.S.; Averna, M.; Elias, N.; Yue, P.; Ackerman, J. Fatty liver in familial hypobetalipoproteinemia: Triglyceride assembly into VLDL particles is affected by the extent of hepatic steatosis. J. Lipid Res. 2003, 44, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, N.; Cefalu, A.B.; Noto, D.; Schonfeld, G.; Averna, M.; Srivastava, R.A. The production of 85 kDa N-terminal fragment of apolipoprotein B in mutant HepG2 cells generated by targeted modification of apoB gene occurs by ALLN-inhibitable protease cleavage during translocation. Biochem. Biophys. Res. Commun. 2010, 398, 665–670. [Google Scholar] [CrossRef] [PubMed]
- Noto, D.; Cefalù, A.B.; Cannizzaro, A.; Minà, M.; Fayer, F.; Valenti, V.; Barbagallo, C.M.; Tuttolomondo, A.; Pinto, A.; Sciumè, C.; et al. Familial hypobetalipoproteinemia due to apolipoprotein B R463W mutation causes intestinal fat accumulation and low postprandial lipemia. Atherosclerosis 2009, 206, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Noto, D.; Arca, M.; Tarugi, P.; Cefalù, A.B.; Barbagallo, C.M.; Averna, M.R. Association between familial hypobetalipoproteinemia and the risk of diabetes. Is this the other side of the cholesterol-diabetes connection? A systematic review of literature. Acta Diabetol. 2017, 54, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Olkkonen, V.M.; Sinisalo, J.; Jauhiainen, M. New medications targeting triglyceride-rich lipoproteins: Can inhibition of ANGPTL3 or apoC-III reduce the residual cardiovascular risk? Atherosclerosis 2018, 272, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, K.; Koishi, R.; Shimizugawa, T.; Ando, Y. Angptl3-null mice show low plasma lipid concentrations by enhanced lipoprotein lipase activity. Exp. Anim. 2006, 55, 27–34. [Google Scholar] [CrossRef]
- Larsson, M.; Allan, C.M.; Jung, R.S.; Heizer, P.J.; Beigneux, A.P.; Young, S.G.; Fong, L.G. Apolipoprotein C-III inhibits triglyceride hydrolysis by GPIHBP1-bound LPL. J. Lipid Res. 2017, 58, 1893–1902. [Google Scholar] [CrossRef]
- Borén, J.; Packard, C.J.; Taskinen, M.R. The Roles of ApoC-III on the Metabolism of Triglyceride-Rich Lipoproteins in Humans. Front. Endocrinol. 2020, 11, 474. [Google Scholar] [CrossRef]
- Chen, Y.Q.; Pottanat, T.G.; Zhen, E.Y.; Siegel, R.W.; Ehsani, M.; Qian, Y.W.; Konrad, R.J. ApoA5 lowers triglyceride levels via suppression of ANGPTL3/8-mediated LPL inhibition. J. Lipid Res. 2021, 62, 100068. [Google Scholar] [CrossRef]
- Ooi, E.M.; Barrett, P.H.; Chan, D.C.; Watts, G.F. Apolipoprotein C-III: Understanding an emerging cardiovascular risk factor. Clin. Sci. 2008, 114, 611–624. [Google Scholar] [CrossRef]
- Goldberg, I.J.; Scheraldi, C.A.; Yacoub, L.K.; Saxena, U.; Bisgaier, C.L. Lipoprotein ApoC-II activation of lipoprotein lipase. Modulation by apolipoprotein A-IV. J. Biol. Chem. 1990, 265, 4266–4272. [Google Scholar] [CrossRef]
- Maeda, N.; Li, H.; Lee, D.; Oliver, P.; Quarfordt, S.H.; Osada, J. Targeted disruption of the apolipoprotein C-III gene in mice results in hypotriglyceridemia and protection from postprandial hypertriglyceridemia. J. Biol. Chem. 1994, 269, 23610–23616. [Google Scholar] [CrossRef]
- Digenio, A.; Dunbar, R.L.; Alexander, V.J.; Hompesch, M.; Morrow, L.; Lee, R.G.; Graham, M.J.; Hughes, S.G.; Yu, R.; Singleton, W.; et al. Antisense-Mediated Lowering of Plasma Apolipoprotein C-III by Volanesorsen Improves Dyslipidemia and Insulin Sensitivity in Type 2 Diabetes. Diabetes Care 2016, 39, 1408–1415. [Google Scholar] [CrossRef]
- Fogacci, F.; Norata, G.D.; Toth, P.P.; Arca, M.; Cicero, A.F.G. Efficacy and Safety of Volanesorsen (ISIS 304801): The Evidence from Phase 2 and 3 Clinical Trials. Curr. Atheroscler. Rep. 2020, 22, 18. [Google Scholar] [CrossRef]
- Li, H.; Han, Y.; Qi, R.; Wang, Y.; Zhang, X.; Yu, M.; Tang, Y.; Wang, M.; Shu, Y.N.; Huang, W.; et al. Aggravated restenosis and atherogenesis in ApoCIII transgenic mice but lack of protection in ApoCIII knockouts: The effect of authentic triglyceride-rich lipoproteins with and without ApoCIII. Cardiovasc. Res. 2015, 107, 579–589. [Google Scholar] [CrossRef]
- Pollin, T.I.; Damcott, C.M.; Shen, H.; Ott, S.H.; Shelton, J.; Horenstein, R.B.; Post, W.; McLenithan, J.C.; Bielak, L.F.; Peyser, P.A.; et al. A null mutation in human APOC3 confers a favorable plasma lipid profile and apparent cardioprotection. Science 2008, 322, 1702–1705. [Google Scholar] [CrossRef] [PubMed]
- Crawford, D.C.; Dumitrescu, L.; Goodloe, R.; Brown-Gentry, K.; Boston, J.; McClellan, B., Jr.; Sutcliffe, C.; Wiseman, R.; Baker, P.; Pericak-Vance, M.A.; et al. Rare variant APOC3 R19X is associated with cardio-protective profiles in a diverse population-based survey as part of the Epidemiologic Architecture for Genes Linked to Environment Study. Circ. Cardiovasc. Genet. 2014, 7, 848–853. [Google Scholar] [CrossRef]
