
Association between MGMT Enhancer Methylation and MGMT Promoter Methylation, MGMT Protein Expression, and Overall Survival in Glioblastoma
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples and Cell Culturing
2.2. DNA Extraction and Bisulfite Conversion
2.3. Primer Design and PCR Conditions for DNA Methylation Analysis
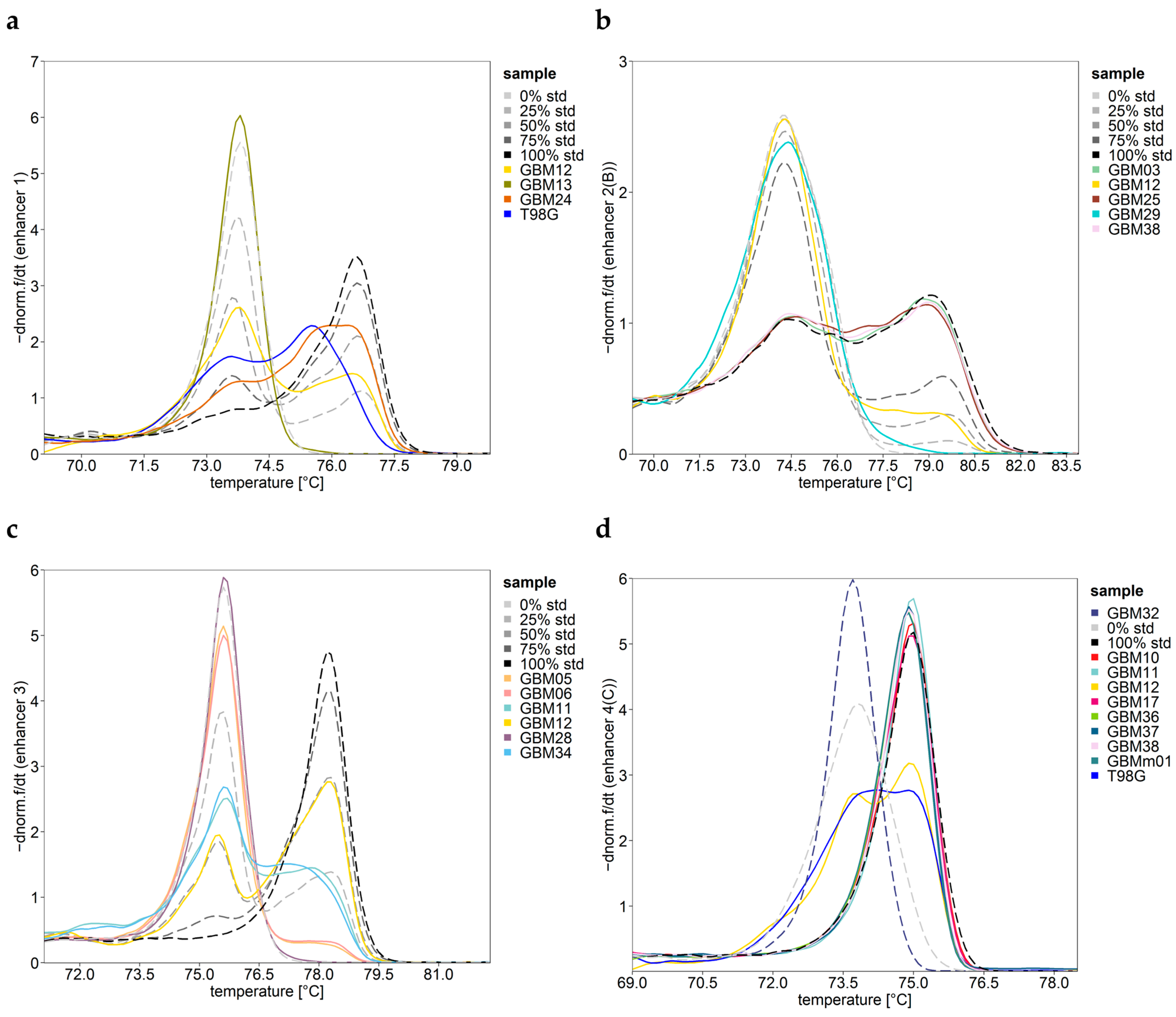
2.4. PSQ of PCR Products
2.5. Data Analysis and Statistics
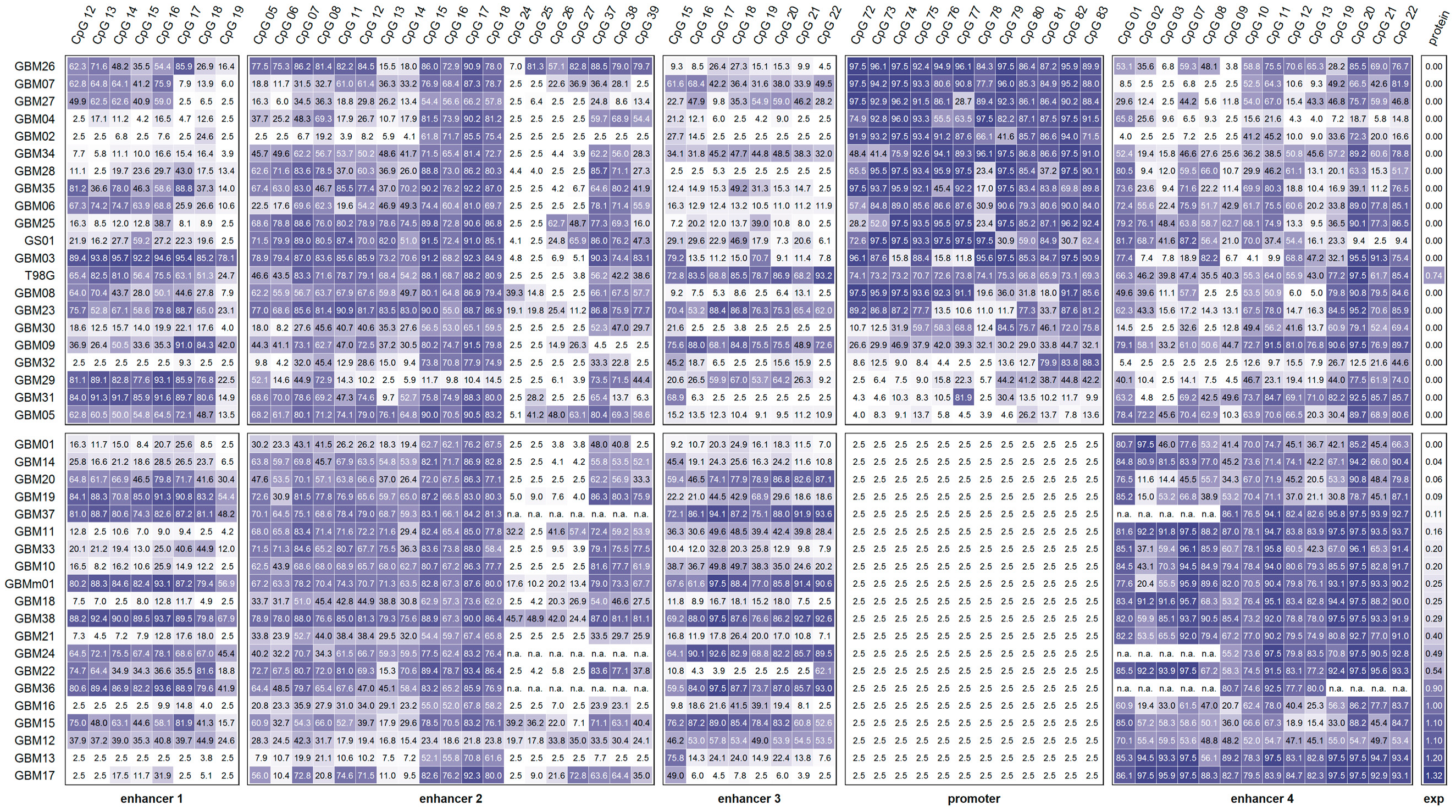
3. Results
3.1. MGMT Promoter and Enhancer Methylation
3.1.1. Promoter Methylation
3.1.2. Enhancer 1 Methylation
3.1.3. Enhancer 2 Methylation
3.1.4. Enhancer 3 Methylation
3.1.5. Enhancer 4 Methylation
3.2. Association between the MGMT Promoter and/or Enhancer Methylation
3.3. Association of the MGMT Promoter and Enhancer Methylation with MGMT Protein Expression
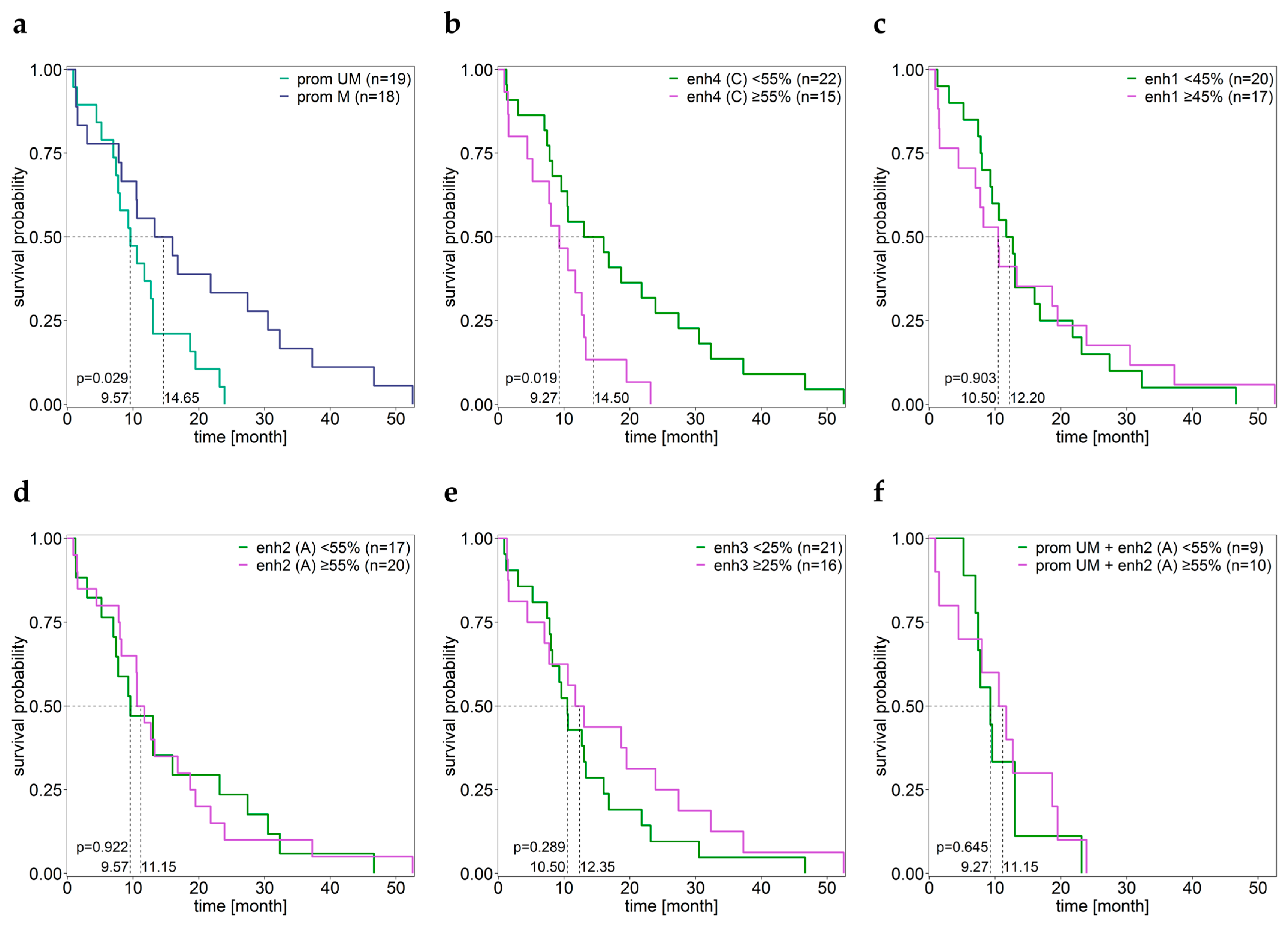
3.4. Association between the MGMT Promoter and Enhancer Methylation and Overall Survival
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO classification of tumors of the central nervous system: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; Van Den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef] [Green Version]
- Soll, J.M.; Sobol, R.W.; Mosammaparast, N. Regulation of DNA Alkylation Damage Repair: Lessons and Therapeutic Opportunities. Trends Biochem. Sci. 2017, 42, 206–218. [Google Scholar] [CrossRef] [Green Version]
- Esteller, M.; Hamilton, S.R.; Burger, P.C.; Baylin, S.B.; Herman, J.G. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res. 1999, 59, 793–797. [Google Scholar]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [Green Version]
