

Vascular Inflammatory Diseases and Endothelial Phenotypes

{kind=link}
{kind=link}
Abstract
:1. Introduction
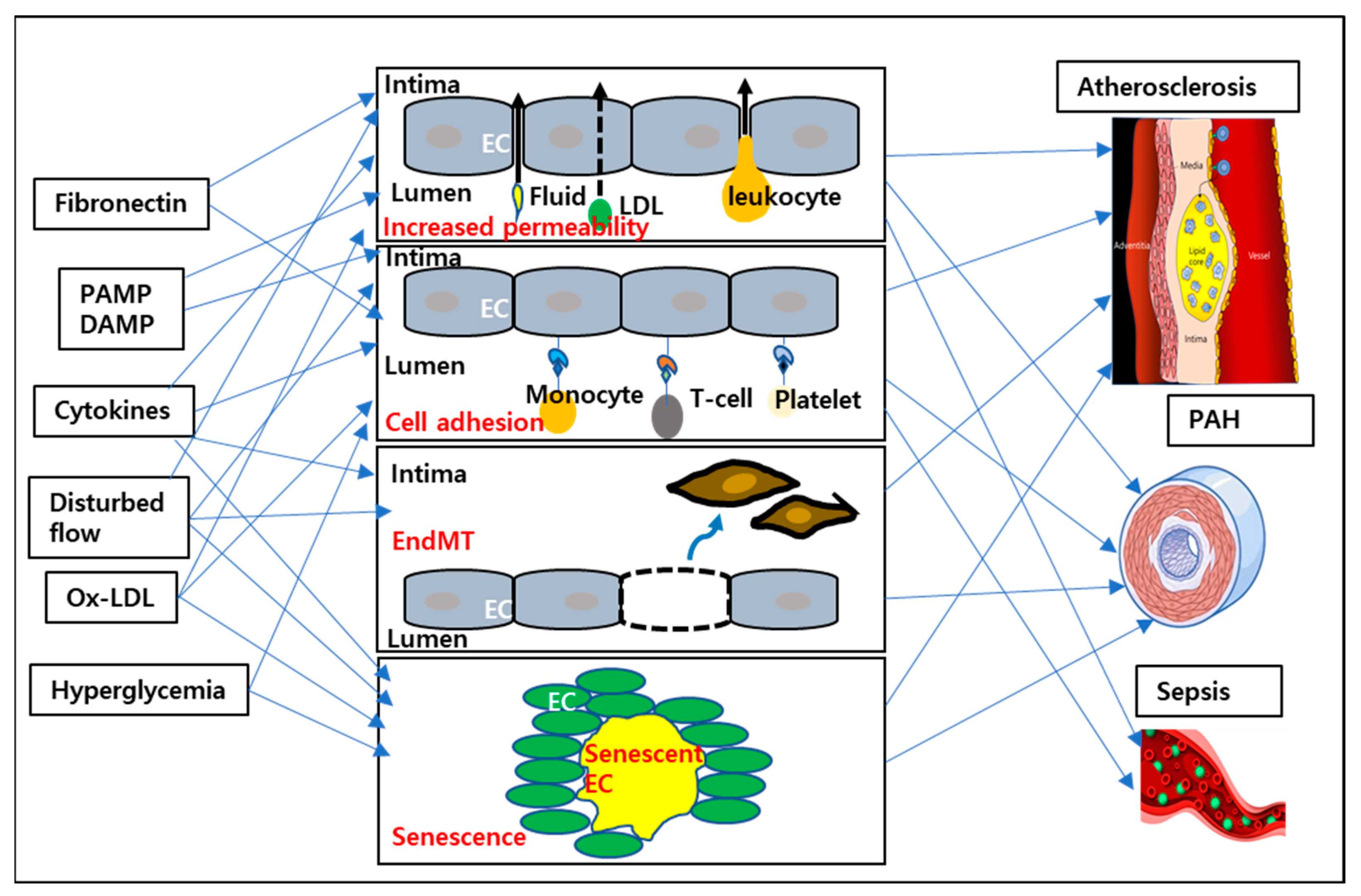
2. Endothelial Proinflammatory Phenotype
2.1. Cell Surface Adhesion Molecule Expression
2.2. Increased Endothelial Permeability
2.3. EndMT
2.4. Senescence
3. EC Proinflammatory Stimuli
3.1. Type I Endothelial Activation Ligands
3.2. Type II Endothelial Activation Ligands
3.3. High Glucose
3.4. Ox-LDLs
3.5. PAMP and DAMP
3.6. ECM (Extracellular Matrix)
3.7. Disturbed Flow
4. The Effect of Shear Stress on the Vascular Endothelium
4.1. Effects of Shear Stress on Leukocyte Adhesion
4.2. Effects of Shear Stress on Vascular Permeability
4.3. Effects of Disturbed Flow on EndMT
4.4. Effects of Disturbed Flow on Glucose Metabolism
5. Vascular Inflammatory Diseases
5.1. Atherosclerosis
5.1.1. Plaque Instability and Endothelium
5.1.2. Plaque Calcification and Endothelium
5.2. Pulmonary Arterial Hypertension (PAH)
5.3. Sepsis
5.3.1. Vascular Leakage
5.3.2. Thrombosis
5.3.3. Vascular Tone
6. Conclusions
6.1. Targeting ECs in Inflammatory Diseases
6.2. Identifying Disease-Specific EC Phenotypes or EC Subpopulations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Krüger-Genge, A.; Blocki, A.; Franke, R.P.; Jung, F. Vascular Endothelial Cell Biology: An Update. Int. J. Mol. Sci. 2019, 20, 4411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marziano, C.; Genet, G.; Hirschi, K.K. Vascular endothelial cell specification in health and disease. Angiogenesis 2021, 24, 213–236. [Google Scholar] [CrossRef] [PubMed]
- Sturtzel, C. Endothelial Cells. Adv. Exp. Med. Biol. 2017, 1003, 71–91. [Google Scholar] [PubMed]
- Conway, D.E.; Schwartz, M.A. Flow-dependent cellular mechanotransduction in atherosclerosis. J. Cell Sci. 2013, 126, 5101–5109. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, K. Platelet endothelial cell adhesion molecule-1 and mechanotransduction in vascular endothelial cells. J. Intern. Med. 2006, 259, 373–380. [Google Scholar] [CrossRef]
- Lertkiatmongkol, P.; Liao, D.; Mei, H.; Hu, Y.; Newman, P.J. Endothelial functions of platelet/endothelial cell adhesion molecule-1 (CD31). Curr. Opin. Hematol. 2016, 23, 253–259. [Google Scholar] [CrossRef] [Green Version]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Effects of shear stress on endothelial cells: Go with the flow. Acta Physiol. 2017, 219, 382–408. [Google Scholar] [CrossRef]
- Cyr, A.R.; Huckaby, L.V.; Shiva, S.S.; Zuckerbraun, B.S. Nitric Oxide and Endothelial Dysfunction. Crit. Care Clin. 2020, 36, 307–321. [Google Scholar] [CrossRef]
- Shaito, A.; Aramouni, K.; Assaf, R.; Parenti, A.; Orekhov, A.; Yazbi, A.E.; Pintus, G.; Eid, A.H. Oxidative Stress-Induced Endothelial Dysfunction in Cardiovascular Diseases. Front. Biosci. 2022, 27, 105. [Google Scholar] [CrossRef]
- Wang, K.-C.; Yeh, Y.-T.; Nguyen, P.; Limqueco, E.; Lopez, J.; Thorossian, S.; Guan, K.-L.; Li, Y.-S.J.; Chien, S. Flow-dependent YAP/TAZ activities regulate endothelial phenotypes and atherosclerosis. Proc. Natl. Acad. Sci. USA 2016, 113, 11525–11530. [Google Scholar] [CrossRef] [Green Version]
- Milošević, N.; Rütter, M.; David, A. Endothelial Cell Adhesion Molecules- (un)Attainable Targets for Nanomedicines. Front. Med. Technol. 2022, 4, 846065. [Google Scholar] [CrossRef]
- Ebnet, K.; Suzuki, A.; Ohno, S.; Vestweber, D. Junctional adhesion molecules (JAMs): More molecules with dual functions? J. Cell Sci. 2004, 117, 19–29. [Google Scholar] [CrossRef] [Green Version]
- Borsig, L. Selectins in cancer immunity. Glycobiology 2018, 28, 648–655. [Google Scholar] [CrossRef] [Green Version]
- Nimrichter, L.; Burdick, M.M.; Aoki, K.; Laroy, W.; Fierro, M.A.; Hudson, S.A.; Von Seggern, C.E.; Cotter, R.J.; Bochner, B.S.; Tiemeyer, M.; et al. E-selectin receptors on human leukocytes. Blood 2008, 112, 3744–3752. [Google Scholar] [CrossRef] [Green Version]
- Speyer, C.L.; Ward, P.A. Role of Endothelial Chemokines and Their Receptors during Inflammation. J. Investig. Surg. 2011, 24, 18–27. [Google Scholar] [CrossRef]
- Yadav, A.; Saini, V.; Arora, S. MCP-1: Chemoattractant with a role beyond immunity: A review. Clin. Chim. Acta 2010, 411, 1570–1579. [Google Scholar] [CrossRef] [PubMed]
- Andreone, B.J.; Chow, B.W.; Tata, A.; Lacoste, B.; Ben-Zvi, A.; Bullock, K.; Deik, A.A.; Ginty, D.D.; Clish, C.B.; Gu, C. Blood-Brain Barrier Permeability Is Regulated by Lipid Transport-Dependent Suppression of Caveolae-Mediated Transcytosis. Neuron 2017, 94, 581–594.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutz, S.E.; Smith, J.R.; Kim, D.H.; Olson, C.V.L.; Ellefsen, K.; Bates, J.M.; Gandhi, S.P.; Agalliu, D. Caveolin1 Is Required for Th1 Cell Infiltration, but Not Tight Junction Remodeling, at the Blood-Brain Barrier in Autoimmune Neuroinflammation. Cell Rep. 2017, 21, 2104–2117. [Google Scholar] [CrossRef] [Green Version]
