1. Introduction
Connexins (Cx) are a large family of transmembrane proteins that are primarily localized in the plasma membrane, but also in intracellular organelles, such as mitochondria [
1,
2]. In mice, 20 different isotypes of Cx have been identified [
3]; they are widely expressed in all organs and tissues starting from early embryonic development [
4]. All Cx share the same structural features consisting of an amino- and carboxy-terminus located in the cytosol, one intracellular loop and four transmembrane domains. Six Cx form a connexon, and two adjacent connexons in the plasma membrane of neighboring cells can form functional intercellular channels, the gap junctions [
5]. Numerous gap junctions form clusters, called plaques [
6]. Cx biology is complex, as different Cx isotypes can assemble and form heteromeric connexons and/or even heterotypic gap junctions [
7]. More recent work proposes the existence of hemichannels, which are thought to open under pathophysiological conditions [
8].
Gap junctions are known to have pleiotropic functions, such as the exchange of nutrients, signaling components, and ions below a MW of 1.5 kDa. Therefore, these proteins mediate electrical and metabolic cell-to-cell coupling. This explains their vital role and the early embryonic lethality upon knocking out specific Cx genes, e.g., Cx45 [
9]. In addition, germ line and even somatic mutations of Cx encoding genes can result in various diseases. In fact, mutations in 10 different human Cx have been linked to 28 distinct genetic disorders, e.g., Cx26-related deafness [
10], Cx46-related cataracts, and Cx32-related X-linked Charcot–Marie–Tooth neuropathy [
11].
One of the most widely distributed and studied members of the Cx family is Cx43, which is known to be strongly expressed in the heart [
12]. The heart forms an electrical syncytium, and the electrical conduction is critically dependent on the expression and function of Cx43 in cardiomyocytes of the atrium and ventricle, whereas Cx40 is preferentially expressed in atrial cardiomyocytes and Cx45 in cells of the sinus and AV node [
13]. In the healthy human heart, Cx43 gap junctions are primarily located at intercalated discs connecting two neighboring cardiomyocytes providing orderly propagation of the electrical conduction throughout the working myocardium. This critical role of gap junctions for heart function is exemplified by mutations of Cx, which are strongly expressed in the heart, causing diseases such as Cx40-related atrial fibrillation [
14] and Cx43-related oculodentodigital dysplasia [
15]. Altered expression, distribution, and modulation of Cx43 channels and/or gap junctions are thought to occur in pathophysiological situations and to be potentially involved in the generation of life-threatening ventricular arrhythmias. In addition, myocardial ischemia and infarction have been reported to result in an altered distribution pattern of Cx43, e.g., atypical cellular localization in the lateral cardiomyocyte membrane, named lateralization [
16,
17]; the pathophysiological significance is still under debate [
18,
19,
20]. Furthermore, hypoxia and high cytosolic Ca
2+ have been reported to change cardiac Cx43 gap junction gating and, consequently, electrical conduction properties [
21].
Given the lack of highly specific pharmacological agonists and blockers of Cx, small Cx43 mimetic peptides with either blocking (e.g., Gap26, Gap27) or agonist functions (Gap134, αCT1) have been developed and tested for the targeting of heart disease [
22]. Besides specificity and efficacy issues, the in vivo delivery of these compounds needs to be further developed [
23,
24,
25]. Most of the actual mechanistic understanding of Cx43 function for heart development and disease has been obtained from knockout (KO) mouse models [
21]. These have revealed that null mutations of the Cx43 gene result in morphogenic malformations of the outflow tract of the heart and perinatal death because of asphyxia [
26]. This phenotype was primarily caused by Cx43-related defects of neural crest cells [
27]. Cell type-specific and inducible deletion of Cx43 in cardiomyocytes resulted in severe ventricular arrhythmias, underscoring its critical role in propagating electrical conduction in the heart. These studies also revealed that only 5–10% of Cx43 protein is required to maintain its normal function [
28]. However, many aspects of the biology and function of Cx43 in cardiomyocytes and in the heart are still poorly understood, in particular Cx43’s contribution to cardiomyocyte development, its effects in disease and malfunctioning, the relevance of its lateralization, and the formation and functional significance of hemichannels. To address these aspects, we have taken a different approach from KOs by generating an inducible Cx43 overexpression model in pluripotent mouse embryonic stem (ES) cells. This strategy was chosen, since earlier experiments in mice illustrated that ubiquitous overexpression of Cx43 resulted in embryonic and postnatal morbidity and reduced postnatal viability [
29]. We reasoned that Cx43 overexpression could also have (adverse) effects on the propagation, development, and differentiation of native cells and/or ES cell-derived cell populations in vitro. Therefore, we considered it a clear advantage to be able to induce Cx43 overexpression at later stages of the in vitro differentiation of (ES) cells.