- Wick, W.; Weller, M.; Van Den Bent, M.; Sanson, M.; Weiler, M.; Von Deimling, A.; Plass, C.; Hegi, M.; Platten, M.; Reifenberger, G. MGMT testing—The challenges for biomarker-based glioma treatment. Nat. Rev. Neurol. 2014, 10, 372–385. [Google Scholar] [CrossRef] [Green Version]
- Harris, L.C.; Remack, J.S.; Brent, T.P. Identification of a 59 bp enhancer located at the first exon/intron boundary of the human O6-methylguanine DNA methyltransferease gene. Nucleic Acids Res. 1994, 22, 4614–4619. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T.; Hamou, M.-F.; De Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [Green Version]
- Brandes, A.A.; Franceschi, E.; Tosoni, A.; Benevento, F.; Scopece, L.; Mazzocchi, V.; Bacci, A.; Agati, R.; Calbucci, F.; Ermani, M. Temozolomide concomitant and adjuvant to radiotherapy in elderly patients with glioblastoma: Correlation with MGMT promoter methylation status. Cancer 2009, 115, 3512–3518. [Google Scholar] [CrossRef]
- Malmström, A.; Grønberg, B.H.; Marosi, C.; Stupp, R.; Frappaz, D.; Schultz, H.; Abacioglu, U.; Tavelin, B.; Lhermitte, B.; Hegi, M.E.; et al. Temozolomide versus standard 6-week radiotherapy versus hypofractionated radiotherapy in patients older than 60 years with glioblastoma: The Nordic randomised, phase 3 trial. Lancet Oncol. 2012, 13, 916–926. [Google Scholar] [CrossRef]
- Wick, W.; Platten, M.; Meisner, C.; Felsberg, J.; Tabatabai, G.; Simon, M.; Nikkhah, G.; Papsdorf, K.; Steinbach, J.P.; Sabel, M.; et al. Temozolomide chemotherapy alone versus radiotherapy alone for malignant astrocytoma in the elderly: The NOA-08 randomised, phase 3 trial. Lancet Oncol. 2012, 13, 707–715. [Google Scholar] [CrossRef] [Green Version]
- Reifenberger, G.; Hentschel, B.; Felsberg, J.; Schackert, G.; Simon, M.; Schnell, O.; Westphal, M.; Wick, W.; Pietsch, T.; Loeffler, M.; et al. Predictive impact of MGMT promoter methylation in glioblastoma of the elderly. Int. J. Cancer 2012, 131, 1342–1350. [Google Scholar] [CrossRef] [PubMed]