- Vestweber, D. Relevance of endothelial junctions in leukocyte extravasation and vascular permeability. Ann. N. Y. Acad. Sci. 2012, 1257, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Sluiter, T.J.; van Buul, J.D.; Huveneers, S.; Quax, P.H.A.; de Vries, M.R. Endothelial Barrier Function and Leukocyte Transmigration in Atherosclerosis. Biomedicines 2021, 9, 328. [Google Scholar] [CrossRef]
- Takakura, Y.; Mahato, R.I.; Hashida, M. Extravasation of macromolecules. Adv. Drug Deliv. Rev. 1998, 34, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Nordborg, C.; Sokrab, T.E.O.; Johansson, B.B. The relationship between plasma protein extravasation and remote tissue changes after experimental brain infarction. Acta Neuropathol. 1991, 82, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Kraehling, J.R.; Chidlow, J.H.; Rajagopal, C.; Sugiyama, M.G.; Fowler, J.W.; Lee, M.Y.; Zhang, X.; Ramírez, C.M.; Park, E.J.; Tao, B.; et al. Genome-wide RNAi screen reveals ALK1 mediates LDL uptake and transcytosis in endothelial cells. Nat. Commun. 2016, 7, 13516. [Google Scholar] [CrossRef]
- Wiklund, O.; Carew, T.E.; Steinberg, D. Role of the low density lipoprotein receptor in penetration of low density lipoprotein into rabbit aortic wall. Arteriosclerosis 1985, 5, 135–141. [Google Scholar] [CrossRef] [Green Version]
- Ryu, J.K.; McLarnon, J.G. A leaky blood-brain barrier, fibrinogen infiltration and microglial reactivity in inflamed Alzheimer’s disease brain. J. Cell. Mol. Med. 2009, 13, 2911–2925. [Google Scholar] [CrossRef] [Green Version]
- Souilhol, C.; Harmsen, M.C.; Evans, P.C.; Krenning, G. Endothelial–mesenchymal transition in atherosclerosis. Cardiovasc. Res. 2018, 114, 565–577. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.-Y.; Qin, L.; Baeyens, N.; Li, G.; Afolabi, T.; Budatha, M.; Tellides, G.; Schwartz, M.A.; Simons, M. Endothelial-to-mesenchymal transition drives atherosclerosis progression. J. Clin. Investig. 2015, 125, 4514–4528. [Google Scholar] [CrossRef] [Green Version]
- Evrard, S.M.; Lecce, L.; Michelis, K.C.; Nomura-Kitabayashi, A.; Pandey, G.; Purushothaman, K.R.; d’Escamard, V.; Li, J.R.; Hadri, L.; Fujitani, K.; et al. Endothelial to mesenchymal transition is common in atherosclerotic lesions and is associated with plaque instability. Nat. Commun. 2016, 7, 11853. [Google Scholar] [CrossRef] [Green Version]
- Pérez, L.; Muñoz-Durango, N.; Riedel, C.A.; Echeverría, C.; Kalergis, A.M.; Cabello-Verrugio, C.; Simon, F. Endothelial-to-mesenchymal transition: Cytokine-mediated pathways that determine endothelial fibrosis under inflammatory conditions. Cytokine Growth Factor Rev. 2017, 33, 41–54. [Google Scholar] [CrossRef]
- Huang, Q.; Gan, Y.; Yu, Z.; Wu, H.; Zhong, Z. Endothelial to Mesenchymal Transition: An Insight in Atherosclerosis. Front. Cardiovasc. Med. 2021, 8, 734550. [Google Scholar] [CrossRef] [PubMed]
- Min, E.; Schwartz, M.A. Translocating transcription factors in fluid shear stress-mediated vascular remodeling and disease. Exp. Cell Res. 2019, 376, 92–97. [Google Scholar] [CrossRef]
- Bloom, S.I.; Islam, M.T.; Lesniewski, L.A.; Donato, A.J. Mechanisms and consequences of endothelial cell senescence. Nat. Rev. Cardiol. 2023, 20, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Del Pinto, R.; Ferri, C. Inflammation-Accelerated Senescence and the Cardiovascular System: Mechanisms and Perspectives. Int. J. Mol. Sci. 2018, 19, 3701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pober, J.S. Warner-Lambert/Parke-Davis award lecture. Cytokine-mediated activation of vascular endothelium. Physiology and pathology. Am. J. Pathol. 1988, 133, 426–433. [Google Scholar] [PubMed]
- Pober, J.S.; Sessa, W.C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007, 7, 803–815. [Google Scholar] [CrossRef] [PubMed]
- Sprague, A.H.; Khalil, R.A. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem. Pharmacol. 2009, 78, 539–552. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Castejon, G.; Brough, D. Understanding the mechanism of IL-1β secretion. Cytokine Growth Factor Rev. 2011, 22, 189–195. [Google Scholar] [CrossRef]
- Xiao, H.; Lu, M.; Lin, T.Y.; Chen, Z.; Chen, G.; Wang, W.-C.; Marin, T.; Shentu, T.-P.; Wen, L.; Gongol, B.; et al. Sterol Regulatory Element Binding Protein 2 Activation of NLRP3 Inflammasome in Endothelium Mediates Hemodynamic-Induced Atherosclerosis Susceptibility. Circulation 2013, 128, 632–642. [Google Scholar] [CrossRef] [Green Version]
