
Redox Implications of Extreme Task Performance: The Case in Driver Athletes

Abstract
1. Introduction
2. Physical Work
3. Vibration
4. Noise
5. Heat
6. Cardiovascular Demand
7. Mental Factors
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reid, M.B.; Lightfoot, J.T. The physiology of auto racing. Med. Sci. Sports Exerc. 2019, 51, 2548–2562. [Google Scholar] [CrossRef] [PubMed]
- Bjugstad, K.B.; Gutowski, P.; Pekarek, J.; Bourg, P.; Mains, C.W.; Bar-Or, D. Redox changes in amateur race car drivers before and after racing. Sports Med. Int. Open 2017, 1, E212–E219. [Google Scholar] [CrossRef][Green Version]
- Pruett, M. You Think Driving an Indy Car Is Easy? Available online: https://www.roadandtrack.com/motorsports/news/a18270/you-think-driving-an-indy-car-is-easy/ (accessed on 25 January 2022).
- Ebben, W.P.; Suchomel, T.J. Physical demands, injuries, and conditioning practices of stock car drivers. J. Strength Cond. Res. 2012, 26, 1188–1198. [Google Scholar] [CrossRef] [PubMed]
- Altmann, D.; Benbow, S.; Bouchard, J. Medicine in Motor Sport; FIA Institute for Motor Sport Safety and Sustainability: London, UK, 2011; pp. 60–69. [Google Scholar]
- Harding, R.M. Human Respiratory Responses during High Performance Flight; Advisory Group for Aerospace Research & Development: Hampshire, UK, 1987. [Google Scholar]
- Norton, C. Formula One Drivers Feel the g-Force. Available online: https://www.telegraph.co.uk/motoring/motorsport/7681665/Formula-One-drivers-feel-the-G-force.html (accessed on 25 January 2022).
- Beaune, B.; Durand, S.; Mariot, J.P. Open-wheel race car driving: Energy cost for pilots. J. Strength Cond. Res. 2010, 24, 2927–2932. [Google Scholar] [CrossRef]
- Lighthall, J.W.; Pierce, J.; Olvey, S. A physiological profile of high-performance race car drivers. SAE Int. 1994, 1, 55–63. [Google Scholar] [CrossRef]
- Ainsworth, B.E.; Haskell, W.L.; Herrmann, S.D.; Meckes, N.; Bassett, D.R., Jr.; Tudor-Locke, C.; Greer, J.L.; Vezina, J.; Whitt-Glover, M.C.; Leon, A.S. 2011 Compendium of physical activities: A second update of codes and MET values. Med. Sci. Sports Exerc. 2011, 43, 1575–1581. [Google Scholar] [CrossRef]
- Jacobs, P.L.; Olvey, S.E.; Johnson, B.M.; Cohn, K. Physiological responses to high-speed, open-wheel racecar driving. Med. Sci. Sports Exerc. 2002, 34, 2085–2090. [Google Scholar] [CrossRef] [PubMed]
- Schwaberger, G. Heart rate, metabolic and hormonal responses to maximal psycho-emotional and physical stress in motor car racing drivers. Int. Arch. Occup. Environ. Health 1987, 59, 579–604. [Google Scholar] [CrossRef]
- Taggart, P.; Carruthers, M. Endogenous hyperlipidaemia induced by emotional stress of racing driving. Lancet 1971, 1, 363–366. [Google Scholar] [CrossRef]
- Collins, V.P. Physiologic Observations on Race Car Drivers. NASA CR-570; NASA Contractor Report; NASA CR: Washington, DC, USA, 1966; pp. 1–114.
- Backman, J.; Hakkinen, K.; Ylinen, J.; Hakkinen, A.; Kyrolainen, H. Neuromuscular performance characteristics of open-wheel and rally drivers. J. Strength Cond. Res. 2005, 19, 777–784. [Google Scholar] [CrossRef]
- Del Rosso, S.; Abreu, L.; Webb, H.E.; Zouhal, H.; Boullosa, D.A. Stress markers during a rally car competition. J. Strength Cond. Res. 2016, 30, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Azizan, M.A.; Fard, M. The influence of vibrations on vehicle occupant fatigue. In Proceedings of the inter.noise 2014. 43rd International Congress on Noise Control Engineering, Melbourne, Australia, 16–19 November 2014; Volume 249, pp. 1767–1777. [Google Scholar]
- Baroody, N.B., Jr.; Thomason, J.M. Blistering the track—and drivers—in stock racing. Phys. Sportsmed. 1975, 3, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Tsopanakis, C.; Tsopanakis, A. Stress hormonal factors, fatigue, and antioxidant responses to prolonged speed driving. Pharmacol. Biochem. Behav. 1998, 60, 747–751. [Google Scholar] [CrossRef]
- Reid, M.B.; Haack, K.E.; Franchek, K.M.; Valberg, P.A.; Kobzik, L.; West, M.S. Reactive oxygen in skeletal muscle. I. Intracellular oxidant kinetics and fatigue in vitro. J. Appl. Physiol. 1992, 73, 1797–1804. [Google Scholar] [CrossRef]
- Reid, M.B.; Shoji, T.; Moody, M.R.; Entman, M.L. Reactive oxygen in skeletal muscle. II. Extracellular release of free radicals. J. Appl. Physiol 1992, 73, 1805–1809. [Google Scholar] [CrossRef]
- Barbieri, E.; Sestili, P. Reactive oxygen species in skeletal muscle signaling. J. Signal Transduct. 2012, 2012, 982794. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef]
- Sakellariou, G.K.; Jackson, M.J.; Vasilaki, A. Redefining the major contributors to superoxide production in contracting skeletal muscle. The role of NAD(P)H oxidases. Free Radic. Res. 2014, 48, 12–29. [Google Scholar] [CrossRef]
- Kobzik, L.; Reid, M.B.; Bredt, D.S.; Stamler, J.S. Nitric oxide in skeletal muscle. Nature 1994, 372, 546–548. [Google Scholar] [CrossRef]
- Kobzik, L.; Stringer, B.; Balligand, J.L.; Reid, M.B.; Stamler, J.S. Endothelial type nitric oxide synthase in skeletal muscle fibers: Mitochondrial relationships. Biochem. Biophys. Res. Commun. 1995, 211, 375–381. [Google Scholar] [CrossRef]
- Jones, A.M.; Vanhatalo, A.; Seals, D.R.; Rossman, M.J.; Piknova, B.; Jonvik, K.L. Dietary nitrate and nitric oxide metabolism: Mouth, circulation, skeletal muscle, and exercise performance. Med. Sci. Sports Exerc. 2021, 53, 280–294. [Google Scholar] [CrossRef] [PubMed]
- Morrison, R.J.; Miller, C.C., 3rd; Reid, M.B. Nitric oxide effects on force-velocity characteristics of the rat diaphragm. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 1998, 119, 203–209. [Google Scholar] [CrossRef]
- Brown, G.C. Nitric oxide regulates mitochondrial respiration and cell functions by inhibiting cytochrome oxidase. FEBS Lett. 1995, 369, 136–139. [Google Scholar] [CrossRef]
- McConell, G.K.; Rattigan, S.; Lee-Young, R.S.; Wadley, G.D.; Merry, T.L. Skeletal muscle nitric oxide signaling and exercise: A focus on glucose metabolism. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E301–E307. [Google Scholar] [CrossRef] [PubMed]
- Reid, M.B.; Kobzik, L.; Bredt, D.S.; Stamler, J.S. Nitric oxide modulates excitation-contraction coupling in the diaphragm. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 1998, 119, 211–218. [Google Scholar] [CrossRef]
- Andrade, F.H.; Reid, M.B.; Allen, D.G.; Westerblad, H. Effect of nitric oxide on single skeletal muscle fibres from the mouse. J. Physiol. 1998, 509 Pt 2, 577–586. [Google Scholar] [CrossRef]
- Andrade, F.H.; Reid, M.B.; Allen, D.G.; Westerblad, H. Effect of hydrogen peroxide and dithiothreitol on contractile function of single skeletal muscle fibres from the mouse. J. Physiol. 1998, 509 Pt 2, 565–575. [Google Scholar] [CrossRef]
- Wolin, M.S.; Hintze, T.H.; Shen, W.; Mohazzab, H.K.; Xie, Y.W. Involvement of reactive oxygen and nitrogen species in signalling mechanisms that control tissue respiration in muscle. Biochem. Soc. Trans. 1997, 25, 934–939. [Google Scholar] [CrossRef]
- Reid, M.B. Nitric oxide, reactive oxygen species, and skeletal muscle contraction. Med. Sci. Sports Exerc. 2001, 33, 371–376. [Google Scholar] [CrossRef]
- Ferreira, L.F.; Reid, M.B. Muscle-derived ROS and thiol regulation in muscle fatigue. J. Appl. Physiol. 2008, 104, 853–860. [Google Scholar] [CrossRef]
- Cocco, L.G.P.; Rapuano, S.; Rossi, L. Vibrations measurements of Formula One car electronic devices. roceedings of the 16th IMEKO TC4 Symposium, Exploring New Frontiers of Instrumentation and Methods for Electrical and Electronic Measurements, Florence, Italy, 22–24 September 2008; 2008. [Google Scholar]
- Leslie-Pelecky, D. The Science of Vibration Harmonics. Available online: https://buildingspeed.org/2008/03/06/the_science_of_vibration_harmonics_6/ (accessed on 25 January 2022).