- Crawford, D.C.; Restrepo, N.A.; Diggins, K.E.; Farber-Eger, E.; Wells, Q.S. Frequency and phenotype consequence of APOC3 rare variants in patients with very low triglyceride levels. BMC Med. Genom. 2018, 11, 66. [Google Scholar] [CrossRef] [PubMed]
- Wulff, A.B.; Nordestgaard, B.G.; Tybjærg-Hansen, A. APOC3 Loss-of-Function Mutations, Remnant Cholesterol, Low-Density Lipoprotein Cholesterol, and Cardiovascular Risk: Mediation- and Meta-Analyses of 137 895 Individuals. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 660–668. [Google Scholar] [CrossRef] [PubMed]
- Shamsudeen, I.; Hegele, R.A. Safety and efficacy of therapies for chylomicronemia. Expert Rev. Clin. Pharmacol. 2022, 15, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Shimamura, M.; Matsuda, M.; Yasumo, H.; Okazaki, M.; Fujimoto, K.; Kono, K.; Shimizugawa, T.; Ando, Y.; Koishi, R.; Kohama, T.; et al. Angiopoietin-like protein3 regulates plasma HDL cholesterol through suppression of endothelial lipase. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 366–372. [Google Scholar] [CrossRef]
- Koishi, R.; Ando, Y.; Ono, M.; Shimamura, M.; Yasumo, H.; Fujiwara, T.; Horikoshi, H.; Furukawa, H. Angptl3 regulates lipid metabolism in mice. Nat. Genet. 2002, 30, 151–157. [Google Scholar] [CrossRef]
- Mattijssen, F.; Kersten, S. Regulation of triglyceride metabolism by Angiopoietin-like proteins. Biochim. Biophys. Acta 2012, 1821, 782–789. [Google Scholar] [CrossRef]
- Shan, L.; Yu, X.C.; Liu, Z.; Hu, Y.; Sturgis, L.T.; Miranda, M.L.; Liu, Q. The angiopoietin-like proteins ANGPTL3 and ANGPTL4 inhibit lipoprotein lipase activity through distinct mechanisms. J. Biol. Chem. 2009, 284, 1419–1424. [Google Scholar] [CrossRef] [PubMed]
- Tikka, A.; Jauhiainen, M. The role of ANGPTL3 in controlling lipoprotein metabolism. Endocrine 2016, 52, 187–193. [Google Scholar] [CrossRef]
- Wang, Y.; Gusarova, V.; Banfi, S.; Gromada, J.; Cohen, J.C.; Hobbs, H.H. Inactivation of ANGPTL3 reduces hepatic VLDL-triglyceride secretion. J. Lipid Res. 2015, 56, 1296–1307. [Google Scholar] [CrossRef]
- Martín-Campos, J.M.; Roig, R.; Mayoral, C.; Martinez, S.; Martí, G.; Arroyo, J.A.; Julve, J.; Blanco-Vaca, F. Identification of a novel mutation in the ANGPTL3 gene in two families diagnosed of familial hypobetalipoproteinemia without APOB mutation. Clin. Chim. Acta 2012, 413, 552–555. [Google Scholar] [CrossRef] [PubMed]
- Pirillo, A.; Catapano, A.L.; Norata, G.D. Monoclonal Antibodies in the Management of Familial Hypercholesterolemia: Focus on PCSK9 and ANGPTL3 Inhibitors. Curr. Atheroscler. Rep. 2021, 23, 79. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.T.; Yamamoto, T.; Goldstein, J.L.; Brown, M.S. Increased mRNA for low density lipoprotein receptor in livers of rabbits treated with 17 alpha-ethinyl estradiol. Proc. Natl. Acad. Sci. USA 1986, 83, 792–796. [Google Scholar] [CrossRef]
- Erickson, S.K.; Jaeckle, S.; Lear, S.R.; Brady, S.M.; Havel, R.J. Regulation of hepatic cholesterol and lipoprotein metabolism in ethinyl estradiol-treated rats. J. Lipid Res. 1989, 30, 1763–1771. [Google Scholar] [CrossRef]
- Srivastava, R.A.; Baumann, D.; Schonfeld, G. In vivo regulation of low-density lipoprotein receptors by estrogen differs at the post-transcriptional level in rat and mouse. Eur. J. Biochem. 1993, 216, 527–538. [Google Scholar] [CrossRef]
- Srivastava, R.A.; Jiao, S.; Tang, J.J.; Pfleger, B.A.; Kitchens, R.T.; Schonfeld, G. In vivo regulation of low-density lipoprotein receptor and apolipoprotein B gene expressions by dietary fat and cholesterol in inbred strains of mice. Biochim. Biophys. Acta 1991, 1086, 29–43. [Google Scholar] [CrossRef] [PubMed]
- Getz, G.S.; Reardon, C.A. Animal models of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1104–1115. [Google Scholar] [CrossRef] [PubMed]
- Kapourchali, F.R.; Surendiran, G.; Chen, L.; Uitz, E.; Bahadori, B.; Moghadasian, M.H. Animal models of atherosclerosis. World J. Clin. Cases 2014, 2, 126–132. [Google Scholar] [CrossRef]
- Hofmann, S.L.; Russell, D.W.; Brown, M.S.; Goldstein, J.L.; Hammer, R.E. Overexpression of low density lipoprotein (LDL) receptor eliminates LDL from plasma in transgenic mice. Science 1988, 239, 1277–1281. [Google Scholar] [CrossRef]
- Srivastava, R.A.; Jahagirdar, R.; Azhar, S.; Sharma, S.; Bisgaier, C.L. Peroxisome proliferator-activated receptor-alpha selective ligand reduces adiposity, improves insulin sensitivity and inhibits atherosclerosis in LDL receptor-deficient mice. Mol. Cell. Biochem. 2006, 285, 35–50. [Google Scholar] [CrossRef]