- Ju, J.; Liu, Y.; Liang, H.; Yang, B. The role of pyroptosis in endothelial dysfunction induced by diseases. Front. Immunol. 2023, 13, 1093985. [Google Scholar] [CrossRef]
- Libby, P. Interleukin-1 Beta as a Target for Atherosclerosis Therapy: Biological Basis of CANTOS and Beyond. J. Am. Coll. Cardiol. 2017, 70, 2278–2289. [Google Scholar] [CrossRef]
- Jang, Y.-E.; Immanuel, J.; Lee, J.-R.; Jang, Y.-J.; Kwon, Y.J.; Kwon, H.S.; Shin, J.-W.; Yun, S. Shinjulactone A Blocks Vascular Inflammation and the Endothelial-Mesenchymal Transition. J. Lipid Atheroscler. 2022, 11, 272–279. [Google Scholar] [CrossRef]
- Kondreddy, V.; Keshava, S.; Das, K.; Magisetty, J.; Rao, L.V.M.; Pendurthi, U.R. The Gab2–MALT1 axis regulates thromboinflammation and deep vein thrombosis. Blood 2022, 140, 1549–1564. [Google Scholar] [CrossRef] [PubMed]
- Papadakis, K.A.; Targan, S.R. Tumor necrosis factor: Biology and therapeutic inhibitors. Gastroenterology 2000, 119, 1148–1157. [Google Scholar] [CrossRef] [PubMed]
- Bolik, J.; Krause, F.; Stevanovic, M.; Gandraß, M.; Thomsen, I.; Schacht, S.-S.; Rieser, E.; Müller, M.; Schumacher, N.; Fritsch, J.; et al. Inhibition of ADAM17 impairs endothelial cell necroptosis and blocks metastasis. J. Exp. Med. 2021, 219, e20201039. [Google Scholar] [CrossRef]
- McCaffrey, T.A. TGF-beta signaling in atherosclerosis and restenosis. FBS 2009, 1, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, X. The Roles of TGF-β Signaling in Cerebrovascular Diseases. Front. Cell Dev. Biol. 2020, 8, 567682. [Google Scholar] [CrossRef]
- Hiepen, C.; Jatzlau, J.; Hildebrandt, S.; Kampfrath, B.; Goktas, M.; Murgai, A.; Cuellar Camacho, J.L.; Haag, R.; Ruppert, C.; Sengle, G.; et al. BMPR2 acts as a gatekeeper to protect endothelial cells from increased TGFβ responses and altered cell mechanics. PLoS Biol. 2019, 17, e3000557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villar-Fincheira, P.; Sanhueza-Olivares, F.; Norambuena-Soto, I.; Cancino-Arenas, N.; Hernandez-Vargas, F.; Troncoso, R.; Gabrielli, L.; Chiong, M. Role of Interleukin-6 in Vascular Health and Disease. Front. Mol. Biosci. 2021, 8, 641734. [Google Scholar] [CrossRef]
- Pedersen, B.K.; Steensberg, A.; Schjerling, P. Muscle-derived interleukin-6: Possible biological effects. J. Physiol. 2001, 536, 329–337. [Google Scholar] [CrossRef]
- Kang, S.; Kishimoto, T. Interplay between interleukin-6 signaling and the vascular endothelium in cytokine storms. Exp. Mol. Med. 2021, 53, 1116–1123. [Google Scholar] [CrossRef]
- Assar, M.E.; Angulo, J.; Rodríguez-Mañas, L. Diabetes and ageing-induced vascular inflammation. J. Physiol. 2016, 594, 2125–2146. [Google Scholar] [CrossRef] [Green Version]
- Spranger, J.; Kroke, A.; Möhlig, M.; Hoffmann, K.; Bergmann, M.M.; Ristow, M.; Boeing, H.; Pfeiffer, A.F.H. Inflammatory Cytokines and the Risk to Develop Type 2 Diabetes: Results of the Prospective Population-Based European Prospective Investigation into Cancer and Nutrition (EPIC)-Potsdam Study. Diabetes 2003, 52, 812–817. [Google Scholar] [CrossRef] [Green Version]
- Callaghan, M.J.; Ceradini, D.J.; Gurtner, G.C. Hyperglycemia-induced reactive oxygen species and impaired endothelial progenitor cell function. Antioxid. Redox Signal. 2005, 7, 1476–1482. [Google Scholar] [CrossRef]
- Baudoin, L.; Issad, T. O-GlcNAcylation and inflammation: A vast territory to explore. Front. Endocrinol. 2015, 5, 235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mastrogiacomo, L.; Ballagh, R.; Venegas-Pino, D.E.; Kaur, H.; Shi, P.; Werstuck, G.H. The Effects of Hyperglycemia on Early Endothelial Activation and the Initiation of Atherosclerosis. Am. J. Pathol. 2023, 193, 121–133. [Google Scholar] [CrossRef]
- Yoshida, H.; Kisugi, R. Mechanisms of LDL oxidation. Clin. Chim. Acta 2010, 411, 1875–1882. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Zhou, Y.; Nabavi, S.M.; Sahebkar, A.; Little, P.J.; Xu, S.; Weng, J.; Ge, J. Mechanisms of Oxidized LDL-Mediated Endothelial Dysfunction and Its Consequences for the Development of Atherosclerosis. Front. Cardiovasc. Med. 2022, 9, 1442. [Google Scholar] [CrossRef] [PubMed]
- Akhmedov, A.; Sawamura, T.; Chen, C.-H.; Kraler, S.; Vdovenko, D.; Lüscher, T.F. Lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1): A crucial driver of atherosclerotic cardiovascular disease. Eur. Heart J. 2020, 42, 1797–1807. [Google Scholar] [CrossRef]
- Moriyama, K.; Nishida, O. Targeting Cytokines, Pathogen-Associated Molecular Patterns, and Damage-Associated Molecular Patterns in Sepsis via Blood Purification. Int. J. Mol. Sci. 2021, 22, 8882. [Google Scholar] [CrossRef]