- Sorokin, P.; Khryakov, K.; Gommers, M. Analysis of Dallara T12 race car front wing vibrations. Acta Polytech. 2018, 58, 308. [Google Scholar] [CrossRef]
- Fouladi, M.H.; Namasivayam, S.; Raj, F.R.; Mamtaz, H.; Hon, H.J.; Sivanesan, S. Enhancement of vibration comfort for FSAE race car driver. J. Adv. Res. Dyn. Control Syst. 2018, 13, 1320–1327. [Google Scholar]
- Abrams, R. Formula SAE Race Car Analysis: Simulation & Testing of the Engine as a Structural Member; The University of Western Ontario: London, ON, Canada, 2008. [Google Scholar]
- Gray, W. Racing Vehicle Environment—Forces and Vibrations. Available online: http://atlasf1.autosport.com/99/dec08/gray.html (accessed on 25 January 2022).
- Koutras, C.; Buecking, B.; Jaeger, M.; Ruchholtz, S.; Heep, H. Musculoskeletal injuries in auto racing: A retrospective study of 137 drivers. Phys. Sportsmed. 2014, 42, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Hood, W.B., Jr.; Murray, R.H.; Urschel, C.W.; Bowers, J.A.; Clark, J.G. Cardiopulmonary effects of whole-body vibration in man. J. Appl. Physiol. 1966, 21, 1725–1731. [Google Scholar] [CrossRef]
- Masmejean, E.H.; Chavane, H.; Chantegret, A.; Issermann, J.J.; Alnot, J.Y. The wrist of the Formula One driver. Br. J. Sports Med. 1999, 33, 270–273. [Google Scholar] [CrossRef]
- Pacurari, M.; Waugh, S.; Krajnak, K. Acute vibration induces peripheral nerve sensitization in a rat tail model: Possible role of oxidative stress and inflammation. Neuroscience 2019, 398, 263–272. [Google Scholar] [CrossRef]
- Krajnak, K. Frequency-dependent changes in mitochondrial number and generation of reactive oxygen species in a rat model of vibration-induced injury. J. Toxicol. Environ. Health Part A 2020, 83, 20–35. [Google Scholar] [CrossRef]
- Antoshina, L.I.; Pavlovskaia, N.A. Biochemical markers of vibration effects in workers, and their criteria evaluation. Med. Str. Promyshlennaya Ekol. 2009, 12, 22–27. [Google Scholar]
- Krajnak, K.; Miller, G.R.; Waugh, S.; Johnson, C.; Li, S.; Kashon, M.L. Characterization of frequency-dependent responses of the vascular system to repetitive vibration. J. Occup. Environ. Med. 2010, 52, 584–594. [Google Scholar] [CrossRef]
- Krajnak, K.; Waugh, S. Systemic effects of segmental vibration in an animal model of hand-arm vibration syndrome. J. Occup. Environ. Med. 2018, 60, 886–895. [Google Scholar] [CrossRef]
- Delliaux, S.; Brerro-Saby, C.; Steinberg, J.G.; Jammes, Y. Reactive oxygen species and inflammatory mediators enhance muscle spindles mechanosensitivity in rats. Pflügers Arch.-Eur. J. Physiol. 2009, 457, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Rosales-Corral, S.; Reiter, R.J.; Tan, D.X.; Ortiz, G.G.; Lopez-Armas, G. Functional aspects of redox control during neuroinflammation. Antioxid. Redox Signal. 2010, 13, 193–247. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, H.L.; Yang, C.M. Role of redox signaling in neuroinflammation and neurodegenerative diseases. Biomed. Res. Int. 2013, 2013, 484613. [Google Scholar] [CrossRef] [PubMed]
- Kurutas, E.B.; Gumusalan, Y.; Cetinkaya, A.; Dogan, E. Evaluation of method performance for oxidative stress biomarkers in urine and biological variations in urine of patients with type 2 diabetes mellitus and diabetic nephropathy. Biol. Proced. Online 2015, 17, 3. [Google Scholar] [CrossRef] [PubMed]
- Inanir, A.; Sogut, E.; Ayan, M.; Inanir, S. Evaluation of pain intensity and oxidative stress levels in patients with inflammatory and non-inflammatory back pain. Eur. J. Gen. Med. 2013, 10, 185–190. [Google Scholar] [CrossRef]
- Vallbo, A.B.; Hagbarth, K.E.; Torebjork, H.E.; Wallin, B.G. Somatosensory, proprioceptive, and sympathetic activity in human peripheral nerves. Physiol. Rev. 1979, 59, 919–957. [Google Scholar] [CrossRef]
- Bentley, S. Exercise-induced muscle cramp. Proposed mechanisms and management. Sports Med. 1996, 21, 409–420. [Google Scholar] [CrossRef]
- Szalma, J.L.; Hancock, P.A. Noise effects on human performance: A meta-analytic synthesis. Psychol. Bull. 2011, 137, 682–707. [Google Scholar] [CrossRef]
- Lindemann, J.; Brusis, T. Is there a risk of noise-induced hearing loss in automobile drivers and in automobile sport racing? Laryngol. Rhinol. Otol. 1985, 64, 476–480. [Google Scholar] [CrossRef]
- Liu, J.; Ghastine, L.; Um, P.; Rovit, E.; Wu, T. Environmental exposures and sleep outcomes: A review of evidence, potential mechanisms, and implications. Environ. Res. 2021, 196, 110406. [Google Scholar] [CrossRef]
- Jafari, M.J.; Sadeghian, M.; Khavanin, A.; Khodakarim, S.; Jafarpisheh, A.S. Effects of noise on mental performance and annoyance considering task difficulty level and tone components of noise. J. Environ. Health Sci. Eng. 2019, 17, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Wilking, M.; Ndiaye, M.; Mukhtar, H.; Ahmad, N. Circadian rhythm connections to oxidative stress: Implications for human health. Antioxid. Redox Signal. 2013, 19, 192–208. [Google Scholar] [CrossRef] [PubMed]
- Munzel, T.; Gori, T.; Babisch, W.; Basner, M. Cardiovascular effects of environmental noise exposure. Eur. Heart J. 2014, 35, 829–836. [Google Scholar] [CrossRef] [PubMed]
- Munzel, T.; Sorensen, M.; Schmidt, F.; Schmidt, E.; Steven, S.; Kroller-Schon, S.; Daiber, A. The adverse effects of environmental noise exposure on oxidative stress and cardiovascular risk. Antioxid. Redox Signal. 2018, 28, 873–908. [Google Scholar] [CrossRef] [PubMed]
- Neiemeier, R.; Dames, B. Recommendations for Occupational Safety and Health: Compendium of Policy Documents and Statements; National Institute for Occupational Safety and Health (NIOSH): Washington, DC, USA, 1992; pp. 1–217. [Google Scholar]