- Binabaj, M.M.; Bahrami, A.; ShahidSales, S.; Joodi, M.; Joudi Mashhad, M.; Hassanian, S.M.; Anvari, K.; Avan, A. The prognostic value of MGMT promoter methylation in glioblastoma: A meta-analysis of clinical trials. J. Cell. Physiol. 2018, 233, 378–386. [Google Scholar] [CrossRef]
- Brown, N.F.; Ottaviani, D.; Tazare, J.; Gregson, J.; Kitchen, N.; Brandner, S.; Fersht, N.; Mulholland, P. Survival Outcomes and Prognostic Factors in Glioblastoma. Cancers 2022, 14, 3161. [Google Scholar] [CrossRef]
- Kreth, S.; Thon, N.; Eigenbrod, S.; Lutz, J.; Ledderose, C.; Egensperger, R.; Tonn, J.C.; Kretzschmar, H.A.; Hinske, L.C.; Kreth, F.W. O6-methylguanine-DNA methyltransferase (MGMT) mRNA expression predicts outcome in malignant glioma independent of MGMT promoter methylation. PLoS ONE 2011, 6, e17156. [Google Scholar] [CrossRef] [Green Version]
- Butler, M.; Pongor, L.; Su, Y.-T.; Xi, L.; Raffeld, M.; Quezado, M.; Trepel, J.; Aldape, K.; Pommier, Y.; Wu, J. MGMT Status as a Clinical Biomarker in Glioblastoma. Trends Cancer 2020, 6, 380–391. [Google Scholar] [CrossRef]
- Kim, T.-K.; Shiekhattar, R. Architectural and Functional Commonalities between Enhancers and Promoters. Cell 2015, 162, 948–959. [Google Scholar] [CrossRef] [Green Version]
- Bulger, M.; Groudine, M. Functional and mechanistic diversity of distal transcription enhancers. Cell 2011, 144, 327–339. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.; Shilatifard, A. Enhancer biology and enhanceropathies. Nat. Struct. Mol. Biol. 2014, 21, 210–219. [Google Scholar] [CrossRef]