- Rai, V.; Agrawal, D.K. The role of damage- and pathogen-associated molecular patterns in inflammation-mediated vulnerability of atherosclerotic plaques. Can. J. Physiol. Pharmacol. 2017, 95, 1245–1253. [Google Scholar] [CrossRef]
- Cheng, K.T.; Xiong, S.; Ye, Z.; Hong, Z.; Di, A.; Tsang, K.M.; Gao, X.; An, S.; Mittal, M.; Vogel, S.M.; et al. Caspase-11–mediated endothelial pyroptosis underlies endotoxemia-induced lung injury. J. Clin. Investig. 2017, 127, 4124–4135. [Google Scholar] [CrossRef]
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 2017, 42, 245–254. [Google Scholar] [CrossRef]
- Yun, S.; Budatha, M.; Dahlman, J.E.; Coon, B.G.; Cameron, R.T.; Langer, R.; Anderson, D.G.; Baillie, G.; Schwartz, M.A. Interaction between integrin α5 and PDE4D regulates endothelial inflammatory signalling. Nat. Cell Biol. 2016, 18, 1043–1053. [Google Scholar] [CrossRef]
- Chen, M.; Cavinato, C.; Hansen, J.; Tanaka, K.; Ren, P.; Hassab, A.; Li, D.S.; Youshao, E.; Tellides, G.; Iyengar, R.; et al. FN (Fibronectin)-Integrin α5 Signaling Promotes Thoracic Aortic Aneurysm in a Mouse Model of Marfan Syndrome. Arterioscler. Thromb. Vasc. Biol. 2023, 43, e132–e150. [Google Scholar] [CrossRef] [PubMed]
- Gubán, B.; Vas, K.; Balog, Z.; Manczinger, M.; Bebes, A.; Groma, G.; Széll, M.; Kemény, L.; Bata-Csörgő, Z. Abnormal regulation of fibronectin production by fibroblasts in psoriasis. Br. J. Dermatol. 2016, 174, 533–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Yafeai, Z.; Yurdagul, A.; Peretik, J.M.; Alfaidi, M.; Murphy, P.A.; Orr, A.W. Endothelial FN (Fibronectin) Deposition by α5β1 Integrins Drives Atherogenic Inflammation. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2601–2614. [Google Scholar] [CrossRef]
- Hahn, C.; Schwartz, M.A. Mechanotransduction in vascular physiology and atherogenesis. Nat. Rev. Mol. Cell Biol. 2009, 10, 53–62. [Google Scholar] [CrossRef] [Green Version]
- Baeyens, N.; Bandyopadhyay, C.; Coon, B.G.; Yun, S.; Schwartz, M.A. Endothelial fluid shear stress sensing in vascular health and disease. J. Clin. Investig. 2016, 126, 821–828. [Google Scholar] [CrossRef]
- Koskinas, K.C.; Chatzizisis, Y.S.; Antoniadis, A.P.; Giannoglou, G.D. Role of Endothelial Shear Stress in Stent Restenosis and Thrombosis. J. Am. Coll. Cardiol. 2012, 59, 1337–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Endothelial Barrier and Its Abnormalities in Cardiovascular Disease. Front. Physiol. 2015, 6, 365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, R.M.; Yang, L.; Garcia-Cardena, G.; Luscinskas, F.W. Endothelial-dependent mechanisms of leukocyte recruitment to the vascular wall. Circ. Res. 2007, 101, 234–247. [Google Scholar] [CrossRef] [Green Version]
- Yurdagul, A., Jr.; Orr, A.W. Blood Brothers: Hemodynamics and Cell-Matrix Interactions in Endothelial Function. Antioxid. Redox Signal. 2016, 25, 415–434. [Google Scholar] [CrossRef] [Green Version]
- Yun, S.; Hu, R.; Schwaemmle, M.E.; Scherer, A.N.; Zhuang, Z.; Koleske, A.J.; Pallas, D.C.; Schwartz, M.A. Integrin α5β1 regulates PP2A complex assembly through PDE4D in atherosclerosis. J. Clin. Investig. 2019, 129, 4863–4874. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Luo, J.-Y.; Li, B.; Tian, X.Y.; Chen, L.-J.; Huang, Y.; Liu, J.; Deng, D.; Lau, C.W.; Wan, S.; et al. Integrin-YAP/TAZ-JNK cascade mediates atheroprotective effect of unidirectional shear flow. Nature 2016, 540, 579–582. [Google Scholar] [CrossRef]
- Chen, Z.; Tzima, E. PECAM-1 is necessary for flow-induced vascular remodeling. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1067–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitt, M.M.; Megens, R.T.; Zernecke, A.; Bidzhekov, K.; van den Akker, N.M.; Rademakers, T.; van Zandvoort, M.A.; Hackeng, T.M.; Koenen, R.R.; Weber, C. Endothelial junctional adhesion molecule-a guides monocytes into flow-dependent predilection sites of atherosclerosis. Circulation 2014, 129, 66–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeVerse, J.S.; Sandhu, A.S.; Mendoza, N.; Edwards, C.M.; Sun, C.; Simon, S.I.; Passerini, A.G. Shear stress modulates VCAM-1 expression in response to TNF-alpha and dietary lipids via interferon regulatory factor-1 in cultured endothelium. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1149–H1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, K.S.; Fujiwara, K.; Abe, J. Shear stress and atherosclerosis. Mol. Cells 2014, 37, 435–440. [Google Scholar] [CrossRef] [Green Version]