- Gwin, K.K.; Wallingford, K.M.; Morata, T.C.; Van Campen, L.E.; Dallaire, J.; Alvarez, F.J. Ototoxic occupational exposures for a stock car racing team: II. Chemical surveys. J. Occup. Environ. Hyg. 2005, 2, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Rose, A.S.; Ebert, C.S., Jr.; Prazma, J.; Pillsbury, H.C., 3rd. Noise exposure levels in stock car auto racing. Ear Nose Throat J. 2008, 87, 689–692. [Google Scholar] [CrossRef]
- Magara, K.; Jansz, J.; Bertolattia, D. Noise exposure assessment for racing car drivers. World Saf. J. 2016, XXV, 345. [Google Scholar]
- Kardous, F.; Morata, T.C. Occupational and recreational noise exposures at stock car racing circuits: An exploratory survey of three professional race tracks. Noise Control Eng. J. 2010, 58, 54–61. [Google Scholar] [CrossRef]
- Spoendlin, H. Anatomy of cochlear innervation. Am. J. Otolaryngol. 1985, 6, 453–467. [Google Scholar] [CrossRef]
- Gates, G.A.; Mills, J.H. Presbycusis. Lancet 2005, 366, 1111–1120. [Google Scholar] [CrossRef]
- Shi, L.; Guo, X.; Shen, P.; Liu, L.; Tao, S.; Li, X.; Song, Q.; Yu, Z.; Yin, S.; Wang, J. Noise-induced damage to ribbon synapses without permanent threshold shifts in neonatal mice. Neuroscience 2015, 304, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Kujawa, S.G.; Liberman, M.C. Adding insult to injury: Cochlear nerve degeneration after “temporary” noise-induced hearing loss. J. Neurosci. 2009, 29, 14077–14085. [Google Scholar] [CrossRef] [PubMed]
- Santi, P.A.; Duvall, A.J., 3rd. Stria vascularis pathology and recovery following noise exposure. Otolaryngology 1978, 86, ORL354–ORL361. [Google Scholar] [CrossRef] [PubMed]
- Kurabi, A.; Keithley, E.M.; Housley, G.D.; Ryan, A.F.; Wong, A.C. Cellular mechanisms of noise-induced hearing loss. Hear. Res. 2017, 349, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Kumar, U.A.; Ameenudin, S.; Sangamanatha, A.V. Temporal and speech processing skills in normal hearing individuals exposed to occupational noise. Noise Health 2012, 14, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Shore, S.E.; Roberts, L.E.; Langguth, B. Maladaptive plasticity in tinnitus-triggers, mechanisms and treatment. Nat. Rev. Neurol. 2016, 12, 150–160. [Google Scholar] [CrossRef]
- Henderson, D.; Bielefeld, E.C.; Harris, K.C.; Hu, B.H. The role of oxidative stress in noise-induced hearing loss. Ear Hear. 2006, 27, 1–19. [Google Scholar] [CrossRef]
- Wong, A.C.; Ryan, A.F. Mechanisms of sensorineural cell damage, death and survival in the cochlea. Front. Aging Neurosci. 2015, 7, 58. [Google Scholar] [CrossRef]
- Yamane, H.; Nakai, Y.; Takayama, M.; Iguchi, H.; Nakagawa, T.; Kojima, A. Appearance of free radicals in the guinea pig inner ear after noise-induced acoustic trauma. Eur. Arch. Otorhinolaryngol. 1995, 252, 504–508. [Google Scholar] [CrossRef]
- Wu, F.; Xiong, H.; Sha, S. Noise-induced loss of sensory hair cells is mediated by ROS/AMPKalpha pathway. Redox Biol. 2020, 29, 101406. [Google Scholar] [CrossRef]
- Kil, J.; Pierce, C.; Tran, H.; Gu, R.; Lynch, E.D. Ebselen treatment reduces noise induced hearing loss via the mimicry and induction of glutathione peroxidase. Hear. Res. 2007, 226, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, C.; Chen, G.D.; Hu, B.H.; Chi, L.H.; Li, M.; Zheng, G.; Bielefeld, E.C.; Jamesdaniel, S.; Coling, D.; Henderson, D. The effects of acoustic environment after traumatic noise exposure on hearing and outer hair cells. Hear. Res. 2009, 250, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Xiong, M.; Lai, H.; Yang, C.; Huang, W.; Wang, J.; Fu, X.; He, Q. Comparison of the protective effects of radix astragali, alpha-lipoic acid, and vitamin E on acute acoustic trauma. Clin. Med. Insights Ear Nose Throat 2012, 5, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Pourbakht, A.; Yamasoba, T. Ebselen attenuates cochlear damage caused by acoustic trauma. Hear. Res. 2003, 181, 100–108. [Google Scholar] [CrossRef]
- Ohinata, Y.; Yamasoba, T.; Schacht, J.; Miller, J.M. Glutathione limits noise-induced hearing loss. Hear. Res. 2000, 146, 28–34. [Google Scholar] [CrossRef]
- Kopke, R.D.; Weisskopf, P.A.; Boone, J.L.; Jackson, R.L.; Wester, D.C.; Hoffer, M.E.; Lambert, D.C.; Charon, C.C.; Ding, D.L.; McBride, D. Reduction of noise-induced hearing loss using L-NAC and salicylate in the chinchilla. Hear. Res. 2000, 149, 138–146. [Google Scholar] [CrossRef]
- Seidman, M.D.; Shivapuja, B.G.; Quirk, W.S. The protective effects of allopurinol and superoxide dismutase on noise-induced cochlear damage. Otolaryngol. Head Neck Surg. 1993, 109, 1052–1056. [Google Scholar] [CrossRef]
- Choi, C.H.; Chen, K.; Vasquez-Weldon, A.; Jackson, R.L.; Floyd, R.A.; Kopke, R.D. Effectiveness of 4-hydroxy phenyl N-tert-butylnitrone (4-OHPBN) alone and in combination with other antioxidant drugs in the treatment of acute acoustic trauma in chinchilla. Free Radic. Biol. Med. 2008, 44, 1772–1784. [Google Scholar] [CrossRef]
- Bottger, E.C.; Schacht, J. The mitochondrion: A perpetrator of acquired hearing loss. Hear. Res. 2013, 303, 12–19. [Google Scholar] [CrossRef]
- Vlajkovic, S.M.; Lin, S.C.; Wong, A.C.; Wackrow, B.; Thorne, P.R. Noise-induced changes in expression levels of NADPH oxidases in the cochlea. Hear. Res. 2013, 304, 145–152. [Google Scholar] [CrossRef]
- Mukherjea, D.; Jajoo, S.; Sheehan, K.; Kaur, T.; Sheth, S.; Bunch, J.; Perro, C.; Rybak, L.P.; Ramkumar, V. NOX3 NADPH oxidase couples transient receptor potential vanilloid 1 to signal transducer and activator of transcription 1-mediated inflammation and hearing loss. Antioxid. Redox Signal. 2011, 14, 999–1010. [Google Scholar] [CrossRef] [PubMed]