- Babaev, V.R.; Ishiguro, H.; Ding, L.; Yancey, P.G.; Dove, D.E.; Kovacs, W.J.; Semenkovich, C.F.; Fazio, S.; Linton, M.F. Macrophage expression of peroxisome proliferator-activated receptor-alpha reduces atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation 2007, 116, 1404–1412. [Google Scholar] [CrossRef]
- Bojic, L.A.; Burke, A.C.; Chhoker, S.S.; Telford, D.E.; Sutherland, B.G.; Edwards, J.Y.; Sawyez, C.G.; Tirona, R.G.; Yin, H.; Pickering, J.G.; et al. Peroxisome proliferator-activated receptor delta agonist GW1516 attenuates diet-induced aortic inflammation, insulin resistance, and atherosclerosis in low-density lipoprotein receptor knockout mice. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 52–60. [Google Scholar] [CrossRef]
- Arai, T.; Wang, N.; Bezouevski, M.; Welch, C.; Tall, A.R. Decreased atherosclerosis in heterozygous low density lipoprotein receptor-deficient mice expressing the scavenger receptor BI transgene. J. Biol. Chem. 1999, 274, 2366–2371. [Google Scholar] [CrossRef]
- Teupser, D.; Kretzschmar, D.; Tennert, C.; Burkhardt, R.; Wilfert, W.; Fengler, D.; Naumann, R.; Sippel, A.E.; Thiery, J. Effect of macrophage overexpression of murine liver X receptor-alpha (LXR-alpha) on atherosclerosis in LDL-receptor deficient mice. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 2009–2015. [Google Scholar] [CrossRef]
- Zhang, S.H.; Reddick, R.L.; Piedrahita, J.A.; Maeda, N. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science 1992, 258, 468–471. [Google Scholar] [CrossRef]
- Plump, A.S.; Smith, J.D.; Hayek, T.; Aalto-Setala, K.; Walsh, A.; Verstuyft, J.G.; Rubin, E.M.; Breslow, J.L. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell 1992, 71, 343–353. [Google Scholar] [CrossRef]
- Meurs, I.; Lammers, B.; Zhao, Y.; Out, R.; Hildebrand, R.B.; Hoekstra, M.; Van Berkel, T.J.; Van Eck, M. The effect of ABCG1 deficiency on atherosclerotic lesion development in LDL receptor knockout mice depends on the stage of atherogenesis. Atherosclerosis 2012, 221, 41–47. [Google Scholar] [CrossRef]
- Yin, K.; Tang, S.L.; Yu, X.H.; Tu, G.H.; He, R.F.; Li, J.F.; Xie, D.; Gui, Q.J.; Fu, Y.C.; Jiang, Z.S.; et al. Apolipoprotein A-I inhibits LPS-induced atherosclerosis in ApoE(−/−) mice possibly via activated STAT3-mediated upregulation of tristetraprolin. Acta Pharmacol. Sin. 2013, 34, 837–846. [Google Scholar] [CrossRef] [PubMed]
- Baldan, A.; Pei, L.; Lee, R.; Tarr, P.; Tangirala, R.K.; Weinstein, M.M.; Frank, J.; Li, A.C.; Tontonoz, P.; Edwards, P.A. Impaired development of atherosclerosis in hyperlipidemic Ldlr−/− and ApoE−/− mice transplanted with Abcg1−/− bone marrow. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2301–2307. [Google Scholar] [CrossRef] [PubMed]
- Sithu, S.D.; Malovichko, M.V.; Riggs, K.A.; Wickramasinghe, N.S.; Winner, M.G.; Agarwal, A.; Hamed-Berair, R.E.; Kalani, A.; Riggs, D.W.; Bhatnagar, A.; et al. Atherogenesis and metabolic dysregulation in LDL receptor-knockout rats. JCI Insight 2017, 2, e86442. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, A.; Taghibiglou, C.; Leung, N.; Szeto, L.; Van Iderstine, S.C.; Uffelman, K.D.; Buckingham, R.; Adeli, K.; Lewis, G.F. Ameliorated hepatic insulin resistance is associated with normalization of microsomal triglyceride transfer protein expression and reduction in very low density lipoprotein assembly and secretion in the fructose-fed hamster. J. Biol. Chem. 2002, 277, 28795–28802. [Google Scholar] [CrossRef]
- Srivastava, R.A.; He, S. Anti-hyperlipidemic and insulin sensitizing activities of fenofibrate reduces aortic lipid deposition in hyperlipidemic Golden Syrian hamster. Mol. Cell. Biochem. 2010, 345, 197–206. [Google Scholar] [CrossRef]
- Nistor, A.; Bulla, A.; Filip, D.A.; Radu, A. The hyperlipidemic hamster as a model of experimental atherosclerosis. Atherosclerosis 1987, 68, 159–173. [Google Scholar] [CrossRef]
- Srivastava, R.A. Evaluation of anti-atherosclerotic activities of PPAR-alpha, PPAR-gamma, and LXR agonists in hyperlipidemic atherosclerosis-susceptible F(1)B hamsters. Atherosclerosis 2011, 214, 86–93. [Google Scholar] [CrossRef]
- Liu, G.L.; Fan, L.M.; Redinger, R.N. The association of hepatic apoprotein and lipid metabolism in hamsters and rats. Comp. Biochem. Physiol. A Comp. Physiol. 1991, 99, 223–228. [Google Scholar] [CrossRef]
- Srivastava, R.A.; Tang, J.; Baumann, D.; Schonfeld, G. Hormonal and nutritional stimuli modulate apolipoprotein B mRNA editing in mouse liver. Biochem. Biophys. Res. Commun. 1992, 188, 135–141. [Google Scholar] [CrossRef]