- Pu, L.; Meng, Q.; Li, S.; Wang, Y.; Sun, B.; Liu, B.; Li, F. Laminar shear stress alleviates monocyte adhesion and atherosclerosis development via miR-29b-3p/CX3CL1 axis regulation. J. Cell Sci. 2022, 135, jcs259696. [Google Scholar] [CrossRef]
- Yao, L.; Romero, M.J.; Toque, H.A.; Yang, G.; Caldwell, R.B.; Caldwell, R.W. The role of RhoA/Rho kinase pathway in endothelial dysfunction. J. Cardiovasc. Dis. Res. 2010, 1, 165–170. [Google Scholar]
- Orr, A.W.; Stockton, R.; Simmers, M.B.; Sanders, J.M.; Sarembock, I.J.; Blackman, B.R.; Schwartz, M.A. Matrix-specific p21-activated kinase activation regulates vascular permeability in atherogenesis. J. Cell Biol. 2007, 176, 719–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gavard, J.; Gutkind, J.S. VEGF controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of VE-cadherin. Nat. Cell Biol. 2006, 8, 1223–1234. [Google Scholar] [CrossRef] [PubMed]
- Yoshimatsu, Y.; Watabe, T. Emerging roles of inflammation-mediated endothelial-mesenchymal transition in health and disease. Inflamm. Regen. 2022, 42, 9. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Sanchez-Duffhues, G.; Goumans, M.-J.; Ten Dijke, P. TGF-β-Induced Endothelial to Mesenchymal Transition in Disease and Tissue Engineering. Front. Cell Dev. Biol. 2020, 8, 260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, T.L.; Lee, P.L.; Lee, D.Y.; Wang, W.L.; Wei, S.Y.; Lee, C.I.; Chiu, J.J. Differential regulations of fibronectin and laminin in Smad2 activation in vascular endothelial cells in response to disturbed flow. J. Biomed. Sci. 2018, 25, 1. [Google Scholar] [CrossRef] [Green Version]
- Wang, E.; Wang, H.; Chakrabarti, S. Endothelial-to-mesenchymal transition: An underappreciated mediator of diabetic complications. Front. Endocrinol. 2023, 14, 1050540. [Google Scholar] [CrossRef]
- Liang, G.; Wang, S.; Shao, J.; Jin, Y.J.; Xu, L.; Yan, Y.; Gunther, S.; Wang, L.; Offermanns, S. Tenascin-X Mediates Flow-Induced Suppression of EndMT and Atherosclerosis. Circ. Res. 2022, 130, 1647–1659. [Google Scholar] [CrossRef]
- Sun, X.; Feinberg, M.W. Regulation of endothelial cell metabolism: Just go with the flow. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 13–15. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.; Wang, X.; Du, J.; Cui, Q.; Huang, Y.; Jin, H. Metabolic Reprogramming of Vascular Endothelial Cells: Basic Research and Clinical Applications. Front. Cell Dev. Biol. 2021, 9, 626047. [Google Scholar] [CrossRef]
- Xiao, W.; Oldham, W.M.; Priolo, C.; Pandey, A.K.; Loscalzo, J. Immunometabolic Endothelial Phenotypes: Integrating Inflammation and Glucose Metabolism. Circ. Res. 2021, 129, 9–29. [Google Scholar] [CrossRef]
- Halvorsen, B.; Otterdal, K.; Dahl, T.B.; Skjelland, M.; Gullestad, L.; Øie, E.; Aukrust, P. Atherosclerotic Plaque Stability—What Determines the Fate of a Plaque? Prog. Cardiovasc. Dis. 2008, 51, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Seneviratne, A.; Hulsmans, M.; Holvoet, P.; Monaco, C. Biomechanical factors and macrophages in plaque stability. Cardiovasc. Res. 2013, 99, 284–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, A.A.C.; Serbulea, V.; Baylis, R.A.; Shankman, L.S.; Bradley, X.; Alencar, G.F.; Owsiany, K.; Deaton, R.A.; Karnewar, S.; Shamsuzzaman, S.; et al. Multiple cell types contribute to the atherosclerotic lesion fibrous cap by PDGFRβ and bioenergetic mechanisms. Nat. Metab. 2021, 3, 166–181. [Google Scholar] [CrossRef] [PubMed]
- Lecce, L.; Xu, Y.; V’Gangula, B.; Chandel, N.; Pothula, V.; Caudrillier, A.; Santini, M.P.; d’Escamard, V.; Ceholski, D.K.; Gorski, P.A.; et al. Histone deacetylase 9 promotes endothelial-mesenchymal transition and an unfavorable atherosclerotic plaque phenotype. J. Clin. Investig. 2021, 131, e131178. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Tkachenko, S.; Chaklader, M.; Pletz, C.; Singh, K.; Bulut, G.B.; Han Ym Mitchell, K.; Baylis, R.A.; Kuzmin, A.A.; Hu, B.; et al. Endothelial OCT4 is atheroprotective by preventing metabolic and phenotypic dysfunction. Cardiovasc. Res. 2022, 118, 2458–2477. [Google Scholar] [CrossRef]
- Lee, K.Y.; Chang, K. Understanding Vulnerable Plaques: Current Status and Future Directions. Korean Circ. J. 2019, 49, 1115–1122. [Google Scholar] [CrossRef]
- Andrews, J.P.M.; Fayad, Z.A.; Dweck, M.R. New methods to image unstable atherosclerotic plaques. Atherosclerosis 2018, 272, 118–128. [Google Scholar] [CrossRef] [Green Version]
- Kelsey, L.J.; Bellinge, J.W.; Majeed, K.; Parker, L.P.; Richards, S.; Schultz, C.J.; Doyle, B.J. Low Endothelial Shear Stress Is Associated With High-Risk Coronary Plaque Features and Microcalcification Activity. JACC Cardiovasc. Imaging 2021, 14, 2262–2264. [Google Scholar] [CrossRef]