- Bielefeld, E.C. Reduction in impulse noise-induced permanent threshold shift with intracochlear application of an NADPH oxidase inhibitor. J. Am. Acad. Audiol. 2013, 24, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Jareño, A.; De La Serna, J.; Cercas, A.; Lobato, A.; Uyá, A. Heat stroke in motor car racing drivers. Br. J. Sports Med. 1987, 21, 48. [Google Scholar] [CrossRef][Green Version]
- Yanagida, R.; Takahashi, K.; Miura, M.; Nomura, M.; Ogawa, Y.; Aoki, K.; Iwasaki, K.I. Speed ratio but cabin temperature positively correlated with increased heart rates among professional drivers during car races. Environ. Health Prev. Med. 2016, 21, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Cheung, S.S.; McLellan, T.M.; Tenaglia, S. The thermophysiology of uncompensable heat stress. Physiological manipulations and individual characteristics. Sports Med. 2000, 29, 329–359. [Google Scholar] [CrossRef] [PubMed]
- Carlson, L.A.; Ferguson, D.P.; Kenefick, R.W. Physiological strain of stock car drivers during competitive racing. J. Therm. Biol. 2014, 44, 20–26. [Google Scholar] [CrossRef]
- Carlson, L.A.; Lawrence, M.A.; Kenefick, R.W. Hydration status and thermoregulatory responses in drivers during competitive racing. J. Strength Cond. Res. 2018, 32, 2061–2065. [Google Scholar] [CrossRef]
- Walker, S.M.; Dawson, B.; Ackland, T.R. Performance enhancement in rally car drivers via heat acclimation and race simulation. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2001, 128, 701–707. [Google Scholar] [CrossRef]
- Brearley, M.B.; Finn, J.P. Responses of motor-sport athletes to V8 supercar racing in hot conditions. Int. J. Sports Physiol. Perform. 2007, 2, 182–191. [Google Scholar] [CrossRef]
- Potkanowicz, E.S. A real-time case study in driver science: Physiological strain and related variables. Int. J. Sports Physiol. Perform. 2015, 10, 1058–1060. [Google Scholar] [CrossRef]
- Lieberman, H.R. Hydration and cognition: A critical review and recommendations for future research. J. Am. Coll. Nutr. 2007, 26, 555S–561S. [Google Scholar] [CrossRef] [PubMed]
- Carneiro Rodrigues, L.O.; de Castro Megalhães, F. Car racing: In the heat of competition. Rev. Bras. Med. Esporte 2004, 10, 216–219. [Google Scholar]
- McAnulty, S.R.; McAnulty, L.; Pascoe, D.D.; Gropper, S.S.; Keith, R.E.; Morrow, J.D.; Gladden, L.B. Hyperthermia increases exercise-induced oxidative stress. Int. J. Sports Med. 2005, 26, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.K.; Lin, C.W.; Chang, C.C.; Chen, P.F.; Wang, C.J.; Hsueh, Y.M.; Chiang, H.C. Heat acclimation decreased oxidative DNA damage resulting from exposure to high heat in an occupational setting. Eur. J. Appl. Physiol. 2012, 112, 4119–4126. [Google Scholar] [CrossRef] [PubMed]
- Mourtakos, S.; Philippou, A.; Papageorgiou, A.; Lembessis, P.; Zaharinova, S.; Hasanova, Y.; Koynova, R.; Bersimis, F.; Tenchov, B.; Geladas, N.; et al. The effect of prolonged intense physical exercise of special forces volunteers on their plasma protein denaturation profile examined by differential scanning calorimetry. J. Therm. Biol. 2021, 96, 102860. [Google Scholar] [CrossRef] [PubMed]
- Barros, B.; Oliveira, M.; Morais, S. Firefighters’ occupational exposure: Contribution from biomarkers of effect to assess health risks. Environ. Int. 2021, 156, 106704. [Google Scholar] [CrossRef]
- Byrne, J.; Ludington-Hoe, S.M.; Voss, J.G. Occupational Heat stress, thermal comfort, and cognitive performance in the OR: An integrative review. AORN J. 2020, 111, 536–545. [Google Scholar] [CrossRef]
- Gharibi, V.; Khanjani, N.; Heidari, H.; Ebrahimi, M.H.; Hosseinabadi, M.B. The effect of heat stress on hematological parameters and oxidative stress among bakery workers. Toxicol. Ind. Health 2020, 36, 1–10. [Google Scholar] [CrossRef]
- Banfi, G.; Malavazos, A.; Iorio, E.; Dolci, A.; Doneda, L.; Verna, R.; Corsi, M.M. Plasma oxidative stress biomarkers, nitric oxide and heat shock protein 70 in trained elite soccer players. Eur. J. Appl. Physiol. 2006, 96, 483–486. [Google Scholar] [CrossRef]
- Knez, W.L.; Periard, J.D. The impact of match-play tennis in a hot environment on indirect markers of oxidative stress and antioxidant status. Br. J. Sports Med. 2014, 48 (Suppl. 1), i59–i63. [Google Scholar] [CrossRef][Green Version]
- Sureda, A.; Mestre-Alfaro, A.; Banquells, M.; Riera, J.; Drobnic, F.; Camps, J.; Joven, J.; Tur, J.A.; Pons, A. Exercise in a hot environment influences plasma anti-inflammatory and antioxidant status in well-trained athletes. J. Therm. Biol. 2015, 47, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Pilch, W.; Szygula, Z.; Tyka, A.K.; Palka, T.; Tyka, A.; Cison, T.; Pilch, P.; Teleglow, A. Disturbances in pro-oxidant-antioxidant balance after passive body overheating and after exercise in elevated ambient temperatures in athletes and untrained men. PLoS ONE 2014, 9, e85320. [Google Scholar] [CrossRef] [PubMed]
- Laitano, O.; Kalsi, K.K.; Pook, M.; Oliveira, A.R.; Gonzalez-Alonso, J. Separate and combined effects of heat stress and exercise on circulatory markers of oxidative stress in euhydrated humans. Eur. J. Appl. Physiol. 2010, 110, 953–960. [Google Scholar] [CrossRef] [PubMed]
- Quindry, J.; Miller, L.; McGinnis, G.; Kliszczewiscz, B.; Slivka, D.; Dumke, C.; Cuddy, J.; Ruby, B. Environmental temperature and exercise-induced blood oxidative stress. Int. J. Sport Nutr. Exerc. Metab. 2013, 23, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Hillman, A.R.; Vince, R.V.; Taylor, L.; McNaughton, L.; Mitchell, N.; Siegler, J. Exercise-induced dehydration with and without environmental heat stress results in increased oxidative stress. Appl. Physiol. Nutr. Metab. 2011, 36, 698–706. [Google Scholar] [CrossRef]