- Spady, D.K.; Dietschy, J.M. Interaction of dietary cholesterol and triglycerides in the regulation of hepatic low density lipoprotein transport in the hamster. J. Clin. Investig. 1988, 81, 300–309. [Google Scholar] [CrossRef]
- He, K.; Wang, J.; Shi, H.; Yu, Q.; Zhang, X.; Guo, M.; Sun, H.; Lin, X.; Wu, Y.; Wang, L.; et al. An interspecies study of lipid profiles and atherosclerosis in familial hypercholesterolemia animal models with low-density lipoprotein receptor deficiency. Am. J. Transl. Res. 2019, 11, 3116–3127. [Google Scholar]
- Guo, X.; Gao, M.; Wang, Y.; Lin, X.; Yang, L.; Cong, N.; An, X.; Wang, F.; Qu, K.; Yu, L.; et al. LDL Receptor Gene-ablated Hamsters: A Rodent Model of Familial Hypercholesterolemia with Dominant Inheritance and Diet-induced Coronary Atherosclerosis. eBioMedicine 2018, 27, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; He, K.; Yang, C.; Lin, X.; Zhang, X.; Wang, Y.; Liu, G.; Xian, X. Dietary Cholesterol Is Highly Associated with Severity of Hyperlipidemia and Atherosclerotic Lesions in Heterozygous LDLR-Deficient Hamsters. Int. J. Mol. Sci. 2019, 20, 3515. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Xu, M.J.; Cao, Z.; Yang, C.; Wang, J.; Wang, B.; Liu, J.; Wang, Y.; Xian, X.; Zhang, F.; et al. Heterozygous Ldlr-Deficient Hamster as a Model to Evaluate the Efficacy of PCSK9 Antibody in Hyperlipidemia and Atherosclerosis. Int. J. Mol. Sci. 2019, 20, 5936. [Google Scholar] [CrossRef] [PubMed]
- Di Taranto, M.D.; Giacobbe, C.; Fortunato, G. Familial hypercholesterolemia: A complex genetic disease with variable phenotypes. Eur. J. Med. Genet. 2020, 63, 103831. [Google Scholar] [CrossRef]
- Streja, D.; Steiner, G.; Kwiterovich, P.O., Jr. Plasma high-density lipoproteins and ischemic heart disease: Studies in a large kindred with familial hypercholesterolemia. Ann. Intern. Med. 1978, 89, 871–880. [Google Scholar] [CrossRef]
- Brousseau, M.E.; Wang, J.; Demosky, S.J., Jr.; Vaisman, B.L.; Talley, G.D.; Santamarina-Fojo, S.; Brewer, H.B., Jr.; Hoeg, J.M. Correction of hypoalphalipoproteinemia in LDL receptor-deficient rabbits by lecithin:cholesterol acyltransferase. J. Lipid Res. 1998, 39, 1558–1567. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y. Serial inbreeding of rabbits with hereditary hyperlipidemia (WHHL-rabbit). Atherosclerosis 1980, 36, 261–268. [Google Scholar] [CrossRef]
- Tanzawa, K.; Shimada, Y.; Kuroda, M.; Tsujita, Y.; Arai, M.; Watanabe, H. WHHL-rabbit: A low density lipoprotein receptor-deficient animal model for familial hypercholesterolemia. FEBS Lett. 1980, 118, 81–84. [Google Scholar] [CrossRef]
- Yamamoto, T.; Bishop, R.W.; Brown, M.S.; Goldstein, J.L.; Russell, D.W. Deletion in cysteine-rich region of LDL receptor impedes transport to cell surface in WHHL rabbit. Science 1986, 232, 1230–1237. [Google Scholar] [CrossRef]
- Kume, N.; Kita, T.; Mikami, A.; Yokode, M.; Ishii, K.; Nagano, Y.; Kawai, C. Induction of mRNA for low-density lipoprotein receptors in heterozygous Watanabe heritable hyperlipidemic rabbits treated with CS-514 (Pravastatin) and cholestyramine. Circulation 1989, 79, 1084–1090. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.T.; Wang, X.J.; Rohret, J.A.; Struzynski, J.T.; Merricks, E.P.; Bellinger, D.A.; Rohret, F.A.; Nichols, T.C.; Rogers, C.S. Targeted disruption of LDLR causes hypercholesterolemia and atherosclerosis in Yucatan miniature pigs. PLoS ONE 2014, 9, e93457. [Google Scholar] [CrossRef] [PubMed]
- Amuzie, C.; Swart, J.R.; Rogers, C.S.; Vihtelic, T.; Denham, S.; Mais, D.E. A Translational Model for Diet-related Atherosclerosis: Effect of Statins on Hypercholesterolemia and Atherosclerosis in a Minipig. Toxicol. Pathol. 2016, 44, 442–449. [Google Scholar] [CrossRef]
- Li, Y.; Fuchimoto, D.; Sudo, M.; Haruta, H.; Lin, Q.F.; Takayama, T.; Morita, S.; Nochi, T.; Suzuki, S.; Sembon, S.; et al. Development of Human-Like Advanced Coronary Plaques in Low-Density Lipoprotein Receptor Knockout Pigs and Justification for Statin Treatment Before Formation of Atherosclerotic Plaques. J. Am. Heart Assoc. 2016, 5, e002779. [Google Scholar] [CrossRef] [PubMed]
- Burke, A.C.; Telford, D.E.; Sutherland, B.G.; Edwards, J.Y.; Sawyez, C.G.; Barrett, P.H.R.; Newton, R.S.; Pickering, J.G.; Huff, M.W. Bempedoic Acid Lowers Low-Density Lipoprotein Cholesterol and Attenuates Atherosclerosis in Low-Density Lipoprotein Receptor-Deficient (LDLR(+/−) and LDLR(−/−)) Yucatan Miniature Pigs. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1178–1190. [Google Scholar] [CrossRef]