- Zhang, L.; Li, L.; Li, Y.; Jiang, H.; Sun, Z.; Zang, G.; Qian, Y.; Shao, C.; Wang, Z. Disruption of COMMD1 accelerates diabetic atherosclerosis by promoting glycolysis. Diabetes Vasc. Dis. Res. 2023, 20, 14791641231159009. [Google Scholar] [CrossRef]
- Poels, K.; Schnitzler, J.G.; Waissi, F.; Levels, J.H.M.; Stroes, E.S.G.; Daemen, M.; Lutgens, E.; Pennekamp, A.M.; De Kleijn, D.P.V.; Seijkens, T.T.P.; et al. Inhibition of PFKFB3 Hampers the Progression of Atherosclerosis and Promotes Plaque Stability. Front. Cell Dev. Biol. 2020, 8, 581641. [Google Scholar] [CrossRef]
- Tawakol, A.; Singh, P.; Mojena, M.; Pimentel-Santillana, M.; Emami, H.; MacNabb, M.; Rudd, J.H.; Narula, J.; Enriquez, J.A.; Través, P.G.; et al. HIF-1α and PFKFB3 Mediate a Tight Relationship Between Proinflammatory Activation and Anerobic Metabolism in Atherosclerotic Macrophages. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1463–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parathath, S.; Yang, Y.; Mick, S.; Fisher, E.A. Hypoxia in murine atherosclerotic plaques and its adverse effects on macrophages. Trends Cardiovasc. Med. 2013, 23, 80–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libby, P.; Pasterkamp, G.; Crea, F.; Jang, I.K. Reassessing the Mechanisms of Acute Coronary Syndromes. Circ. Res. 2019, 124, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Durham, A.L.; Speer, M.Y.; Scatena, M.; Giachelli, C.M.; Shanahan, C.M. Role of smooth muscle cells in vascular calcification: Implications in atherosclerosis and arterial stiffness. Cardiovasc. Res. 2018, 114, 590–600. [Google Scholar] [CrossRef] [Green Version]
- Shi, X.; Gao, J.; Lv, Q.; Cai, H.; Wang, F.; Ye, R.; Liu, X. Calcification in Atherosclerotic Plaque Vulnerability: Friend or Foe? Front. Physiol. 2020, 11, 56. [Google Scholar] [CrossRef]
- Shi, X.; Han, Y.; Li, M.; Yin, Q.; Liu, R.; Wang, F.; Xu, X.; Xiong, Y.; Ye, R.; Liu, X. Superficial Calcification with Rotund Shape Is Associated with Carotid Plaque Rupture: An Optical Coherence Tomography Study. Front. Neurol. 2020, 11, 563334. [Google Scholar] [CrossRef]
- Russo, G.; Pedicino, D.; Chiastra, C.; Vinci, R.; Lodi Rizzini, M.; Genuardi, L.; Sarraf, M.; d’Aiello, A.; Bologna, M.; Aurigemma, C.; et al. Coronary artery plaque rupture and erosion: Role of wall shear stress profiling and biological patterns in acute coronary syndromes. Int. J. Cardiol. 2023, 370, 356–365. [Google Scholar] [CrossRef]
- Demer, L.L.; Tintut, Y. Vascular calcification: Pathobiology of a multifaceted disease. Circulation 2008, 117, 2938–2948. [Google Scholar] [CrossRef]
- Chen, W.; Dilsizian, V. Targeted PET/CT imaging of vulnerable atherosclerotic plaques: Microcalcification with sodium fluoride and inflammation with fluorodeoxyglucose. Curr. Cardiol. Rep. 2013, 15, 364. [Google Scholar] [CrossRef]
- Sánchez-Duffhues, G.; García de Vinuesa, A.; van de Pol, V.; Geerts, M.E.; de Vries, M.R.; Janson, S.G.; van Dam, H.; Lindeman, J.H.; Goumans, M.J.; Ten Dijke, P. Inflammation induces endothelial-to-mesenchymal transition and promotes vascular calcification through downregulation of BMPR2. J. Pathol. 2019, 247, 333–346. [Google Scholar] [CrossRef] [Green Version]
- Vengrenyuk, Y.; Carlier, S.; Xanthos, S.; Cardoso, L.; Ganatos, P.; Virmani, R.; Einav, S.; Gilchrist, L.; Weinbaum, S. A hypothesis for vulnerable plaque rupture due to stress-induced debonding around cellular microcalcifications in thin fibrous caps. Proc. Natl. Acad. Sci. USA. 2006, 103, 14678–14683. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Pu, Y.; Zhang, H.; Xie, L.; He, L.; Zhang, C.L.; Cheng, C.K.; Huo, Y.; Wan, S.; Chen, S.; et al. KLF2 Mediates the Suppressive Effect of Laminar Flow on Vascular Calcification by Inhibiting Endothelial BMP/SMAD1/5 Signaling. Circ. Res. 2021, 129, e87–e100. [Google Scholar] [CrossRef]
- Zhu, Y.; Ji, J.J.; Wang, X.D.; Sun, X.J.; Li, M.; Wei, Q.; Ren, L.Q.; Liu, N.F. Periostin promotes arterial calcification through PPARγ-related glucose metabolism reprogramming. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H2222–h2239. [Google Scholar] [CrossRef]
- Niu, J.; Wu, C.; Zhang, M.; Yang, Z.; Liu, Z.; Fu, F.; Li, J.; Feng, N.; Gu, X.; Zhang, S.; et al. κ-opioid receptor stimulation alleviates rat vascular smooth muscle cell calcification via PFKFB3-lactate signaling. Aging 2021, 13, 14355–14371. [Google Scholar] [CrossRef]