2. Materials and Methods
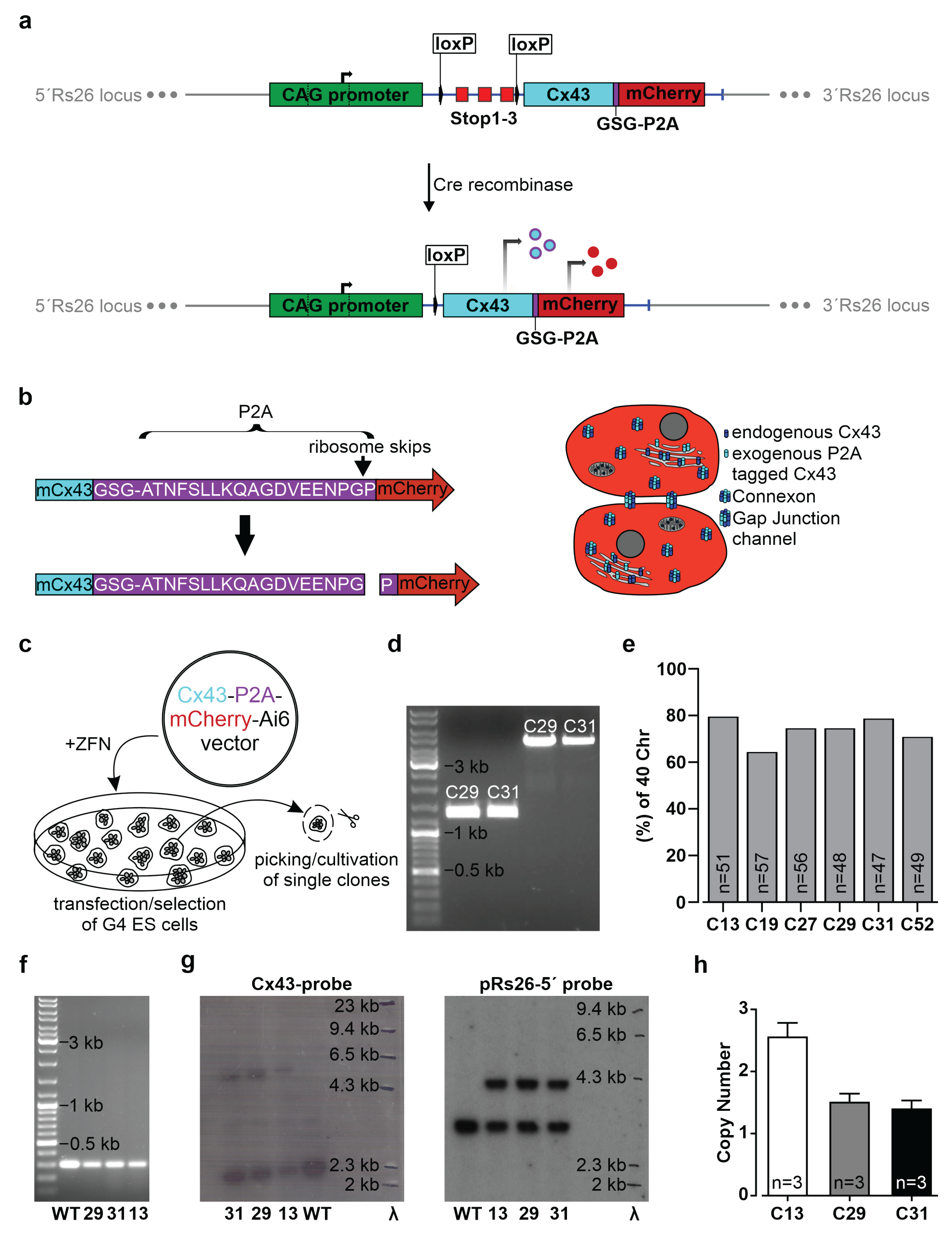
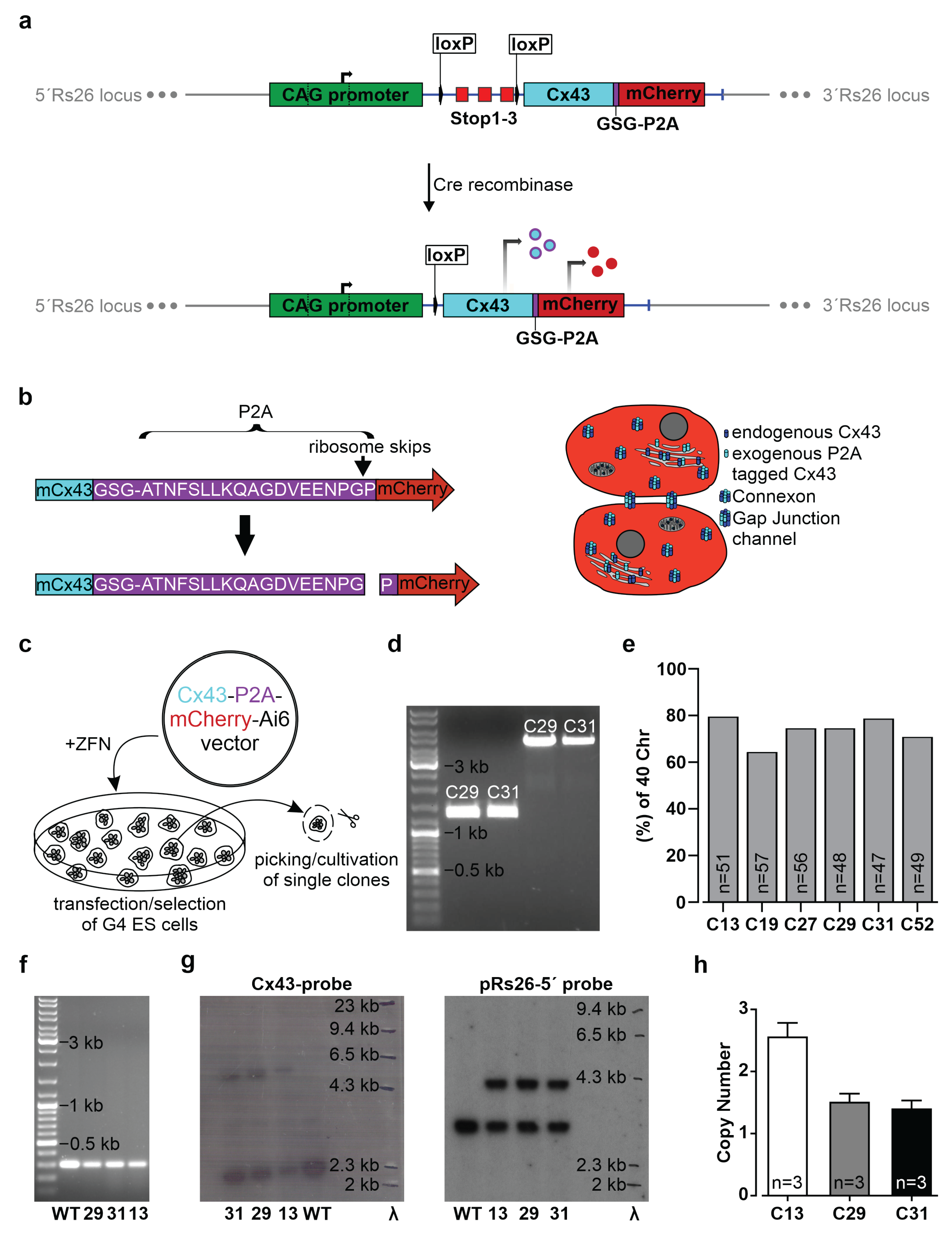
2.1. Generation of the CAG-floxSTOP-Cx43-P2A-mCherry-Ai6 Construct
The sequence of the mouse Cx43 cDNA, followed in frame by the DNA sequence of the GSG-P2A peptide and the mCherry cDNA, was synthesized together with flanking FseI sites by GeneArt Gene Synthesis (Thermo Fisher Scientific, Waltham, MA, USA) and subcloned into the pMK-RQ standard vector from Thermo Fisher. The customized 1968 bp DNA fragment was cut out by digestion with FseI, isolated and ligated downstream of the CAG promoter and a floxed stop cassette into the FseI sites of the Ai6 vector (Addgene, Watertown, MA, USA, #22798) [
30], thereby exchanging the zsGreen reporter. The Ai6 vector is a Rosa26 (Rs26) targeting vector that allows homologous integration of expression and NeoR/KanR cassettes into the mouse Rs26 locus by co-transfection with specific zinc finger nucleases. The Ai6 vector contains the post-transcriptional regulatory element of woodchuck hepatitis virus (WPRE) to increase transgene expression [
31]. All DNAs were prepared using the EndoFree Plasmid Maxi Kit from Qiagen (Venlo, The Netherlands). To verify correct cloning of the CAG-floxSTOP-Cx43-P2A-mCherry-Ai6 construct, restriction analyses and sequencing (Eurofins Genomics, Ebersberg, Germany) were performed. For molecular cloning procedures and sequence analyses, SnapGene Viewer 5.3.2 (from Insightful Science, available at snapgene.com, San Diego, CA, USA) and Clone Manager Professional 9 (Sci Ed Software LLC, Denver, CO, USA) were used.