- Chan, J.C.; Piper, D.E.; Cao, Q.; Liu, D.; King, C.; Wang, W.; Tang, J.; Liu, Q.; Higbee, J.; Xia, Z.; et al. A proprotein convertase subtilisin/kexin type 9 neutralizing antibody reduces serum cholesterol in mice and nonhuman primates. Proc. Natl. Acad. Sci. USA 2009, 106, 9820–9825. [Google Scholar] [CrossRef]
- Kumar, S.; Kang, D.W.; Rezvan, A.; Jo, H. Accelerated atherosclerosis development in C57Bl6 mice by overexpressing AAV-mediated PCSK9 and partial carotid ligation. Lab. Investig. 2017, 97, 935–945. [Google Scholar] [CrossRef]
- Small, A.M.; Huffman, J.E.; Klarin, D.; Lynch, J.A.; Assimes, T.; DuVall, S.; Sun, Y.V.; Shere, L.; Natarajan, P.; Gaziano, M.; et al. PCSK9 loss of function is protective against extra-coronary atherosclerotic cardiovascular disease in a large multi-ethnic cohort. PLoS ONE 2020, 15, e0239752. [Google Scholar] [CrossRef]
- Bayona, A.; Arrieta, F.; Rodríguez-Jiménez, C.; Cerrato, F.; Rodríguez-Nóvoa, S.; Fernández-Lucas, M.; Gómez-Coronado, D.; Mata, P. Loss-of-function mutation of PCSK9 as a protective factor in the clinical expression of familial hypercholesterolemia: A case report. Medicine 2020, 99, e21754. [Google Scholar] [CrossRef]
- Sánchez-Hernández, R.M.; Di Taranto, M.D.; Benito-Vicente, A.; Uribe, K.B.; Lamiquiz-Moneo, I.; Larrea-Sebal, A.; Jebari, S.; Galicia-Garcia, U.; Nóvoa, F.J.; Boronat, M.; et al. The Arg499His gain-of-function mutation in the C-terminal domain of PCSK9. Atherosclerosis 2019, 289, 162–172. [Google Scholar] [CrossRef]
- Pećin, I.; Reiner, Ž. Novel Experimental Agents for the Treatment of Hypercholesterolemia. J. Exp. Pharmacol. 2021, 13, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Musunuru, K.; Chadwick, A.C.; Mizoguchi, T.; Garcia, S.P.; DeNizio, J.E.; Reiss, C.W.; Wang, K.; Iyer, S.; Dutta, C.; Clendaniel, V.; et al. In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates. Nature 2021, 593, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.L.; Shiu, S.W.; Cheung, C.L.; Yu-Hung Leung, A.; Tan, K.C. Effects of statins on the inducible degrader of low-density lipoprotein receptor in familial hypercholesterolemia. Endocr. Connect. 2022, 11, e220019. [Google Scholar] [CrossRef] [PubMed]
- Somanathan, S.; Jacobs, F.; Wang, Q.; Hanlon, A.L.; Wilson, J.M.; Rader, D.J. AAV vectors expressing LDLR gain-of-function variants demonstrate increased efficacy in mouse models of familial hypercholesterolemia. Circ. Res. 2014, 115, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Wada, F.; Yamamoto, T.; Kobayashi, T.; Tachibana, K.; Ito, K.R.; Hamasaki, M.; Kayaba, Y.; Terada, C.; Yamayoshi, A.; Obika, S.; et al. Drug discovery and development scheme for liver-targeting bridged nucleic acid antisense oligonucleotides. Mol. Ther. Nucleic Acids 2021, 26, 957–969. [Google Scholar] [CrossRef] [PubMed]
- Zelcer, N.; Hong, C.; Boyadjian, R.; Tontonoz, P. LXR regulates cholesterol uptake through Idol-dependent ubiquitination of the LDL receptor. Science 2009, 325, 100–104. [Google Scholar] [CrossRef]
- Liang, C.; Wang, X.; Peng, K.; Lai, P.; Liu, Z.; Ma, J.; Chen, X.; Liu, G.; Zheng, M.; Wang, Y.; et al. Idol Depletion Protects against Spontaneous Atherosclerosis in a Hamster Model of Familial Hypercholesterolemia. Oxid. Med. Cell. Longev. 2022, 2022, 1889632. [Google Scholar] [CrossRef]
- Sasaki, M.; Terao, Y.; Ayaori, M.; Uto-Kondo, H.; Iizuka, M.; Yogo, M.; Hagisawa, K.; Takiguchi, S.; Yakushiji, E.; Nakaya, K.; et al. Hepatic overexpression of idol increases circulating protein convertase subtilisin/kexin type 9 in mice and hamsters via dual mechanisms: Sterol regulatory element-binding protein 2 and low-density lipoprotein receptor-dependent pathways. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1171–1178. [Google Scholar] [CrossRef] [PubMed]
- Maestro, S.; Weber, N.D.; Zabaleta, N.; Aldabe, R.; Gonzalez-Aseguinolaza, G. Novel vectors and approaches for gene therapy in liver diseases. JHEP Rep. 2021, 3, 100300. [Google Scholar] [CrossRef] [PubMed]
- Ramms, B.; Patel, S.; Nora, C.; Pessentheiner, A.R.; Chang, M.W.; Green, C.R.; Golden, G.J.; Secrest, P.; Krauss, R.M.; Metallo, C.M.; et al. ApoC-III ASO promotes tissue LPL activity in the absence of apoE-mediated TRL clearance. J. Lipid Res. 2019, 60, 1379–1395. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, A.B.; Frikke-Schmidt, R.; Nordestgaard, B.G.; Tybjærg-Hansen, A. Loss-of-function mutations in APOC3 and risk of ischemic vascular disease. N. Engl. J. Med. 2014, 371, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Crosby, J.; Peloso, G.M.; Auer, P.L.; Crosslin, D.R.; Stitziel, N.O.; Lange, L.A.; Lu, Y.; Tang, Z.Z.; Zhang, H.; Hindy, G.; et al. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N. Engl. J. Med. 2014, 371, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Liu, A.; Wang, S.; Wang, T.; Hu, D.; Wu, S.; Peng, D. ApoCIII enrichment in HDL impairs HDL-mediated cholesterol efflux capacity. Sci. Rep. 2017, 7, 2312. [Google Scholar] [CrossRef]
