
Viral Interference of Hepatitis C and E Virus Replication in Novel Experimental Co-Infection Systems

 , , , , ,
, , , , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Plasmids
2.2. Compounds and Reagents
2.3. Cell Culture
2.4. In Vitro Transcription and Electroporation
2.5. Luciferase Assay
2.6. Immunofluorescence Staining
2.7. Quantification of HCV and HEV-Positive Cells in Fluorescence Microscopy Pictures
2.8. VSV Infection
2.9. Flow Cytometry
2.10. Pseudoparticle Production for Production of Transduced Cell Lines
2.11. HCVcc and HEVcc Production
2.12. HEV Super-Transfection Experiments
2.13. HEV Infection Experiments
2.14. HCV Super-Infection on HEV Expressing Cells
2.15. Determination of Viral Titers
2.16. Production and Infection of Human Liver Chimeric Mice
2.17. Statistical Analysis and Graphics
3. Results
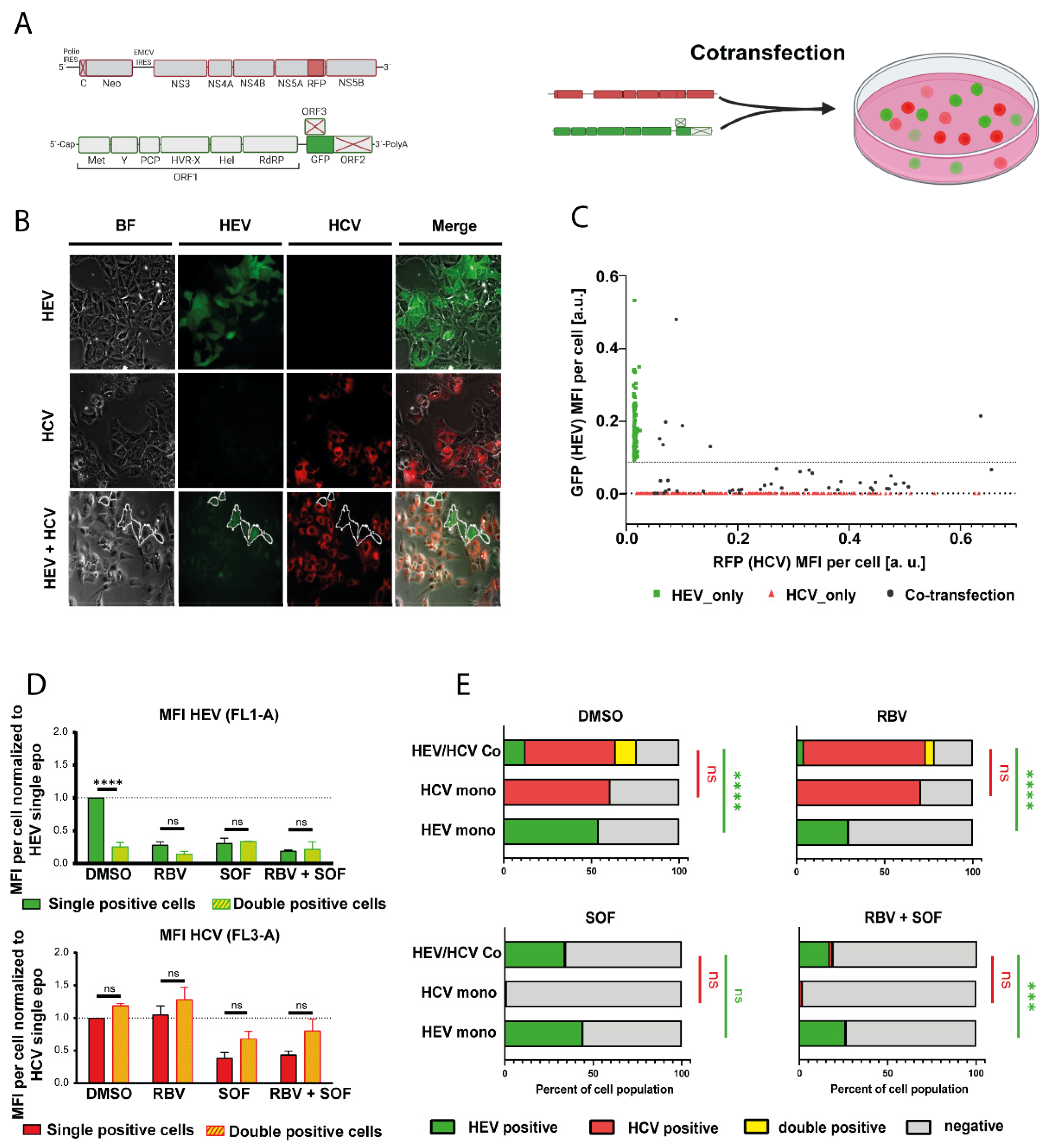
3.1. HCV Impairs HEV Replication in Subgenomic Reporter Co-Transfection Assays
3.2. HCV Treatment Restores HEV Replication Capacity
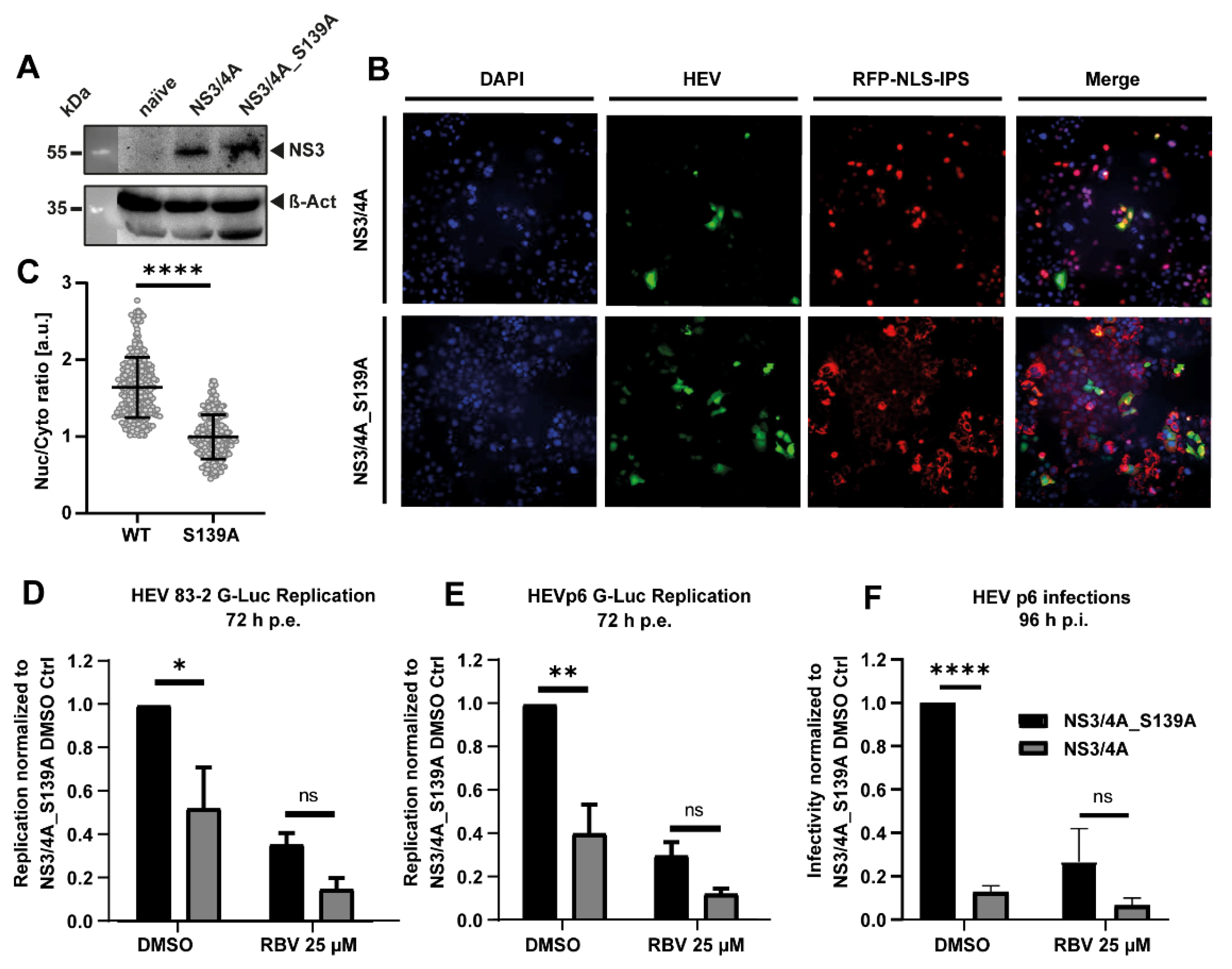
3.3. The HCV Protease NS3/4A Inhibits HEV Replication
3.4. HCV Super-Infection of HEV-Replicating Cells
3.5. HEV or HCV-Positive Human Liver Chimeric Mice Showed Reduced Viral Loads in Individual Mice after Super-Infection
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rein, D.B.; Stevens, G.A.; Theaker, J.; Wittenborn, J.S.; Wiersma, S.T. The global burden of Hepatitis E virus genotypes 1 and 2 in 2005. Hepatology 2012, 55, 988–997. [Google Scholar] [CrossRef] [PubMed]
- Wedemeyer, H.; Pischke, S.; Manns, M.P. Pathogenesis and Treatment of Hepatitis E Virus Infection. Gastroenterology 2012, 142, 1388–1397. [Google Scholar] [CrossRef] [PubMed]
- Kamar, N.; Rostaing, L.; Izopet, J. Hepatitis E virus infection in immunosuppressed patients: Natural history and therapy. Semin. Liver Dis. 2013, 33, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Dalton, H.R.; Kamar, N.; Baylis, S.A.; Moradpour, D.; Wedemeyer, H.; Negro, F.; European Association for the Study of the Liver. EASL Clinical Practice Guidelines on hepatitis E virus infection. J. Hepatol. 2018, 68, 1256–1271. [Google Scholar] [CrossRef] [PubMed]
- Kamar, N.; Izopet, J.; Tripon, S.; Bismuth, M.; Hillaire, S.; Dumortier, J.; Radenne, S.; Coilly, A.; Garrigue, V.; D’Alteroche, L.; et al. Ribavirin for chronic hepatitis E virus infection in transplant recipients. N. Engl. J. Med. 2014, 370, 1111–1120. [Google Scholar] [CrossRef]