- Stadler, M.B.; Murr, R.; Burger, L.; Ivanek, R.; Lienert, F.; Schöler, A.; Van Nimwegen, E.; Wirbelauer, C.; Oakeley, E.J.; Gaidatzis, D.; et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 2011, 480, 490–495. [Google Scholar] [CrossRef]
- Aran, D.; Sabato, S.; Hellman, A. DNA methylation of distal regulatory sites characterizes dysregulation of cancer genes. Genome Biol. 2013, 14, R21. [Google Scholar] [CrossRef] [Green Version]
- Kreibich, E.; Kleinendorst, R.; Barzaghi, G.; Kaspar, S.; Krebs, A.R. Single-molecule footprinting identifies context-dependent regulation of enhancers by DNA methylation. Mol. Cell 2023, 83, 787–802. [Google Scholar] [CrossRef]
- Bell, R.E.; Golan, T.; Sheinboim, D.; Malcov, H.; Amar, D.; Salamon, A.; Liron, T.; Gelfman, S.; Gabet, Y.; Shamir, R.; et al. Enhancer methylation dynamics contribute to cancer plasticity and patient mortality. Genome Res. 2016, 26, 601–611. [Google Scholar] [CrossRef] [Green Version]
- Stone, A.; Zotenko, E.; Locke, W.J.; Korbie, D.; Millar, E.K.A.; Pidsley, R.; Stirzaker, C.; Graham, P.; Trau, M.; Musgrove, E.A.; et al. DNA methylation of oestrogen-regulated enhancers defines endocrine sensitivity in breast cancer. Nat. Commun. 2015, 6, 7558. [Google Scholar] [CrossRef] [Green Version]
- Wahlberg, P.; Lundmark, A.; Nordlund, J.; Busche, S.; Raine, A.; Tandre, K.; Rönnblom, L.; Sinnett, D.; Forestier, E.; Pastinen, T.; et al. DNA methylome analysis of acute lymphoblastic leukemia cells reveals stochastic de novo DNA methylation in CpG islands. Epigenomics 2016, 8, 1367–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baek, S.-J.; Kim, M.; Bae, D.-H.; Kim, J.-H.; Kim, H.-J.; Han, M.-E.; Oh, S.-O.; Kim, Y.S.; Kim, S.-Y. Integrated epigenomic analyses of enhancer as well as promoter regions in gastric cancer. Oncotarget 2016, 7, 25620–25631. [Google Scholar] [CrossRef]