- Wang, S.; Yu, H.; Gao, J.; Chen, J.; He, P.; Zhong, H.; Tan, X.; Staines, K.A.; Macrae, V.E.; Fu, X.; et al. PALMD regulates aortic valve calcification via altered glycolysis and NF-κB-mediated inflammation. J. Biol. Chem. 2022, 298, 101887. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.-S.; He, Q.-Z.; Qin, C.H.; Little, P.J.; Weng, J.-P.; Xu, S.-W. Therapeutic potential of colchicine in cardiovascular medicine: A pharmacological review. Acta Pharmacol. Sin. 2022, 43, 2173–2190. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.P.; Chen, C.Y.; Lin, T.W.; Kuo, C.S.; Huang, H.L.; Huang, P.H.; Lin, S.J. Fibroblast growth factor 21 reverses high-fat diet-induced impairment of vascular function via the anti-oxidative pathway in ApoE knockout mice. J. Cell. Mol. Med. 2022, 26, 2451–2461. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; He, S.; Wang, C.; Jian, W.; Shen, X.; Shi, Y.; Liu, J. Fibroblast growth factor 21 inhibits vascular calcification by ameliorating oxidative stress of vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 2023, 650, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.E.; Cober, N.D.; Dai, Z.; Stewart, D.J.; Zhao, Y.Y. Endothelial cells in the pathogenesis of pulmonary arterial hypertension. Eur. Respir. J. 2021, 58. [Google Scholar] [CrossRef] [PubMed]
- Ranchoux, B.; Harvey, L.D.; Ayon, R.J.; Babicheva, A.; Bonnet, S.; Chan, S.Y.; Yuan, J.X.; Perez, V.J. Endothelial dysfunction in pulmonary arterial hypertension: An evolving landscape (2017 Grover Conference Series). Pulm. Circ. 2018, 8, 2045893217752912. [Google Scholar] [CrossRef] [Green Version]
- Budhiraja, R.; Tuder, R.M.; Hassoun, P.M. Endothelial dysfunction in pulmonary hypertension. Circulation 2004, 109, 159–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurakula, K.; Smolders, V.; Tura-Ceide, O.; Jukema, J.W.; Quax, P.H.A.; Goumans, M.J. Endothelial Dysfunction in Pulmonary Hypertension: Cause or Consequence? Biomedicines 2021, 9, 57. [Google Scholar] [CrossRef]
- Dickinson, M.G.; Bartelds, B.; Borgdorff, M.A.; Berger, R.M. The role of disturbed blood flow in the development of pulmonary arterial hypertension: Lessons from preclinical animal models. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 305, L1–L14. [Google Scholar] [CrossRef] [Green Version]
- Rodor, J.; Chen, S.H.; Scanlon, J.P.; Monteiro, J.P.; Caudrillier, A.; Sweta, S.; Stewart, K.R.; Shmakova, A.; Dobie, R.; Henderson, B.E.P.; et al. Single-cell RNA sequencing profiling of mouse endothelial cells in response to pulmonary arterial hypertension. Cardiovasc. Res. 2022, 118, 2519–2534. [Google Scholar] [CrossRef]
- Culley, M.K.; Zhao, J.; Tai, Y.Y.; Tang, Y.; Perk, D.; Negi, V.; Yu, Q.; Woodcock, C.-S.C.; Handen, A.; Speyer, G.; et al. Frataxin deficiency promotes endothelial senescence in pulmonary hypertension. J. Clin. Investig. 2021, 131, e136459. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Xing, Y.; Zhang, J.; He, M.; Dong, J.; Chen, S.; Wu, H.; Huang, H.-Y.; Chou, C.-H.; Bai, L.; et al. MED1 Regulates BMP/TGF-β in Endothelium: Implication for Pulmonary Hypertension. Circ. Res. 2022, 131, 828–841. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Stenmark, K.R.; Shandas, R.; Tan, W. Effects of pathological flow on pulmonary artery endothelial production of vasoactive mediators and growth factors. J Vasc Res. 2009, 46, 561–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.; Zhang, X.; Wang, L.; Yang, Q.; Ma, Q.; Xu, J.; Wang, J.; Kovacs, L.; Ayon, R.J.; Liu, Z.; et al. PFKFB3-mediated endothelial glycolysis promotes pulmonary hypertension. Proc. Natl. Acad. Sci. USA 2019, 116, 13394–13403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacs, L.; Cao, Y.; Han, W.; Meadows, L.; Kovacs-Kasa, A.; Kondrikov, D.; Verin, A.D.; Barman, S.A.; Dong, Z.; Huo, Y.; et al. PFKFB3 in Smooth Muscle Promotes Vascular Remodeling in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2019, 200, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, X.; Cao, Y.; Ma, Q.; Mao, X.; Xu, J.; Yang, Q.; Zhou, Y.; Lucas, R.; Fulton, D.J.; et al. Mice with a specific deficiency of Pfkfb3 in myeloid cells are protected from hypoxia-induced pulmonary hypertension. Br. J. Pharmacol. 2021, 178, 1055–1072. [Google Scholar] [CrossRef]
- Miyagawa, K.; Shi, M.; Chen, P.I.; Hennigs, J.K.; Zhao, Z.; Wang, M.; Li, C.G.; Saito, T.; Taylor, S.; Sa, S.; et al. Smooth Muscle Contact Drives Endothelial Regeneration by BMPR2-Notch1-Mediated Metabolic and Epigenetic Changes. Circ. Res. 2019, 124, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Hwangbo, C.; Hu, X.; Kang, Y.; Papangeli, I.; Mehrotra, D.; Park, H.; Ju, H.; McLean, D.L.; Comhair, S.A.; et al. Restoration of impaired endothelial myocyte enhancer factor 2 function rescues pulmonary arterial hypertension. Circulation 2015, 131, 190–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joffre, J.; Hellman, J.; Ince, C.; Ait-Oufella, H. Endothelial Responses in Sepsis. Am. J. Respir. Crit. Care Med. 2020, 202, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.C.; Filbin, M.R.; Wang, H.; Ngo, L.; Huang, D.T.; Aird, W.C.; Yealy, D.M.; Angus, D.C.; Kellum, J.A.; Shapiro, N.I. Endothelial Permeability and Hemostasis in Septic Shock: Results from the ProCESS Trial. Chest 2017, 152, 22–31. [Google Scholar] [CrossRef]