- Kamar, N.; Abravanel, F.; Behrendt, P.; Hofmann, J.; Pageaux, G.P.; Barbet, C.; Moal, V.; Couzi, L.; Horvatits, T.; de Man, R.A.; et al. Ribavirin for Hepatitis E Virus Infection After Organ Transplantation: A Large European Retrospective Multicenter Study. Clin. Infect. Dis. 2020, 71, 1204–1211. [Google Scholar] [CrossRef]
- Kinast, V.; Burkard, T.L.; Todt, D.; Steinmann, E. Hepatitis E Virus Drug Development. Viruses 2019, 11, 485. [Google Scholar] [CrossRef]
- Todt, D.; Friesland, M.; Moeller, N.; Praditya, D.; Kinast, V.; Brüggemann, Y.; Knegendorf, L.; Burkard, T.; Steinmann, J.; Burm, R.; et al. Robust Hepatitis E virus infection and transcriptional response in human hepatocytes. Proc. Natl. Acad. Sci. USA 2020, 117, 1731. [Google Scholar] [CrossRef]
- Nimgaonkar, I.; Ding, Q.; Schwartz, R.E.; Ploss, A. Hepatitis E virus: Advances and challenges. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 96–110. [Google Scholar] [CrossRef]
- Meister, T.L.; Bruening, J.; Todt, D.; Steinmann, E. Cell culture systems for the study of Hepatitis E virus. Antivir. Res. 2019, 163, 34–49. [Google Scholar] [CrossRef]
- Gower, E.; Estes, C.; Blach, S.; Razavi-Shearer, K.; Razavi, H. Global epidemiology and genotype distribution of the Hepatitis C virus infection. J. Hepatol. 2014, 61, S45–S57. [Google Scholar] [CrossRef] [PubMed]
- Thrift, A.P.; El-Serag, H.B.; Kanwal, F. Global epidemiology and burden of HCV infection and HCV-related disease. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Manns, M.P.; Buti, M.; Gane, E.; Pawlotsky, J.-M.; Razavi, H.; Terrault, N.; Younossi, Z. Hepatitis C virus infection. Nat. Rev. Dis. Primers 2017, 3, 17006. [Google Scholar] [CrossRef] [PubMed]
- Yamane, D.; McGivern, D.R.; Masaki, T.; Lemon, S.M. Liver Injury and Disease Pathogenesis in Chronic Hepatitis C. In Hepatitis C Virus: From Molecular Virology to Antiviral Therapy; Bartenschlager, R., Ed.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 263–288. ISBN 978-3-642-27340-7. [Google Scholar]
- EASL recommendations on treatment of Hepatitis C: Final update of the series. J. Hepatol. 2020, 73, 1170–1218. [CrossRef]
- Razavi, H.; Sanchez Gonzalez, Y.; Yuen, C.; Cornberg, M. Global timing of Hepatitis C virus elimination in high-income countries. Liver Int. 2020, 40, 522–529. [Google Scholar] [CrossRef]
- Hepatitis C virus prevalence and level of intervention required to achieve the WHO targets for elimination in the European Union by 2030: A modelling study. Lancet Gastroenterol. Hepatol. 2017, 2, 325–336. [CrossRef]