- Apro, J.; Tietge, U.J.; Dikkers, A.; Parini, P.; Angelin, B.; Rudling, M. Impaired Cholesterol Efflux Capacity of High-Density Lipoprotein Isolated From Interstitial Fluid in Type 2 Diabetes Mellitus—Brief Report. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 787–791. [Google Scholar] [CrossRef]
- Chen, P.Y.; Gao, W.Y.; Liou, J.W.; Lin, C.Y.; Wu, M.J.; Yen, J.H. Angiopoietin-Like Protein 3 (ANGPTL3) Modulates Lipoprotein Metabolism and Dyslipidemia. Int. J. Mol. Sci. 2021, 22, 7310. [Google Scholar] [CrossRef]
- Bell, T.A., 3rd; Liu, M.; Donner, A.J.; Lee, R.G.; Mullick, A.E.; Crooke, R.M. Antisense oligonucleotide-mediated inhibition of angiopoietin-like protein 3 increases reverse cholesterol transport in mice. J. Lipid Res. 2021, 62, 100101. [Google Scholar] [CrossRef]
- Srivastava, N.; Cefalu, A.B.; Averna, M.; Srivastava, R.A.K. Lack of Correlation of Plasma HDL with Fecal Cholesterol and Plasma Cholesterol Efflux Capacity Suggests Importance of HDL Functionality in Attenuation of Atherosclerosis. Front. Physiol. 2018, 9, 1222. [Google Scholar] [CrossRef]
- Srivastava, R.A.K.; Cefalu, A.B.; Srivastava, N.S.; Averna, M. NPC1L1 and ABCG5/8 induction explain synergistic fecal cholesterol excretion in ob/ob mice co-treated with PPAR-α and LXR agonists. Mol. Cell. Biochem. 2020, 473, 247–262. [Google Scholar] [CrossRef]
- Defesche, J.C.; Gidding, S.S.; Harada-Shiba, M.; Hegele, R.A.; Santos, R.D.; Wierzbicki, A.S. Familial hypercholesterolaemia. Nat. Rev. Dis. Prim. 2017, 3, 17093. [Google Scholar] [CrossRef] [PubMed]
- Gretarsdottir, S.; Helgason, H.; Helgadottir, A.; Sigurdsson, A.; Thorleifsson, G.; Magnusdottir, A.; Oddsson, A.; Steinthorsdottir, V.; Rafnar, T.; de Graaf, J.; et al. A Splice Region Variant in LDLR Lowers Non-high Density Lipoprotein Cholesterol and Protects against Coronary Artery Disease. PLoS Genet. 2015, 11, e1005379. [Google Scholar] [CrossRef] [PubMed]
- Bjornsson, E.; Gunnarsdottir, K.; Halldorsson, G.H.; Sigurdsson, A.; Arnadottir, G.A.; Jonsson, H.; Olafsdottir, E.F.; Niehus, S.; Kehr, B.; Sveinbjörnsson, G.; et al. Lifelong Reduction in LDL (Low-Density Lipoprotein) Cholesterol due to a Gain-of-Function Mutation in LDLR. Circ. Genom. Precis. Med. 2021, 14, e003029. [Google Scholar] [CrossRef]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef] [PubMed]
- Rosenson, R.S.; Burgess, L.J.; Ebenbichler, C.F.; Baum, S.J.; Stroes, E.S.G.; Ali, S.; Khilla, N.; Hamlin, R.; Pordy, R.; Dong, Y.; et al. Evinacumab in Patients with Refractory Hypercholesterolemia. N. Engl. J. Med. 2020, 383, 2307–2319. [Google Scholar] [CrossRef]
- Hamilton-Craig, I.; Kostner, K.; Colquhoun, D.; Woodhouse, S. Combination therapy of statin and ezetimibe for the treatment of familial hypercholesterolemia. Vasc. Health Risk Manag. 2010, 6, 1023–1037. [Google Scholar] [CrossRef]
- Raal, F.J.; Stein, E.A.; Dufour, R.; Turner, T.; Civeira, F.; Burgess, L.; Langslet, G.; Scott, R.; Olsson, A.G.; Sullivan, D.; et al. PCSK9 inhibition with evolocumab (AMG 145) in heterozygous familial hypercholesterolaemia (RUTHERFORD-2): A randomised, double-blind, placebo-controlled trial. Lancet 2015, 385, 331–340. [Google Scholar] [CrossRef]
- Castilla Cabezas, J.A.; López-Cillero, P.; Jiménez, J.; Fraga, E.; Arizón, J.M.; Briceño, J.; Solórzano, G.; De la Mata, M.; Pera, C. Role of orthotopic liver transplant in the treatment of homozygous familial hypercholesterolemia. Rev. Esp. Enferm. Dig. 2000, 92, 601–608. [Google Scholar]
- Goldberg, A.C.; Hopkins, P.N.; Toth, P.P.; Ballantyne, C.M.; Rader, D.J.; Robinson, J.G.; Daniels, S.R.; Gidding, S.S.; de Ferranti, S.D.; Ito, M.K.; et al. Familial hypercholesterolemia: Screening, diagnosis and management of pediatric and adult patients: Clinical guidance from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J. Clin. Lipidol. 2011, 5, S1–S8. [Google Scholar] [CrossRef]
- Grossman, M.; Rader, D.J.; Muller, D.W.; Kolansky, D.M.; Kozarsky, K.; Clark, B.J., 3rd; Stein, E.A.; Lupien, P.J.; Brewer, H.B., Jr.; Raper, S.E.; et al. A pilot study of ex vivo gene therapy for homozygous familial hypercholesterolaemia. Nat. Med. 1995, 1, 1148–1154. [Google Scholar] [CrossRef]