- Zhao, X.; Ji, J.; Wang, S.; Wang, R.; Yu, Q.; Li, D. The regulatory pattern of target gene expression by aberrant enhancer methylation in glioblastoma. BMC Bioinform. 2021, 22, 420. [Google Scholar] [CrossRef]
- Visel, A.; Minovitsky, S.; Dubchak, I.; Pennacchio, L.A. VISTA Enhancer Browser—a database of tissue-specific human enhancers. Nucleic Acids Res. 2007, 35, D88–D92. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, M.; Gan, H.; Wang, H.; Lee, J.H.; Fang, D.; Kitange, G.J.; He, L.; Hu, Z.; Parney, I.F.; et al. A novel enhancer regulates MGMT expression and promotes temozolomide resistance in glioblastoma. Nat. Commun. 2018, 9, 2949. [Google Scholar] [CrossRef] [Green Version]
- Switzeny, O.J.; Christmann, M.; Renovanz, M.; Giese, A.; Sommer, C.; Kaina, B. MGMT promoter methylation determined by HRM in comparison to MSP and pyrosequencing for predicting high-grade glioma response. Clin. Epigenet. 2016, 8, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiegl-Kreinecker, S.; Pirker, C.; Marosi, C.; Buchroithner, J.; Pichler, J.; Silye, R.; Fischer, J.; Micksche, M.; Berger, W. Dynamics of chemosensitivity and chromosomal instability in recurrent glioblastoma. Br. J. Cancer 2007, 96, 960–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiegl-Kreinecker, S.; Pirker, C.; Filipits, M.; Lötsch, D.; Buchroithner, J.; Pichler, J.; Silye, R.; Weis, S.; Micksche, M.; Fischer, J.; et al. O6-Methylguanine DNA methyltransferase protein expression in tumor cells predicts outcome of temozolomide therapy in glioblastoma patients. Neuro-Oncology 2010, 12, 28–36. [Google Scholar] [CrossRef] [Green Version]
- Genbank. Available online: https://www.ncbi.nlm.nih.gov/nucleotide/ (accessed on 12 June 2022).