- Yeo, Y.H.; Kam, L.Y.; Le, M.H.; Jeong, D.; Dang, N.; Henry, L.; Cheung, R.; Nguyen, M.H. A population-based US study of Hepatitis C diagnosis rate. Eur. J. Gastroenterol. Hepatol. 2021, 33, e471–e477. [Google Scholar] [CrossRef]
- Hartl, J.; Otto, B.; Madden, R.G.; Webb, G.; Woolson, K.L.; Kriston, L.; Vettorazzi, E.; Lohse, A.W.; Dalton, H.R.; Pischke, S. Hepatitis E Seroprevalence in Europe: A Meta-Analysis. Viruses 2016, 8, 211. [Google Scholar] [CrossRef]
- Boland, F.; Martinez, A.; Pomeroy, L.; O’Flaherty, N. Blood Donor Screening for Hepatitis E Virus in the European Union. Transfus. Med. Hemother. 2019, 46, 95–103. [Google Scholar] [CrossRef]
- de Martin, E.; Antonini, T.M.; Coilly, A.; Pittau, G.; Vibert, E.; Duclos-Vallée, J.-C.; Samuel, D.; Roque-Afonso, A.-M. HCV and HEV recurrence after liver transplantation: One antiviral therapy for two viruses. Transpl. Int. 2017, 30, 318–319. [Google Scholar] [CrossRef]
- Donnelly, M.C.; Imlach, S.N.; Abravanel, F.; Ramalingam, S.; Johannessen, I.; Petrik, J.; Fraser, A.R.; Campbell, J.D.M.; Bramley, P.; Dalton, H.R.; et al. Sofosbuvir and Daclatasvir Anti-Viral Therapy Fails to Clear HEV Viremia and Restore Reactive T Cells in a HEV/HCV Co-Infected Liver Transplant Recipient. Gastroenterology 2017, 152, 300–301. [Google Scholar] [CrossRef] [PubMed]
- Wahid, B. Successful treatment of HBV, HCV, & HEV, with 12-week long use of tenofovir, sofosbuvir, daclatasvir, and ribavirin: A case report. J. Infect. Public Health 2020, 13, 149–150. [Google Scholar] [CrossRef] [PubMed]
- Vrolijk, J.M.; Kaul, A.; Hansen, B.E.; Lohmann, V.; Haagmans, B.L.; Schalm, S.W.; Bartenschlager, R. A replicon-based bioassay for the measurement of interferons in patients with chronic Hepatitis C. J. Virol. Methods 2003, 110, 201–209. [Google Scholar] [CrossRef]
- Schaller, T.; Appel, N.; Koutsoudakis, G.; Kallis, S.; Lohmann, V.; Pietschmann, T.; Bartenschlager, R. Analysis of Hepatitis C Virus Superinfection Exclusion by Using Novel Fluorochrome Gene-Tagged Viral Genomes. J. Virol. 2007, 81, 4591. [Google Scholar] [CrossRef] [PubMed]
- Pietschmann, T.; Kaul, A.; Koutsoudakis, G.; Shavinskaya, A.; Kallis, S.; Steinmann, E.; Abid, K.; Negro, F.; Dreux, M.; Cosset, F.-L.; et al. Construction and characterization of infectious intragenotypic and intergenotypic Hepatitis C virus chimeras. Proc. Natl. Acad. Sci. USA 2006, 103, 7408–7413. [Google Scholar] [CrossRef]