- Laitano, O.; Kalsi, K.K.; Pearson, J.; Lotlikar, M.; Reischak-Oliveira, A.; Gonzalez-Alonso, J. Effects of graded exercise-induced dehydration and rehydration on circulatory markers of oxidative stress across the resting and exercising human leg. Eur. J. Appl. Physiol. 2012, 112, 1937–1944. [Google Scholar] [CrossRef]
- Georgescu, V.P.; de Souza Junior, T.P.; Behrens, C.; Barros, M.P.; Bueno, C.A.; Utter, A.C.; McAnulty, L.S.; McAnulty, S.R. Effect of exercise-induced dehydration on circulatory markers of oxidative damage and antioxidant capacity. Appl. Physiol. Nutr. Metab. 2017, 42, 694–699. [Google Scholar] [CrossRef]
- McNeely, B.D.; Meade, R.D.; Fujii, N.; Seely, A.J.E.; Sigal, R.J.; Kenny, G.P. Fluid replacement modulates oxidative stress- but not nitric oxide-mediated cutaneous vasodilation and sweating during prolonged exercise in the heat. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2017, 313, R730–R739. [Google Scholar] [CrossRef]
- Zhao, Q.L.; Fujiwara, Y.; Kondo, T. Mechanism of cell death induction by nitroxide and hyperthermia. Free Radic. Biol. Med. 2006, 40, 1131–1143. [Google Scholar] [CrossRef]
- Mujahid, A.; Sato, K.; Akiba, Y.; Toyomizu, M. Acute heat stress stimulates mitochondrial superoxide production in broiler skeletal muscle, possibly via downregulation of uncoupling protein content. Poult. Sci. 2006, 85, 1259–1265. [Google Scholar] [CrossRef]
- Moon, E.J.; Sonveaux, P.; Porporato, P.E.; Danhier, P.; Gallez, B.; Batinic-Haberle, I.; Nien, Y.C.; Schroeder, T.; Dewhirst, M.W. NADPH oxidase-mediated reactive oxygen species production activates hypoxia-inducible factor-1 (HIF-1) via the ERK pathway after hyperthermia treatment. Proc. Natl. Acad. Sci. USA 2010, 107, 20477–20482. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.Y.; Lin, M.T. Oxidative stress in rats with heatstroke-induced cerebral ischemia. Stroke 2002, 33, 790–794. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.B.; Russo, A. Thiols, thiol depletion, and thermosensitivity. Radiat. Res. 1983, 95, 471–485. [Google Scholar] [CrossRef] [PubMed]
- King, M.A.; Clanton, T.L.; Laitano, O. Hyperthermia, dehydration, and osmotic stress: Unconventional sources of exercise-induced reactive oxygen species. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 310, R105–R114. [Google Scholar] [CrossRef] [PubMed]
- Connes, P.; Simmonds, M.J.; Brun, J.F.; Baskurt, O.K. Exercise hemorheology: Classical data, recent findings and unresolved issues. Clin. Hemorheol. Microcirc. 2013, 53, 187–199. [Google Scholar] [CrossRef]
- Lehoux, S. Redox signalling in vascular responses to shear and stretch. Cardiovasc. Res. 2006, 71, 269–279. [Google Scholar] [CrossRef]
- Ajmani, R.S.; Fleg, J.L.; Demehin, A.A.; Wright, J.G.; O’Connor, F.; Heim, J.M.; Tarien, E.; Rifkind, J.M. Oxidative stress and hemorheological changes induced by acute treadmill exercise. Clin. Hemorheol. Microcirc. 2003, 28, 29–40. [Google Scholar]
- Rifkind, J.M.; Ajmani, R.S.; Heim, J. Impaired hemorheology in the aged associated with oxidative stress. Adv. Exp. Med. Biol. 1997, 428, 7–13. [Google Scholar] [CrossRef]
- Ganesan, S.; Summers, C.M.; Pearce, S.C.; Gabler, N.K.; Valentine, R.J.; Baumgard, L.H.; Rhoads, R.P.; Selsby, J.T. Short-term heat stress causes altered intracellular signaling in oxidative skeletal muscle. J. Anim. Sci. 2017, 95, 2438–2451. [Google Scholar] [CrossRef]
- Mujahid, A.; Pumford, N.; Bottje, W.; Nakagawa, K.; Miyazaqa, T.; Akiba, Y.; Toyomizu, M. Mitochondrial oxidative damage in chicken skeletal muscle induced by acute heat stress. J. Poult. Sci. 2007, 439–445. [Google Scholar] [CrossRef]
- Montilla, S.I.; Johnson, T.P.; Pearce, S.C.; Gardan-Salmon, D.; Gabler, N.K.; Ross, J.W.; Rhoads, R.P.; Baumgard, L.H.; Lonergan, S.M.; Selsby, J.T. Heat stress causes oxidative stress but not inflammatory signaling in porcine skeletal muscle. Temperature 2014, 1, 42–50. [Google Scholar] [CrossRef]
- Taggart, P.; Gibbons, D. Motor-car driving and the heart rate. Br. Med. J. 1967, 1, 411–412. [Google Scholar] [CrossRef]
- Taggart, P.; Gibbons, D.; Somerville, W. Some effects of motor-car driving on the normal and abnormal heart. Br. Med. J. 1969, 4, 130–134. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Matsumura, K.; Yamakoshi, T.; Yamakoshi, Y.; Rolfe, P. The effect of competition on heart rate during kart driving: A field study. BMC Res. Notes 2011, 4, 342. [Google Scholar] [CrossRef] [PubMed]
- Mallows, R.J.; Newman, D.G. Cardiovascular data acquisition in a dynamic motion environment. Aviat. Space Environ. Med. 2008, 79, 416–419. [Google Scholar] [CrossRef] [PubMed]
- Watkins, E.S. The physiology and pathology of formula one Grand Prix motor racing. Clin. Neurosurg. 2006, 53, 145–152. [Google Scholar]
- Hawkins, S. Extreme G-Forces Prompt Race Cancellation. Available online: https://abcnews.go.com/US/story?id=93412&page=1 (accessed on 25 January 2022).
- Moran, D.S.; Shitzer, A.; Pandolf, K.B. A physiological strain index to evaluate heat stress. Am. J. Physiol. 1998, 275, R129–R134. [Google Scholar] [CrossRef]
- Turner, A.P.; Richards, H. Physiological and selective attention demands during an international rally motor sport event. Biomed. Res. Int. 2015, 2015, 638659. [Google Scholar] [CrossRef]
- Martinelli, M. Kasey Kahne Opens up about Severe Dehydration, Heat Exhaustion. Available online: https://ftw.usatoday.com/2018/09/kasey-kahne-nascar-health-dehydration-heat-exhaustion-indy (accessed on 25 January 2022).
- Raymond, C. Dehydrated and Hurt: Sergio Perez Is Proof That Formula 1 Drivers Are Super Athletes. Available online: https://www.news24.com/wheels/formulaone/dehydrated-and-hurt-sergio-perez-is-proof-that-formula-1-drivers-are-super-athletes-20211027 (accessed on 25 January 2022).