- Oertel, M.; Rosencrantz, R.; Chen, Y.Q.; Thota, P.N.; Sandhu, J.S.; Dabeva, M.D.; Pacchia, A.L.; Adelson, M.E.; Dougherty, J.P.; Shafritz, D.A. Repopulation of rat liver by fetal hepatoblasts and adult hepatocytes transduced ex vivo with lentiviral vectors. Hepatology 2003, 37, 994–1005. [Google Scholar] [CrossRef]
- Hytönen, E.; Laurema, A.; Kankkonen, H.; Miyanohara, A.; Kärjä, V.; Hujo, M.; Laham-Karam, N.; Ylä-Herttuala, S. Bile-duct proliferation as an unexpected side-effect after AAV2-LDLR gene transfer to rabbit liver. Sci. Rep. 2019, 9, 6934. [Google Scholar] [CrossRef]
- Leggiero, E.; Labruna, G.; Iaffaldano, L.; Lombardo, B.; Greco, A.; Fiorenza, D.; Gramanzini, M.; Montanaro, D.; Baldi, A.; Cerullo, V.; et al. Helper-dependent adenovirus-mediated gene transfer of a secreted LDL receptor/transferrin chimeric protein reduces aortic atherosclerosis in LDL receptor-deficient mice. Gene Ther. 2019, 26, 121–130. [Google Scholar] [CrossRef]
- Kassim, S.H.; Li, H.; Vandenberghe, L.H.; Hinderer, C.; Bell, P.; Marchadier, D.; Wilson, A.; Cromley, D.; Redon, V.; Yu, H.; et al. Gene therapy in a humanized mouse model of familial hypercholesterolemia leads to marked regression of atherosclerosis. PLoS ONE 2010, 5, e13424. [Google Scholar] [CrossRef]
- Li, Z.; Zhao, P.; Zhang, Y.; Wang, J.; Wang, C.; Liu, Y.; Yang, G.; Yuan, L. Exosome-based Ldlr gene therapy for familial hypercholesterolemia in a mouse model. Theranostics 2021, 11, 2953–2965. [Google Scholar] [CrossRef]
- Kassim, S.H.; Li, H.; Bell, P.; Somanathan, S.; Lagor, W.; Jacobs, F.; Billheimer, J.; Wilson, J.M.; Rader, D.J. Adeno-associated virus serotype 8 gene therapy leads to significant lowering of plasma cholesterol levels in humanized mouse models of homozygous and heterozygous familial hypercholesterolemia. Hum. Gene Ther. 2013, 24, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Muthuramu, I.; Somanathan, S.; Zhang, H.; Bell, P.; He, Z.; Yu, H.; Zhu, Y.; Tretiakova, A.P.; Wilson, J.M. Developing a second-generation clinical candidate AAV vector for gene therapy of familial hypercholesterolemia. Mol. Ther. Methods Clin. Dev. 2021, 22, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Jiao, R.; Guo, X.; Wang, T.; Chen, P.; Wang, D.; Chen, Y.; He, C.Y.; Chen, Z.Y. Construction of minicircle DNA vectors capable of correcting familial hypercholesterolemia phenotype in a LDLR-deficient mouse model. Gene Ther. 2016, 23, 657–663. [Google Scholar] [CrossRef]
- Zhao, H.; Li, Y.; He, L.; Pu, W.; Yu, W.; Li, Y.; Wu, Y.T.; Xu, C.; Wei, Y.; Ding, Q.; et al. In Vivo AAV-CRISPR/Cas9-Mediated Gene Editing Ameliorates Atherosclerosis in Familial Hypercholesterolemia. Circulation 2020, 141, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Lev, M.; Gefen, L.; Levy, Y.; Gotliv, I.; Izhar, L.; Emmanuel, R. CRISPR-based Gene Editing Enhances LDLR Expression and Boosts LDL-C Uptake in Familial Hypercholesterolemia. Mol. Ther. 2022, 30, 466. [Google Scholar]
- Qiu, M.; Glass, Z.; Chen, J.; Haas, M.; Jin, X.; Zhao, X.; Rui, X.; Ye, Z.; Li, Y.; Zhang, F.; et al. Lipid nanoparticle-mediated codelivery of Cas9 mRNA and single-guide RNA achieves liver-specific in vivo genome editing of Angptl3. Proc. Natl. Acad. Sci. USA 2021, 118, e2020401118. [Google Scholar] [CrossRef] [PubMed]
- Chadwick, A.C.; Wang, X.; Musunuru, K. In Vivo Base Editing of PCSK9 (Proprotein Convertase Subtilisin/Kexin Type 9) as a Therapeutic Alternative to Genome Editing. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1741–1747. [Google Scholar] [CrossRef]
- Chadwick, A.C.; Evitt, N.H.; Lv, W.; Musunuru, K. Reduced Blood Lipid Levels With In Vivo CRISPR-Cas9 Base Editing of ANGPTL3. Circulation 2018, 137, 975–977. [Google Scholar] [CrossRef] [PubMed]
- Rothgangl, T.; Dennis, M.K.; Lin, P.J.C.; Oka, R.; Witzigmann, D.; Villiger, L.; Qi, W.; Hruzova, M.; Kissling, L.; Lenggenhager, D.; et al. In vivo adenine base editing of PCSK9 in macaques reduces LDL cholesterol levels. Nat. Biotechnol. 2021, 39, 949–957. [Google Scholar] [CrossRef]
- Khera, A.; Lee, R.; Rohde, E.; Layaram, H.; Kathiresan, S.; Bellinger, A. An in vivo CRISPR base editing therapy to inactivate ANGPTL3: Nomination of a development candidate for VERVE-201. Eur. Heart J. 2022, 43, 3087. [Google Scholar] [CrossRef]
- Omer, L.; Hudson, E.A.; Zheng, S.; Hoying, J.B.; Shan, Y.; Boyd, N.L. CRISPR Correction of a Homozygous Low-Density Lipoprotein Receptor Mutation in Familial Hypercholesterolemia Induced Pluripotent Stem Cells. Hepatol. Commun. 2017, 1, 886–898. [Google Scholar] [CrossRef]
- Ibraheim, R.; Song, C.Q.; Mir, A.; Amrani, N.; Xue, W.; Sontheimer, E.J. All-in-one adeno-associated virus delivery and genome editing by Neisseria meningitidis Cas9 in vivo. Genome Biol. 2018, 19, 137. [Google Scholar] [CrossRef]