- Anggakusuma; Brown, R.J.P.; Banda, D.H.; Todt, D.; Vieyres, G.; Steinmann, E.; Pietschmann, T.; Diamond, M.S. Hepacivirus NS3/4A Proteases Interfere with MAVS Signaling in both Their Cognate Animal Hosts and Humans: Implications for Zoonotic Transmission. J. Virol. 2016, 90, 10670. [Google Scholar] [CrossRef] [PubMed]
- Todt, D.; François, C.; Anggakusuma; Behrendt, P.; Engelmann, M.; Knegendorf, L.; Vieyres, G.; Wedemeyer, H.; Hartmann, R.; Pietschmann, T.; et al. Antiviral Activities of Different Interferon Types and Subtypes against Hepatitis E Virus Replication. Antimicrob. Agents Chemother. 2016, 60, 2132–2139. [Google Scholar] [CrossRef]
- Ding, Q.; Nimgaonkar, I.; Archer, N.F.; Bram, Y.; Heller, B.; Schwartz, R.E.; Ploss, A. Identification of the Intragenomic Promoter Controlling Hepatitis E Virus Subgenomic RNA Transcription. mBio 2018, 9, e00769-18. [Google Scholar] [CrossRef]
- Shiota, T.; Li, T.-C.; Yoshizaki, S.; Kato, T.; Wakita, T.; Ishii, K. The hepatitis E virus capsid C-terminal region is essential for the viral life cycle: Implication for viral genome encapsidation and particle stabilization. J. Virol. 2013, 87, 6031–6036. [Google Scholar] [CrossRef]
- Anggakusuma; Romero-Brey, I.; Berger, C.; Colpitts, C.C.; Boldanova, T.; Engelmann, M.; Todt, D.; Perin, P.M.; Behrendt, P.; Vondran, F.W.R.; et al. Interferon-inducible cholesterol-25-hydroxylase restricts Hepatitis C virus replication through blockage of membranous web formation. Hepatology 2015, 62, 702–714. [Google Scholar] [CrossRef]
- Hoffmann, M.; Wu, Y.-J.; Gerber, M.; Berger-Rentsch, M.; Heimrich, B.; Schwemmle, M.; Zimmer, G. Fusion-active glycoprotein G mediates the cytotoxicity of vesicular stomatitis virus M mutants lacking host shut-off activity. J. Gen. Virol. 2010, 91, 2782–2793. [Google Scholar] [CrossRef] [PubMed]
- Vieyres, G.; Pietschmann, T. Entry and replication of recombinant Hepatitis C viruses in cell culture. Methods 2013, 59, 233–248. [Google Scholar] [CrossRef] [PubMed]
- Meuleman, P.; Libbrecht, L.; de Vos, R.; de Hemptinne, B.; Gevaert, K.; Vandekerckhove, J.; Roskams, T.; Leroux-Roels, G. Morphological and biochemical characterization of a human liver in a uPA-SCID mouse chimera. Hepatology 2005, 41, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Sayed, I.M.; Verhoye, L.; Cocquerel, L.; Abravanel, F.; Foquet, L.; Montpellier, C.; Debing, Y.; Farhoudi, A.; Wychowski, C.; Dubuisson, J.; et al. Study of hepatitis E virus infection of genotype 1 and 3 in mice with humanised liver. Gut 2017, 66, 920. [Google Scholar] [CrossRef]