- Castro-Sepulveda, M.; Cerda-Kohler, H.; Perez-Luco, C.; Monsalves, M.; Andrade, D.C.; Zbinden-Foncea, H.; Baez-San Martin, E.; Ramirez-Campillo, R. Hydration status after exercise affect resting metabolic rate and heart rate variability. Nutr. Hosp. 2014, 31, 1273–1277. [Google Scholar] [CrossRef]
- Heinzel, F.R.; Luo, Y.; Dodoni, G.; Boengler, K.; Petrat, F.; Di Lisa, F.; de Groot, H.; Schulz, R.; Heusch, G. Formation of reactive oxygen species at increased contraction frequency in rat cardiomyocytes. Cardiovasc. Res. 2006, 71, 374–382. [Google Scholar] [CrossRef]
- Saitoh, S.; Zhang, C.; Tune, J.D.; Potter, B.; Kiyooka, T.; Rogers, P.A.; Knudson, J.D.; Dick, G.M.; Swafford, A.; Chilian, W.M. Hydrogen peroxide: A feed-forward dilator that couples myocardial metabolism to coronary blood flow. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2614–2621. [Google Scholar] [CrossRef] [PubMed]
- Maack, C.; Cortassa, S.; Aon, M.A.; Ganesan, A.N.; Liu, T.; O’Rourke, B. Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ. Res. 2006, 99, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Kubin, A.M.; Skoumal, R.; Tavi, P.; Konyi, A.; Perjes, A.; Leskinen, H.; Ruskoaho, H.; Szokodi, I. Role of reactive oxygen species in the regulation of cardiac contractility. J. Mol. Cell. Cardiol. 2011, 50, 884–893. [Google Scholar] [CrossRef]
- Mak, S.; Newton, G.E. Vitamin C augments the inotropic response to dobutamine in humans with normal left ventricular function. Circulation 2001, 103, 826–830. [Google Scholar] [CrossRef] [PubMed]
- Filho, E.; Di Fronso, S.; Mazzoni, C.; Robazza, C.; Bortoli, L.; Bertollo, M. My heart is racing! Psychophysiological dynamics of skilled racecar drivers. J. Sports Sci. 2015, 33, 945–959. [Google Scholar] [CrossRef]
- Andersson, D.C.; Fauconnier, J.; Yamada, T.; Lacampagne, A.; Zhang, S.J.; Katz, A.; Westerblad, H. Mitochondrial production of reactive oxygen species contributes to the beta-adrenergic stimulation of mouse cardiomycytes. J. Physiol. 2011, 589, 1791–1801. [Google Scholar] [CrossRef] [PubMed]
- Perjes, A.; Kubin, A.M.; Konyi, A.; Szabados, S.; Cziraki, A.; Skoumal, R.; Ruskoaho, H.; Szokodi, I. Physiological regulation of cardiac contractility by endogenous reactive oxygen species. Acta Physiol. 2012, 205, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Ahlborg, G.; Weitzberg, E.; Lundberg, J. Metabolic and vascular effects of circulating endothelin-1 during moderately heavy prolonged exercise. J. Appl. Physiol. 1995, 78, 2294–2300. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, H.; Oribe, E.; Oliver, J.A. Plasma endothelin during upright tilt: Relevance for orthostatic hypotension? Lancet 1991, 338, 1542–1545. [Google Scholar] [CrossRef]
- Sovershaev, M.A.; Egorina, E.M.; Andreasen, T.V.; Jonassen, A.K.; Ytrehus, K. Preconditioning by 17beta-estradiol in isolated rat heart depends on PI3-K/PKB pathway, PKC, and ROS. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H1554–H1562. [Google Scholar] [CrossRef]
- Barouch, L.A.; Harrison, R.W.; Skaf, M.W.; Rosas, G.O.; Cappola, T.P.; Kobeissi, Z.A.; Hobai, I.A.; Lemmon, C.A.; Burnett, A.L.; O’Rourke, B.; et al. Nitric oxide regulates the heart by spatial confinement of nitric oxide synthase isoforms. Nature 2002, 416, 337–339. [Google Scholar] [CrossRef] [PubMed]
- Frank, K.; Kranias, E.G. Phospholamban and cardiac contractility. Ann. Med. 2000, 32, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Kohr, M.J.; Traynham, C.J.; Wheeler, D.G.; Janssen, P.M.; Ziolo, M.T. Neuronal nitric oxide synthase signaling within cardiac myocytes targets phospholamban. Am. J. Physiol.-Cell Physiol. 2008, 294, C1566–C1575. [Google Scholar] [CrossRef] [PubMed]
- Champion, H.C.; Georgakopoulos, D.; Takimoto, E.; Isoda, T.; Wang, Y.; Kass, D.A. Modulation of in vivo cardiac function by myocyte-specific nitric oxide synthase-3. Circ. Res. 2004, 94, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Kohr, M.J.; Wheeler, D.G.; Ziolo, M.T. Endothelial nitric oxide synthase decreases beta-adrenergic responsiveness via inhibition of the L-type Ca2+ current. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H1473–H1480. [Google Scholar] [CrossRef] [PubMed]
- Land, M.F.; Tatler, B.W. Steering with the head. The visual strategy of a racing driver. Curr. Biol. 2001, 11, 1215–1220. [Google Scholar] [CrossRef]
- van Leeuwen, P.M.; de Groot, S.; Happee, R.; de Winter, J.C.F. Differences between racing and non-racing drivers: A simulator study using eye-tracking. PLoS ONE 2017, 12, e0186871. [Google Scholar] [CrossRef] [PubMed]
- Land, M.F.; Lee, D.N. Where We Look When We Steer. Nature 1994, 369, 742–744. [Google Scholar] [CrossRef]
- Bernardi, G.; Ricciardi, E.; Sani, L.; Gaglianese, A.; Papasogli, A.; Ceccarelli, R.; Franzoni, F.; Galetta, F.; Santoro, G.; Goebel, R.; et al. How skill expertise shapes the brain functional architecture: An fMRI study of visuo-spatial and motor processing in professional racing-car and naive drivers. PLoS ONE 2013, 8, e77764. [Google Scholar] [CrossRef]