- Jiang, C.; Mei, M.; Li, B.; Zhu, X.; Zu, W.; Tian, Y.; Wang, Q.; Guo, Y.; Dong, Y.; Tan, X. A non-viral CRISPR/Cas9 delivery system for therapeutically targeting HBV DNA and pcsk9 in vivo. Cell Res. 2017, 27, 440–443. [Google Scholar] [CrossRef]
- Okada, H.; Nakanishi, C.; Yoshida, S.; Shimojima, M.; Yokawa, J.; Mori, M.; Tada, H.; Yoshimuta, T.; Hayashi, K.; Yamano, T.; et al. Function and Immunogenicity of Gene-corrected iPSC-derived Hepatocyte-Like Cells in Restoring Low Density Lipoprotein Uptake in Homozygous Familial Hypercholesterolemia. Sci. Rep. 2019, 9, 4695. [Google Scholar] [CrossRef]
- Kuijper, E.C.; Bergsma, A.J.; Pijnappel, W.; Aartsma-Rus, A. Opportunities and challenges for antisense oligonucleotide therapies. J. Inherit. Metab. Dis. 2021, 44, 72–87. [Google Scholar] [CrossRef] [PubMed]
- Golkar, Z. CRISPR: A journey of gene-editing based medicine. Genes Genom. 2020, 42, 1369–1380. [Google Scholar] [CrossRef] [PubMed]
- Witzigmann, D.; Kulkarni, J.A.; Leung, J.; Chen, S.; Cullis, P.R.; van der Meel, R. Lipid nanoparticle technology for therapeutic gene regulation in the liver. Adv. Drug Deliv. Rev. 2020, 159, 344–363. [Google Scholar] [CrossRef]
- Bessis, N.; GarciaCozar, F.J.; Boissier, M.C. Immune responses to gene therapy vectors: Influence on vector function and effector mechanisms. Gene Ther. 2004, 11 (Suppl. S1), S10–S17. [Google Scholar] [CrossRef]
- Kasiewicz, L.N.; Biswas, S.; Beach, A.; Ren, H.; Dutta, C.; Marie, A.; Rohde, M.E.; Chadwick, A.; Cheng, C.; Musunuru, K.; et al. Lipid nanoparticles incorporating a GalNAc ligand enable in vivo liver ANGPTL3 editing in wild-type and somatic LDLR knockout non-human primates. bioRxiv 2021, 10, 1–7. [Google Scholar] [CrossRef]
- Rees, H.A.; Liu, D.R. Publisher Correction: Base editing: Precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018, 19, 801. [Google Scholar] [CrossRef]
- Kantor, A.; McClements, M.E.; MacLaren, R.E. CRISPR-Cas9 DNA Base-Editing and Prime-Editing. Int. J. Mol. Sci. 2020, 21, 6240. [Google Scholar] [CrossRef]
- Kim, E.; Koo, T.; Park, S.W.; Kim, D.; Kim, K.; Cho, H.Y.; Song, D.W.; Lee, K.J.; Jung, M.H.; Kim, S.; et al. In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nat. Commun. 2017, 8, 14500. [Google Scholar] [CrossRef]
- Wang, X.; Raghavan, A.; Chen, T.; Qiao, L.; Zhang, Y.; Ding, Q.; Musunuru, K. CRISPR-Cas9 Targeting of PCSK9 in Human Hepatocytes In Vivo—Brief Report. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 783–786. [Google Scholar] [CrossRef]
- Carreras, A.; Pane, L.S.; Nitsch, R.; Madeyski-Bengtson, K.; Porritt, M.; Akcakaya, P.; Taheri-Ghahfarokhi, A.; Ericson, E.; Bjursell, M.; Perez-Alcazar, M.; et al. In vivo genome and base editing of a human PCSK9 knock-in hypercholesterolemic mouse model. BMC Biol. 2019, 17, 4. [Google Scholar] [CrossRef]





| Preclinical Studies | Drug Class | Primary Endpoint | Mechanism | Refs. |
|---|---|---|---|---|
| Adenoviral | Gene therapy | LDL | LDLR expression | [205,207] |
| AAV | Gene therapy | LDL | LDLR expression | [208] |
| AAV/CRISPR-Cas9 | Gene editing | LDL | LDLR expression | [210] |
| CRISPR-Cas9 | Gene editing | LDL | LDLR expression | [211] |
| LNP/CRISPR-Cas9 | Gene editing | LDL, TG | ANGPTL silencing | [212] |
| CRISPR-Cas9 | Base editing | LDL | PCSK9 silencing | [213] |
| CRISPR-Cas9 | Base editing | LDL, TG | ANGPTL silencing | [214] |
| CRISPR-Cas9 | Base editing | LDL | PCSK9 silencing | [215] |
| CRISPR-Cas9 | Base editing | LDL, TG | ANGPTL silencing | [216] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Srivastava, R.A.K. A Review of Progress on Targeting LDL Receptor-Dependent and -Independent Pathways for the Treatment of Hypercholesterolemia, a Major Risk Factor of ASCVD. Cells 2023, 12, 1648. https://doi.org/10.3390/cells12121648
Srivastava RAK. A Review of Progress on Targeting LDL Receptor-Dependent and -Independent Pathways for the Treatment of Hypercholesterolemia, a Major Risk Factor of ASCVD. Cells. 2023; 12(12):1648. https://doi.org/10.3390/cells12121648
Chicago/Turabian StyleSrivastava, Rai Ajit K. 2023. "A Review of Progress on Targeting LDL Receptor-Dependent and -Independent Pathways for the Treatment of Hypercholesterolemia, a Major Risk Factor of ASCVD" Cells 12, no. 12: 1648. https://doi.org/10.3390/cells12121648
APA StyleSrivastava, R. A. K. (2023). A Review of Progress on Targeting LDL Receptor-Dependent and -Independent Pathways for the Treatment of Hypercholesterolemia, a Major Risk Factor of ASCVD. Cells, 12(12), 1648. https://doi.org/10.3390/cells12121648