- London, N.R.; Zhu, W.; Bozza, F.A.; Smith, M.C.P.; Greif, D.M.; Sorensen, L.K.; Chen, L.; Kaminoh, Y.; Chan, A.C.; Passi, S.F.; et al. Targeting Robo4-Dependent Slit Signaling to Survive the Cytokine Storm in Sepsis and Influenza. Sci. Transl. Med. 2010, 2, ra19–ra23. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Lee, S.-J.; Kim, K.E.; Lee, H.S.; Oh, N.; Park, I.; Ko, E.; Oh, S.J.; Lee, Y.-S.; Kim, D.; et al. Amelioration of sepsis by TIE2 activation–induced vascular protection. Sci. Transl. Med. 2016, 8, ra55–ra335. [Google Scholar] [CrossRef]
- Morita, M.; Yoneda, A.; Tokunoh, N.; Masaki, T.; Shirakura, K.; Kinoshita, M.; Hashimoto, R.; Shigesada, N.; Takahashi, J.; Tachibana, M.; et al. Upregulation of Robo4 expression by SMAD signaling suppresses vascular permeability and mortality in endotoxemia and COVID-19 models. Proc. Natl. Acad. Sci. USA 2023, 120, e2213317120. [Google Scholar] [CrossRef]
- Yang, K.; Fan, M.; Wang, X.; Xu, J.; Wang, Y.; Gill, P.S.; Ha, T.; Liu, L.; Hall, J.V.; Williams, D.L.; et al. Lactate induces vascular permeability via disruption of VE-cadherin in endothelial cells during sepsis. Sci. Adv. 2022, 8, eabm8965. [Google Scholar] [CrossRef]
- Semeraro, N.; Ammollo, C.T.; Semeraro, F.; Colucci, M. Sepsis, thrombosis and organ dysfunction. Thromb. Res. 2012, 129, 290–295. [Google Scholar] [CrossRef]
- Yau, J.W.; Teoh, H.; Verma, S. Endothelial cell control of thrombosis. BMC Cardiovasc. Disord. 2015, 15, 130. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, D.; Assreuy, J. Nitric Oxide and Vascular Reactivity in Sepsis. Shock 2008, 30, 10–13. [Google Scholar] [CrossRef]
- Cauwels, A. Nitric oxide in shock. Kidney Int. 2007, 72, 557–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Hu, R.; Cavinato, C.; Zhuang, Z.W.; Zhang, J.; Yun, S.; Fernandez Tussy, P.; Singh, A.; Murtada, S.-I.; Tanaka, K.; et al. Fibronectin–Integrin α5 Signaling in Vascular Complications of Type 1 Diabetes. Diabetes. 2022, 71, 2020–2033. [Google Scholar] [CrossRef]
- Maharjan, S.; Kim, K.; Agrawal, V.; Choi, H.-J.; Kim, N.-J.; Kim, Y.-M.; Suh, Y.-G.; Kwon, Y.-G. Sac-1004, a novel vascular leakage blocker, enhances endothelial barrier through the cAMP/Rac/cortactin pathway. Biochem. Biophys. Res. Commun. 2013, 435, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Shao, S.; Zhu, Z.; Chen, J.; Hao, J.; Bai, Y.; Li, B.; Dang, E.; Wang, G. An IGFBP7high endothelial cell subset drives T cell extravasation in psoriasis via endothelial glycocalyx degradation. J. Clin. Investig. 2023, 133, e160451. [Google Scholar] [CrossRef] [PubMed]
- Hoeper, M.M.; Badesch, D.B.; Ghofrani, H.A.; Gibbs, J.S.R.; Gomberg-Maitland, M.; McLaughlin, V.V.; Preston, I.R.; Souza, R.; Waxman, A.B.; Grünig, E.; et al. Phase 3 Trial of Sotatercept for Treatment of Pulmonary Arterial Hypertension. N. Engl. J. Med. 2023, 388, 1478–1490. [Google Scholar] [CrossRef]
- Geldhof, V.; de Rooij, L.P.M.H.; Sokol, L.; Amersfoort, J.; De Schepper, M.; Rohlenova, K.; Hoste, G.; Vanderstichele, A.; Delsupehe, A.-M.; Isnaldi, E.; et al. Single cell atlas identifies lipid-processing and immunomodulatory endothelial cells in healthy and malignant breast. Nat. Commun. 2022, 13, 5511. [Google Scholar] [CrossRef]
- Andueza, A.; Kumar, S.; Kim, J.; Kang, D.-W.; Mumme, H.L.; Perez, J.I.; Villa-Roel, N.; Jo, H. Endothelial Reprogramming by Disturbed Flow Revealed by Single-Cell RNA and Chromatin Accessibility Study. Cell Rep. 2020, 33, 108491. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Immanuel, J.; Yun, S. Vascular Inflammatory Diseases and Endothelial Phenotypes. Cells 2023, 12, 1640. https://doi.org/10.3390/cells12121640
Immanuel J, Yun S. Vascular Inflammatory Diseases and Endothelial Phenotypes. Cells. 2023; 12(12):1640. https://doi.org/10.3390/cells12121640
Chicago/Turabian StyleImmanuel, Jenita, and Sanguk Yun. 2023. "Vascular Inflammatory Diseases and Endothelial Phenotypes" Cells 12, no. 12: 1640. https://doi.org/10.3390/cells12121640
APA StyleImmanuel, J., & Yun, S. (2023). Vascular Inflammatory Diseases and Endothelial Phenotypes. Cells, 12(12), 1640. https://doi.org/10.3390/cells12121640