- Nishiyama, T.; Kobayashi, T.; Jirintai, S.; Nagashima, S.; Primadharsini, P.P.; Nishizawa, T.; Okamoto, H. Antiviral candidates against the hepatitis E virus (HEV) and their combinations inhibit HEV growth in in vitro. Antivir. Res. 2019, 170, 104570. [Google Scholar] [CrossRef]
- Dao Thi, V.L.; Debing, Y.; Wu, X.; Rice, C.M.; Neyts, J.; Moradpour, D.; Gouttenoire, J. Sofosbuvir Inhibits Hepatitis E Virus Replication In Vitro and Results in an Additive Effect When Combined with Ribavirin. Gastroenterology 2016, 150, 82–85. [Google Scholar] [CrossRef]
- Cornberg, M.; Pischke, S.; Müller, T.; Behrendt, P.; Piecha, F.; Benckert, J.; Todt, D.; Steinmann, E.; Papkalla, A.; von Karpowitz, M.; et al. Sofosbuvir monotherapy fails to achieve HEV RNA elimination in patients with chronic hepatitis E-The HepNet SofE pilot study. J. Hepatol. 2020, 73, 696–699. [Google Scholar] [CrossRef]
- Jones, C.T.; Catanese, M.T.; Law, L.M.J.; Khetani, S.R.; Syder, A.J.; Ploss, A.; Oh, T.S.; Schoggins, J.W.; MacDonald, M.R.; Bhatia, S.N.; et al. Real-time imaging of Hepatitis C virus infection using a fluorescent cell-based reporter system. Nat. Biotechnol. 2010, 28, 167–171. [Google Scholar] [CrossRef]
- Allweiss, L.; Gass, S.; Giersch, K.; Groth, A.; Kah, J.; Volz, T.; Rapp, G.; Schöbel, A.; Lohse, A.W.; Polywka, S.; et al. Human liver chimeric mice as a new model of chronic hepatitis E virus infection and preclinical drug evaluation. J. Hepatol. 2016, 64, 1033–1040. [Google Scholar] [CrossRef]
- Burm, R.; Collignon, L.; Mesalam, A.A.; Meuleman, P. Animal Models to Study Hepatitis C Virus Infection. Front. Immunol. 2018, 9, 1032. [Google Scholar] [CrossRef]
- Lin, C. HCV NS3-4A Serine Protease. In Hepatitis C Viruses: Genomes and Molecular Biology; Tan, S.L., Ed.; Horizon Bioscience: Norfolk, UK, 2006. [Google Scholar]
- Li, K.; Foy, E.; Ferreon, J.C.; Nakamura, M.; Ferreon, A.C.M.; Ikeda, M.; Ray, S.C.; Gale, M., Jr.; Lemon, S.M. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc. Natl. Acad. Sci. USA 2005, 102, 2992–2997. [Google Scholar] [CrossRef] [PubMed]
- Shiryaev, S.A.; Thomsen, E.R.; Cieplak, P.; Chudin, E.; Cheltsov, A.V.; Chee, M.S.; Kozlov, I.A.; Strongin, A.Y. New details of HCV NS3/4A proteinase functionality revealed by a high-throughput cleavage assay. PLoS ONE 2012, 7, e35759. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Elhendawy, M.; Abo-Ali, L.; Abd-Elsalam, S.; Hagras, M.M.; Kabbash, I.; Mansour, L.; Atia, S.; Esmat, G.; Abo-ElAzm, A.-R.; El-Kalla, F.; et al. HCV and HEV: Two players in an Egyptian village, a study of prevalence, incidence, and co-infection. Environ. Sci. Pollut. Res. Int. 2020, 27, 33659–33667. [Google Scholar] [CrossRef] [PubMed]