- Bernardi, G.; Cecchetti, L.; Handjaras, G.; Sani, L.; Gaglianese, A.; Ceccarelli, R.; Franzoni, F.; Galetta, F.; Santoro, G.; Goebel, R.; et al. It’s not all in your car: Functional and structural correlates of exceptional driving skills in professional racers. Front. Hum. Neurosci. 2014, 8, 888. [Google Scholar] [CrossRef]
- Lappi, O. The racer’s brain—How domain expertise is reflected in the neural substrates of driving. Front. Hum. Neurosci. 2015, 9, 635. [Google Scholar] [CrossRef]
- Tyrance, S. The Psychology of Auto Racing. Available online: https://nationalpsychologist.com/2012/03/the-psychology-of-auto-racing/101641.html (accessed on 25 January 2022).
- Baroody, N.B.; Thomason, J.M.; O’Bryan, E.C., Jr. The heart of the 500 mile race. Am. Fam. Physician 1973, 8, 184–189. [Google Scholar] [PubMed]
- Basner, M.; McGuire, S. WHO environmental noise guidelines for the European region: A systematic review on environmental noise and effects on sleep. Int. J. Environ. Res. Public Health 2018, 15, 519. [Google Scholar] [CrossRef] [PubMed]
- Nollet, M.; Wisden, W.; Franks, N.P. Sleep deprivation and stress: A reciprocal relationship. Interface Focus 2020, 10, 20190092. [Google Scholar] [CrossRef] [PubMed]
- Koch, C.E.; Leinweber, B.; Drengberg, B.C.; Blaum, C.; Oster, H. Interaction between circadian rhythms and stress. Neurobiol. Stress 2017, 6, 57–67. [Google Scholar] [CrossRef]
- Moller, P.; Wallin, H.; Knudsen, L.E. Oxidative stress associated with exercise, psychological stress and life-style factors. Chem Biol Interact 1996, 102, 17–36. [Google Scholar] [CrossRef]
- Aschbacher, K.; O’Donovan, A.; Wolkowitz, O.M.; Dhabhar, F.S.; Su, Y.; Epel, E. Good stress, bad stress and oxidative stress: Insights from anticipatory cortisol reactivity. NPP 2013, 38, 1698–1708. [Google Scholar] [CrossRef]
- Irie, M.; Asami, S.; Nagata, S.; Miyata, M.; Kasai, H. Relationships between perceived workload, stress and oxidative DNA damage. Int. Arch. Occup. Environ. Health 2001, 74, 153–157. [Google Scholar] [CrossRef]
- McAllister, M.J.; Basham, S.A.; Waldman, H.S.; Smith, J.W.; Mettler, J.A.; Butawan, M.B.; Bloomer, R.J. Effects of psychological stress during exercise on markers of oxidative stress in young healthy, trained men. Physiol. Behav. 2019, 198, 90–95. [Google Scholar] [CrossRef]
- Teixeira, K.R.C.; Dos Santos, C.P.; de Medeiros, L.A.; Mendes, J.A.; Cunha, T.M.; De Angelis, K.; Penha-Silva, N.; de Oliveira, E.P.; Crispim, C.A. Night workers have lower levels of antioxidant defenses and higher levels of oxidative stress damage when compared to day workers. Sci. Rep. 2019, 9, 4455. [Google Scholar] [CrossRef]
- Kim, E.; Zhao, Z.; Rzasa, J.R.; Glassman, M.; Bentley, W.E.; Chen, S.; Kelly, D.L.; Payne, G.F. Association of acute psychosocial stress with oxidative stress: Evidence from serum analysis. Redox Biol. 2021, 47, 102138. [Google Scholar] [CrossRef]
- Said, M.A.; El-Gohary, O.A. Effect of noise stress on cardiovascular system in adult male albino rat: Implication of stress hormones, endothelial dysfunction and oxidative stress. Gen. Physiol. Biophys. 2016, 35, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Solanki, N.; Salvi, A.; Patki, G.; Salim, S. Modulating oxidative stress relieves stress-induced behavioral and cognitive impairments in rats. Int. J. Neuropsychopharmacol. 2017, 20, 550–561. [Google Scholar] [CrossRef] [PubMed]
- Suer, C.; Dolu, N.; Artis, A.S.; Sahin, L.; Yilmaz, A.; Cetin, A. The effects of long-term sleep deprivation on the long-term potentiation in the dentate gyrus and brain oxidation status in rats. Neurosci. Res. 2011, 70, 71–77. [Google Scholar] [CrossRef]
- Lv, H.; Zhu, C.; Wu, R.; Ni, H.; Lian, J.; Xu, Y.; Xia, Y.; Shi, G.; Li, Z.; Caldwell, R.B.; et al. Chronic mild stress induced anxiety-like behaviors can be attenuated by inhibition of NOX2-derived oxidative stress. J. Psychiatr. Res. 2019, 114, 55–66. [Google Scholar] [CrossRef]
- Chan, J.Y.H.; Chan, S.H.H. Differential impacts of brain stem oxidative stress and nitrosative stress on sympathetic vasomotor tone. Pharmacol. Ther. 2019, 201, 120–136. [Google Scholar] [CrossRef]
- Acevedo, E.O.; Webb, H.E.; Weldy, M.L.; Fabianke, E.C.; Orndorff, G.R.; Starks, M.A. Cardiorespiratory responses of Hi Fit and Low Fit subjects to mental challenge during exercise. Int. J. Sports Med. 2006, 27, 1013–1022. [Google Scholar] [CrossRef]
- Aoyagi, Y.; McLellan, T.M.; Shephard, R.J. Effects of endurance training and heat acclimation on psychological strain in exercising men wearing protective clothing. Ergonomics 1998, 41, 328–357. [Google Scholar] [CrossRef]