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [Green Version]
- Fishilevich, S.; Nudel, R.; Rappaport, N.; Hadar, R.; Plaschkes, I.; Iny Stein, T.; Rosen, N.; Kohn, A.; Twik, M.; Safran, M.; et al. GeneHancer: Genome-wide integration of enhancers and target genes in GeneCards. Database 2017, 2017, bax028. [Google Scholar] [CrossRef] [Green Version]
- Team., R.C. R: A Language and Environment for Statistical Computing. Available online: https://www.R-project.org/ (accessed on 12 June 2022).
- Office of Pesticide Programs U.S. Environmental Protection Agency. Assigning Values to Non-Detected/Non-Quantified Pesticide Residues in Human Health Food Exposure Assessments; US.EPA: Washington, DC, USA, 2000.
- Spiegl-Kreinecker, S.; Lötsch, D.; Ghanim, B.; Pirker, C.; Mohr, T.; Laaber, M.; Weis, S.; Olschowski, A.; Webersinke, G.; Pichler, J.; et al. Prognostic quality of activating TERT promoter mutations in glioblastoma: Interaction with the rs2853669 polymorphism and patient age at diagnosis. Neuro-Oncology 2015, 17, 1231–1240. [Google Scholar] [CrossRef] [Green Version]
- Candiloro, I.L.M.; Mikeska, T.; Dobrovic, A. Assessing combined methylation-sensitive high resolution melting and pyrosequencing for the analysis of heterogeneous DNA methylation. Epigenetics 2011, 6, 500–507. [Google Scholar] [CrossRef] [Green Version]
- Zappe, K.; Pirker, C.; Miedl, H.; Schreiber, M.; Heffeter, P.; Pfeiler, G.; Hacker, S.; Haslik, W.; Spiegl-Kreinecker, S.; Cichna-Markl, M. Discrimination between 34 of 36 possible combinations of three C>T SNP genotypes in the MGMT promoter by high resolution melting analysis coupled with pyrosequencing using a single primer set. Int. J. Mol. Sci. 2021, 22, 12527. [Google Scholar] [CrossRef]
- da Rocha, S.T.; Gendrel, A.-V. The influence of DNA methylation on monoallelic expression. Essays Biochem. 2019, 63, 663–676. [Google Scholar] [CrossRef]
- Kristensen, L.S.; Michaelsen, S.R.; Dyrbye, H.; Aslan, D.; Grunnet, K.; Christensen, I.J.; Poulsen, H.S.; Grønbæk, K.; Broholm, H. Assessment of quantitative and allelic MGMT methylation patterns as a prognostic marker in glioblastoma. J. Neuropathol. Exp. Neurol. 2016, 75, 246–255. [Google Scholar] [CrossRef] [Green Version]
- Everhard, S.; Tost, J.; El Abdalaoui, H.; Crinière, E.; Busato, F.; Marie, Y.; Gut, I.G.; Sanson, M.; Mokhtari, K.; Laigle-Donadey, F.; et al. Identification of regions correlating MGMT promoter methylation and gene expression in glioblastomas. Neuro-Oncology 2009, 11, 348–356. [Google Scholar] [CrossRef] [Green Version]
- Bady, P.; Sciuscio, D.; Diserens, A.C.; Bloch, J.; Van Den Bent, M.J.; Marosi, C.; Dietrich, P.Y.; Weller, M.; Mariani, L.; Heppner, F.L.; et al. MGMT methylation analysis of glioblastoma on the Infinium methylation BeadChip identifies two distinct CpG regions associated with gene silencing and outcome, yielding a prediction model for comparisons across datasets, tumor grades, and CIMP-status. Acta Neuropathol. 2012, 124, 547–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikeska, T.; Bock, C.; El-Maarri, O.; Hübner, A.; Ehrentraut, D.; Schramm, J.; Felsberg, J.; Kahl, P.; Büttner, R.; Pietsch, T.; et al. Optimization of quantitative MGMT promoter methylation analysis using pyrosequencing and combined bisulfite restriction analysis. J. Mol. Diagn. 2007, 9, 368–381. [Google Scholar] [CrossRef] [Green Version]