- Bayram, A.; Eksi, F.; Mehli, M.; Sözen, E. Prevalence of Hepatitis E Virus Antibodies in Patients with Chronic Hepatitis B and Chronic Hepatitis C. Intervirology 2007, 50, 281–286. [Google Scholar] [CrossRef]
- Mellgren, Å.; Karlsson, M.; Karlsson, M.; Lagging, M.; Wejstål, R.; Norder, H. High seroprevalence against hepatitis E virus in patients with chronic Hepatitis C virus infection. J. Clin. Virol. 2017, 88, 39–45. [Google Scholar] [CrossRef]
- Kuniholm, M.H.; Purcell, R.H.; McQuillan, G.M.; Engle, R.E.; Wasley, A.; Nelson, K.E. Epidemiology of hepatitis E virus in the United States: Results from the Third National Health and Nutrition Examination Survey, 1988–1994. J. Infect. Dis. 2009, 200, 48–56. [Google Scholar] [CrossRef]
- Taremi, M.; Khoshbaten, M.; Gachkar, L.; EhsaniArdakani, M.; Zali, M. Hepatitis E virus infection in hemodialysis patients: A seroepidemiological survey in Iran. BMC Infect. Dis. 2005, 5, 36. [Google Scholar] [CrossRef]
- Rivero-Juarez, A.; Martinez-Dueñas, L.; Martinez-Peinado, A.; Camacho, A.; Cifuentes, C.; Gordon, A.; Frias, M.; Torre-Cisneros, J.; Pineda, J.A.; Rivero, A. High hepatitis E virus seroprevalence with absence of chronic infection in HIV-infected patients. J. Infect. 2015, 70, 624–630. [Google Scholar] [CrossRef]
- Yu, C.; Boon, D.; McDonald, S.L.; Myers, T.G.; Tomioka, K.; Nguyen, H.; Engle, R.E.; Govindarajan, S.; Emerson, S.U.; Purcell, R.H. Pathogenesis of hepatitis E virus and Hepatitis C virus in chimpanzees: Similarities and differences. J. Virol. 2010, 84, 11264–11278. [Google Scholar] [CrossRef]
- Liang, Y.; Shilagard, T.; Xiao, S.-Y.; Snyder, N.; Lau, D.; Cicalese, L.; Weiss, H.; Vargas, G.; Lemon, S.M. Visualizing Hepatitis C virus infections in human liver by two-photon microscopy. Gastroenterology 2009, 137, 1448–1458. [Google Scholar] [CrossRef]
- Kandathil, A.J.; Graw, F.; Quinn, J.; Hwang, H.S.; Torbenson, M.; Perelson, A.S.; Ray, S.C.; Thomas, D.L.; Ribeiro, R.M.; Balagopal, A. Use of laser capture microdissection to map Hepatitis C virus-positive hepatocytes in human liver. Gastroenterology 2013, 145, 1404–1413. [Google Scholar] [CrossRef] [PubMed]
- Lenggenhager, D.; Gouttenoire, J.; Malehmir, M.; Bawohl, M.; Honcharova-Biletska, H.; Kreutzer, S.; Semela, D.; Neuweiler, J.; Hürlimann, S.; Aepli, P.; et al. Visualization of hepatitis E virus RNA and proteins in the human liver. J. Hepatol. 2017, 67, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Boldanova, T.; Suslov, A.; Heim, M.H.; Necsulea, A. Transcriptional response to Hepatitis C virus infection and interferon-alpha treatment in the human liver. EMBO Mol. Med. 2017, 9, 816–834. [Google Scholar] [CrossRef] [PubMed]