- Huang, C.J.; Webb, H.E.; Garten, R.S.; Kamimori, G.H.; Evans, R.K.; Acevedo, E.O. Stress hormones and immunological responses to a dual challenge in professional firefighters. Int. J. Psychophysiol. 2010, 75, 312–318. [Google Scholar] [CrossRef]
- Pacak, K.; Palkovits, M. Stressor specificity of central neuroendocrine responses: Implications for stress-related disorders. Endocr. Rev. 2001, 22, 502–548. [Google Scholar] [CrossRef]
- Radakovic, M.; Borozan, S.; Djelic, N.; Ivanovic, S.; Miladinovic, D.C.; Ristanic, M.; Spremo-Potparevic, B.; Stanimirovic, Z. Nitroso-oxidative stress, acute phase response, and cytogenetic damage in Wistar rats treated with adrenaline. Oxidative Med. Cell. Longev. 2018, 2018, 1805354. [Google Scholar] [CrossRef] [PubMed]
- Majer, A.D.; Fasanello, V.J.; Tindle, K.; Frenz, B.J.; Ziur, A.D.; Fischer, C.P.; Fletcher, K.L.; Seecof, O.M.; Gronsky, S.; Vassallo, B.G.; et al. Is there an oxidative cost of acute stress? Characterization, implication of glucocorticoids and modulation by prior stress experience. Proc. Biol. Sci. 2019, 286, 20191698. [Google Scholar] [CrossRef] [PubMed]
- De, R.; Mazumder, S.; Sarkar, S.; Debsharma, S.; Siddiqui, A.A.; Saha, S.J.; Banerjee, C.; Nag, S.; Saha, D.; Bandyopadhyay, U. Acute mental stress induces mitochondrial bioenergetic crisis and hyper-fission along with aberrant mitophagy in the gut mucosa in rodent model of stress-related mucosal disease. Free Radic. Biol. Med. 2017, 113, 424–438. [Google Scholar] [CrossRef] [PubMed]
- Picard, M.; McManus, M.J.; Gray, J.D.; Nasca, C.; Moffat, C.; Kopinski, P.K.; Seifert, E.L.; McEwen, B.S.; Wallace, D.C. Mitochondrial functions modulate neuroendocrine, metabolic, inflammatory, and transcriptional responses to acute psychological stress. Proc. Natl. Acad. Sci. USA 2015, 112, E6614–E6623. [Google Scholar] [CrossRef] [PubMed]
- Ramzan, R.; Vogt, S.; Kadenbach, B. Stress-mediated generation of deleterious ROS in healthy individuals-role of cytochrome c oxidase. J. Mol. Med. 2020, 98, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Scheeren, T.W.; Arndt, J.O. Different response of oxygen consumption and cardiac output to various endogenous and synthetic catecholamines in awake dogs. Crit. Care Med. 2000, 28, 3861–3868. [Google Scholar] [CrossRef]
- Du, J.; Wang, Y.; Hunter, R.; Wei, Y.; Blumenthal, R.; Falke, C.; Khairova, R.; Zhou, R.; Yuan, P.; Machado-Vieira, R.; et al. Dynamic regulation of mitochondrial function by glucocorticoids. Proc. Natl. Acad. Sci. USA 2009, 106, 3543–3548. [Google Scholar] [CrossRef]
- Zhou, Q.G.; Zhu, L.J.; Chen, C.; Wu, H.Y.; Luo, C.X.; Chang, L.; Zhu, D.Y. Hippocampal neuronal nitric oxide synthase mediates the stress-related depressive behaviors of glucocorticoids by downregulating glucocorticoid receptor. J. Neurosci. 2011, 31, 7579–7590. [Google Scholar] [CrossRef]
- Okamoto, K.; Tanaka, H.; Ogawa, H.; Makino, Y.; Eguchi, H.; Hayashi, S.; Yoshikawa, N.; Poellinger, L.; Umesono, K.; Makino, I. Redox-dependent regulation of nuclear import of the glucocorticoid receptor. J. Biol. Chem. 1999, 274, 10363–10371. [Google Scholar] [CrossRef]



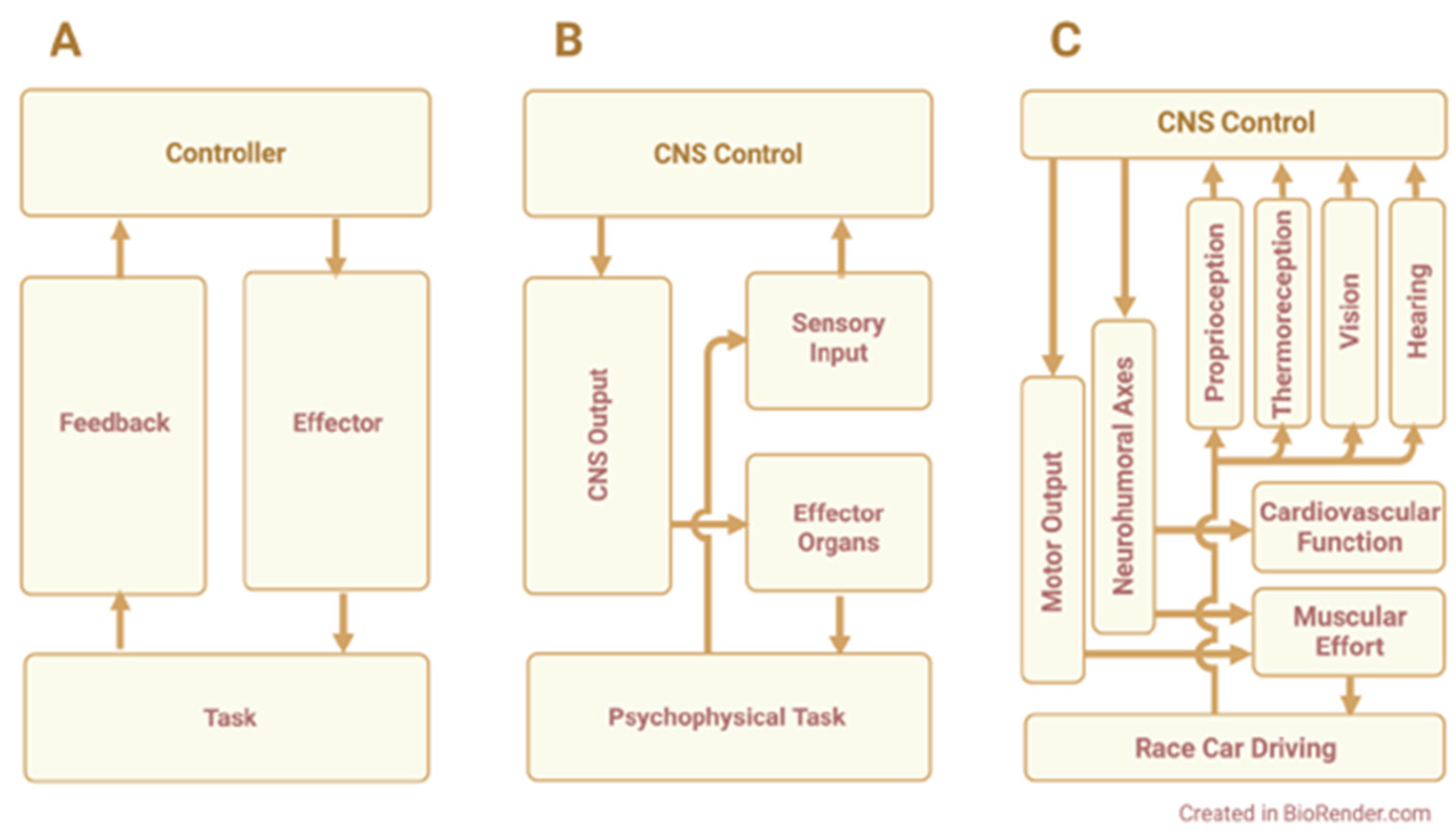{kind=link}
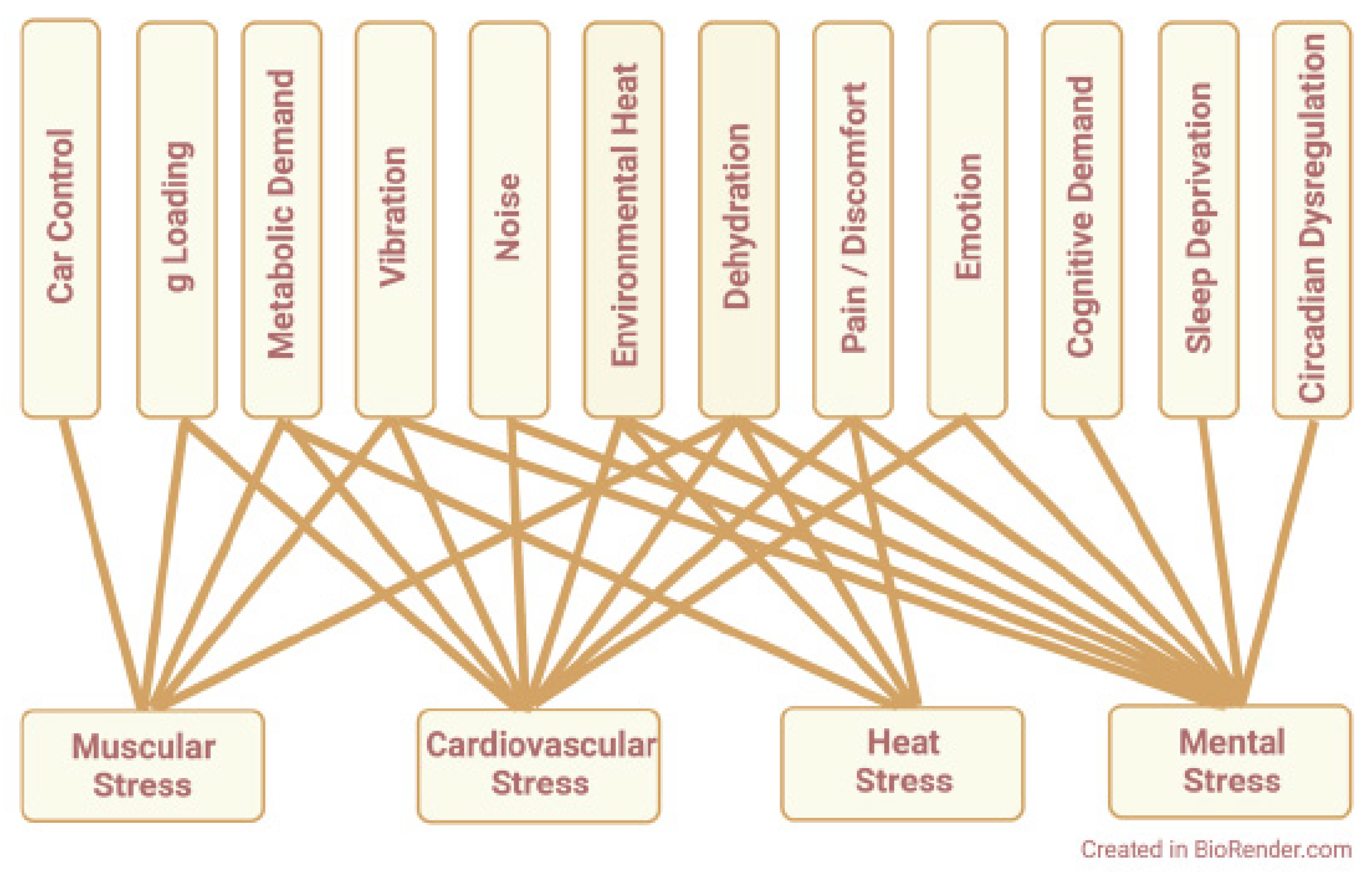{kind=link}
| Physiologic Stimulus | Tissue of Origin | Oxidant Cascade | Intracellular Source(s) | Physiologic Outcome | |
|---|---|---|---|---|---|
| Pathways for Redox Signaling | physical work, g loading, metabolic demand | skeletal muscle myofibers | ROS RNS | mitochondria, NOX, nNOS | muscle weakness, fatigue |
| vibration | skin, blood vessels, sensory receptors | ROS | mitochondria | nerve sensitization, muscle cramps, limb and back pain | |
| noise | cochlea | ROS | mitochondria, NOX3 | tinnitus, hearing loss, sleep disruption | |
| heat, dehydration | heart, liver, aerobic skeletal muscle | ROS RNS | mitochondria, NOX1, eNOS | antioxidant depletion, cellular damage, hemolysis | |
| cardiovascular demand | heart, vascular endothelium | ROS RNS | mitochondria, NOX, nNOS, eNOS | altered contractilityand vascular tone | |
| cognitive load, emotion, discomfort, sleeploss, circadian dysregulation | brain, vasculature, stress hormone-sensitive tissues | ROS | mitochondria | systemic oxidative stress, lucocorticoid desensitization |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reid, M.B. Redox Implications of Extreme Task Performance: The Case in Driver Athletes. Cells 2022, 11, 899. https://doi.org/10.3390/cells11050899
Reid MB. Redox Implications of Extreme Task Performance: The Case in Driver Athletes. Cells. 2022; 11(5):899. https://doi.org/10.3390/cells11050899
Chicago/Turabian StyleReid, Michael B. 2022. "Redox Implications of Extreme Task Performance: The Case in Driver Athletes" Cells 11, no. 5: 899. https://doi.org/10.3390/cells11050899
APA StyleReid, M. B. (2022). Redox Implications of Extreme Task Performance: The Case in Driver Athletes. Cells, 11(5), 899. https://doi.org/10.3390/cells11050899