- Dunn, J.; Baborie, A.; Alam, F.; Joyce, K.; Moxham, M.; Sibson, R.; Crooks, D.; Husband, D.; Shenoy, A.; Brodbelt, A.; et al. Extent of MGMT promoter methylation correlates with outcome in glioblastomas given temozolomide and radiotherapy. Br. J. Cancer 2009, 101, 124–131. [Google Scholar] [CrossRef] [Green Version]
- Karayan-Tapon, L.; Quillien, V.; Guilhot, J.; Wager, M.; Fromont, G.; Saikali, S.; Etcheverry, A.; Hamlat, A.; Loussouarn, D.; Campion, L.; et al. Prognostic value of O6-methylguanine-DNA methyltransferase status in glioblastoma patients, assessed by five different methods. J. Neurooncol 2010, 97, 311–322. [Google Scholar] [CrossRef]
- Håvik, A.B.; Brandal, P.; Honne, H.; Dahlback, H.S.S.; Scheie, D.; Hektoen, M.; Meling, T.R.; Helseth, E.; Heim, S.; Lothe, R.A.; et al. MGMT promoter methylation in gliomas-assessment by pyrosequencing and quantitative methylation-specific PCR. J. Transl. Med. 2012, 10, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quillien, V.; Lavenu, A.; Sanson, M.; Legrain, M.; Dubus, P.; Karayan-Tapon, L.; Mosser, J.; Ichimura, K.; Figarella-Branger, D. Outcome-based determination of optimal pyrosequencing assay for MGMT methylation detection in glioblastoma patients. J. Neurooncol 2014, 116, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Quillien, V.; Lavenu, A.; Ducray, F.; Meyronet, D.; Chinot, O.; Fina, F.; Sanson, M.; Carpentier, C.; Karayan-Tapon, L.; Rivet, P.; et al. Clinical validation of the CE-IVD marked Therascreen MGMT kit in a cohort of glioblastoma patients. Cancer Biomark. 2017, 20, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Kavouridis, V.K.; Ligon, K.L.; Wen, P.Y.; Iorgulescu, J.B. Survival outcomes associated with MGMT promoter methylation and temozolomide in gliosarcoma patients. J. Neurooncol 2022, 158, 111–116. [Google Scholar] [CrossRef]
- Mansouri, A.; Hachem, L.D.; Mansouri, S.; Nassiri, F.; Laperriere, N.J.; Xia, D.; Lindeman, N.I.; Wen, P.Y.; Chakravarti, A.; Mehta, M.P.; et al. MGMT promoter methylation status testing to guide therapy for glioblastoma: Refining the approach based on emerging evidence and current challenges. Neuro-Oncology 2019, 21, 167–178. [Google Scholar] [CrossRef] [Green Version]
- Bienkowski, M.; Berghoff, A.S.; Marosi, C.; Wöhrer, A.; Heinzl, H.; Hainfellner, J.A.; Preusser, M. Clinical Neuropathology practice guide 5-2015: MGMT methylation pyrosequencing in glioblastoma: Unresolved issues and open questions. Clin. Neuropathol. 2015, 34, 250–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Carlo, E.; Gerratana, L.; De Maglio, G.; Buoro, V.; Cortiula, F.; Gurrieri, L.; Isola, M.; Fasola, G.; Puglisi, F.; Pizzolitto, S.; et al. Defining a prognostic score based on O6-methylguanine-DNA methyltransferase cut-off methylation level determined by pyrosequencing in patients with glioblastoma multiforme. J. Neurooncol. 2018, 140, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Gurrieri, L.; De Carlo, E.; Gerratana, L.; De Maglio, G.; Macerelli, M.; Pisa, F.E.; Masiero, E.; Aprile, G.; Follador, A.; Puglisi, F.; et al. MGMT pyrosequencing-based cut-off methylation level and clinical outcome in patients with glioblastoma multiforme. Future Oncol. 2018, 14, 699–707. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | Primer Set | Primer Sequence (5′→3′) | Length [bp] | CpGs Analyzed |