- Sheahan, T.; Imanaka, N.; Marukian, S.; Dorner, M.; Liu, P.; Ploss, A.; Rice, C.M. Interferon lambda alleles predict innate antiviral immune responses and Hepatitis C virus permissiveness. Cell Host Microbe 2014, 15, 190–202. [Google Scholar] [CrossRef] [PubMed]
- Walters, K.-A.; Syder, A.J.; Lederer, S.L.; Diamond, D.L.; Paeper, B.; Rice, C.M.; Katze, M.G. Genomic analysis reveals a potential role for cell cycle perturbation in HCV-mediated apoptosis of cultured hepatocytes. PLoS Pathog. 2009, 5, e1000269. [Google Scholar] [CrossRef]
- Blackham, S.; Baillie, A.; Al-Hababi, F.; Remlinger, K.; You, S.; Hamatake, R.; McGarvey, M.J. Gene expression profiling indicates the roles of host oxidative stress, apoptosis, lipid metabolism, and intracellular transport genes in the replication of Hepatitis C virus. J. Virol. 2010, 84, 5404–5414. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burkard, T.; Proske, N.; Resner, K.; Collignon, L.; Knegendorf, L.; Friesland, M.; Verhoye, L.; Sayed, I.M.; Brüggemann, Y.; Nocke, M.K.; et al. Viral Interference of Hepatitis C and E Virus Replication in Novel Experimental Co-Infection Systems. Cells 2022, 11, 927. https://doi.org/10.3390/cells11060927
Burkard T, Proske N, Resner K, Collignon L, Knegendorf L, Friesland M, Verhoye L, Sayed IM, Brüggemann Y, Nocke MK, et al. Viral Interference of Hepatitis C and E Virus Replication in Novel Experimental Co-Infection Systems. Cells. 2022; 11(6):927. https://doi.org/10.3390/cells11060927
Chicago/Turabian StyleBurkard, Thomas, Nora Proske, Kathrin Resner, Laura Collignon, Leonard Knegendorf, Martina Friesland, Lieven Verhoye, Ibrahim M. Sayed, Yannick Brüggemann, Maximilian K. Nocke, and et al. 2022. "Viral Interference of Hepatitis C and E Virus Replication in Novel Experimental Co-Infection Systems" Cells 11, no. 6: 927. https://doi.org/10.3390/cells11060927
APA StyleBurkard, T., Proske, N., Resner, K., Collignon, L., Knegendorf, L., Friesland, M., Verhoye, L., Sayed, I. M., Brüggemann, Y., Nocke, M. K., Behrendt, P., Wedemeyer, H., Meuleman, P., Todt, D., & Steinmann, E. (2022). Viral Interference of Hepatitis C and E Virus Replication in Novel Experimental Co-Infection Systems. Cells, 11(6), 927. https://doi.org/10.3390/cells11060927