|---|---|---|---|---|
| enhancer 1 | A | F: AGAATGTAGATTTGGGATTAGTTAAT | 229 | 12–19 |
| (hs737) | R: [Btn] TAAAATACAAAATATACCCTCCAACA | |||
| 27 CpGs | S: TATATAAAGAAGGTTGGT | |||
| enhancer 2 | A | F: AGTTAGGAAATTAGAAATGGAATGTTT | 255 | 05–08 |
| (Chen et al.) | R: [Btn] CAAATCACACTCTAAATTCCCAATT | |||
| 46 CpGs | S: TGGTATTAGAGGTTA | |||
| B | F: [Btn] TTAAATAAGTGGTTTAGGTAGAGG | 137 | 11–18 | |
| R: TACTAAACATTCCATTTCTAATTTCC | ||||
| S: CCATTTCTAATTTCCTAACTC | ||||
| C | F: GTTGTAGGGTATATGAGTTTAGAT | 271 | 24–27 | |
| R: [Btn] TTCATAACTCAAATTAACACACACT | ||||
| S: TTTTGTGTTGAATGG | ||||
| D | F: GAGGTTATTTGGAAAGTTGAGAT | 286 | 37–39 | |
| R: [Btn] CTAATAATCCAAACCCTCTATTC | ||||
| S: TTTAGTGTTATGGGAG | ||||
| enhancer 3 | A | F: TGTGTTAGTTTTAGTGGTTTAGA | 138 | 15–22 |
| (hs699) | R: [Btn] TAACACACAAACCAATCTCTC | |||
| 33 CpGs | S: TAGTTTTAGTGGTTTAGAAGT | |||
| enhancer 4 | A | F: [Btn] GGAATGTGTTATTTAATTGGTATGT | 204 | 01–03 |
| (hs696) | R: CAAATCCCACAACAAATCCTTAT | |||
| 26 CpGs | S: TCAAAAAAAAAAAATCACC | |||
| B | F: GAGGTTTGATATAAGTAATGATGG | 131 | 07–08 | |
| R: [Btn] CCTCCTAATCCCACAATACAA | ||||
| S: TAAGTAATGATGGTATG | ||||
| C | F: AGGTTTGATATAAGTAATGATGGTAT | 257 | 09–13 | |
| R: [Btn] CRTATTCTCTCCCACTTCAATA | ||||
| S: GTATTGTGGGATTAGGA | ||||
| D | F: [Btn] GTGTATTGAAGTGGGAGAGAATA | 241 | 19–22 | |
| R: CAATAACAATTTTACAAACACAAATAACTT | ||||
| S: ATAACTTTTCATTCA | ||||
| promoter | A [31] | F: GGATATGTTGGGATAGTT | 98 | 72–83 |
| 98 CpGs | R: [Btn] CCCAAACACTCACCAAAT | |||
| S: GGATATGTTGGGATAGTT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zappe, K.; Pühringer, K.; Pflug, S.; Berger, D.; Böhm, A.; Spiegl-Kreinecker, S.; Cichna-Markl, M. Association between MGMT Enhancer Methylation and MGMT Promoter Methylation, MGMT Protein Expression, and Overall Survival in Glioblastoma. Cells 2023, 12, 1639. https://doi.org/10.3390/cells12121639
Zappe K, Pühringer K, Pflug S, Berger D, Böhm A, Spiegl-Kreinecker S, Cichna-Markl M. Association between MGMT Enhancer Methylation and MGMT Promoter Methylation, MGMT Protein Expression, and Overall Survival in Glioblastoma. Cells. 2023; 12(12):1639. https://doi.org/10.3390/cells12121639
Chicago/Turabian StyleZappe, Katja, Katharina Pühringer, Simon Pflug, Daniel Berger, Andreas Böhm, Sabine Spiegl-Kreinecker, and Margit Cichna-Markl. 2023. "Association between MGMT Enhancer Methylation and MGMT Promoter Methylation, MGMT Protein Expression, and Overall Survival in Glioblastoma" Cells 12, no. 12: 1639. https://doi.org/10.3390/cells12121639
APA StyleZappe, K., Pühringer, K., Pflug, S., Berger, D., Böhm, A., Spiegl-Kreinecker, S., & Cichna-Markl, M. (2023). Association between MGMT Enhancer Methylation and MGMT Promoter Methylation, MGMT Protein Expression, and Overall Survival in Glioblastoma. Cells, 12(12), 1639. https://doi.org/10.3390/cells12121639