2.2. Cell Culture
Undifferentiated murine G4 hybrid embryonic stem (ES) cells [
32] were cultivated in KnockOut Dulbecco’s modified Eagle’s medium (DMEM high glucose, Gibco, Thermo Fisher Scientific, supplemented with 15%
v/
v fetal calf serum (FCS, PAN Biotech, Aidenbach, Germany), 0.1 mM nonessential amino acids (NEAA, Gibco, Thermo Fisher Scientific), 2.4 mM L-glutamine (Thermo Scientific), 100 U/mL/100 µg/mL penicillin/streptomycin (P/S), 500 U/mL leukemia inhibitory factor (LIF), and 0.1 mM β-mercaptoethanol (β-ME, Gibco, Thermo Fisher Scientific)) in tissue culture flasks coated with irradiated neomycin-resistant mouse embryonic fibroblasts (PMEF-NL, Merck, Darmstadt, Germany). ES cells were differentiated using embryoid body (EB) formation via hanging drop method [
33]. For differentiation, cells were harvested and resuspended in Iscove’s MEM high-glucose medium (IMDM, Thermo Fisher) supplemented with 20%
v/
v FCS, 100 U/mL/100 µg/mL P/S, 0.1 mM NEAA, and 0.1 mM β-ME in the absence of LIF. Irradiated fibroblasts were reduced by differential sedimentation, and the ES cell-enriched suspension was dripped (400 cells per 20 µL drop) on the inner side of the lid of a cell culture dish. After two days of incubation, the hanging drops were rinsed off with 20% IMDM and further differentiated in suspension culture on a shaking plate (horizontal shaker, LAUDA-GFL, Burgwedel, Germany).
HPV18-positive HeLa (RRID: CVCL_0030) human cervical carcinoma cells were maintained in DMEM (pH 7.2), plus additives (20% v/v FCS, 120 U/mL/120 µg/mL P/S, 0.1 mM NEAA, 2 mM L-glutamine, 1.2 mM sodium pyruvate (Gibco, Thermo Fisher Scientific), and 0.12 mM β-ME).
NIH/3T3 mouse fibroblasts (3T3, ATCC, Manassas, VA, USA, #CRL1658) were cultivated in DMEM (plus 10% v/v FCS, 100 U/mL/100 µg/mL P/S, 0.1 mM NEAA, and 0.1 mM β-ME).
All cells were cultivated at 37 °C and 5% CO2.
2.3. CAG-floxSTOP-Cx43-P2A-mCherry-Ai6 Transfection
For the generation of transgenic cells, either mouse G4 ES cells (5.4 × 106) or 3T3 mouse fibroblasts (4 × 106) were mixed in 800 µL PBS with 30 µg circular CAG-floxSTOP-Cx43-P2A-mCherry-Ai6 plasmid DNA and 10 µg zinc finger nucleases pCMV-rosaR4-KKR (Addgene, #37199) and pCMV-rosaL6-ELD mutations (Addgene, #37198) and electroporated (ES cells: 240 V, 500 µF, 1 pulse; 3T3-fibroblasts: 180 V, 950 µF, 1 pulse) using the Gene Pulser Xcell Electroporation Systems (Bio-Rad, Hercules, CA, USA). Two days after electroporation, transgenic G4 ES cells and 3T3 cells were selected by adding 225 µg/mL and 800 µg/mL G418 (neomycin, Gibco, Thermo Fisher Scientific), respectively, to the medium for ten (G4 ES cells) or twelve days (3T3 mouse fibroblasts).
HeLa cells (60% confluency) were transfected with the PacI digested linearized CAG-floxSTOP-Cx43-P2A-mCherry-Ai6 construct using the TransIT-HeLaMONSTER Transfection Kit (Mirus Bio LLC, Madison, WI, USA) according to the manufacturer’s instructions. Two days after transfection, transgenic HeLa cells were selected by adding 600 µg/mL G418 to the medium for twelve days.
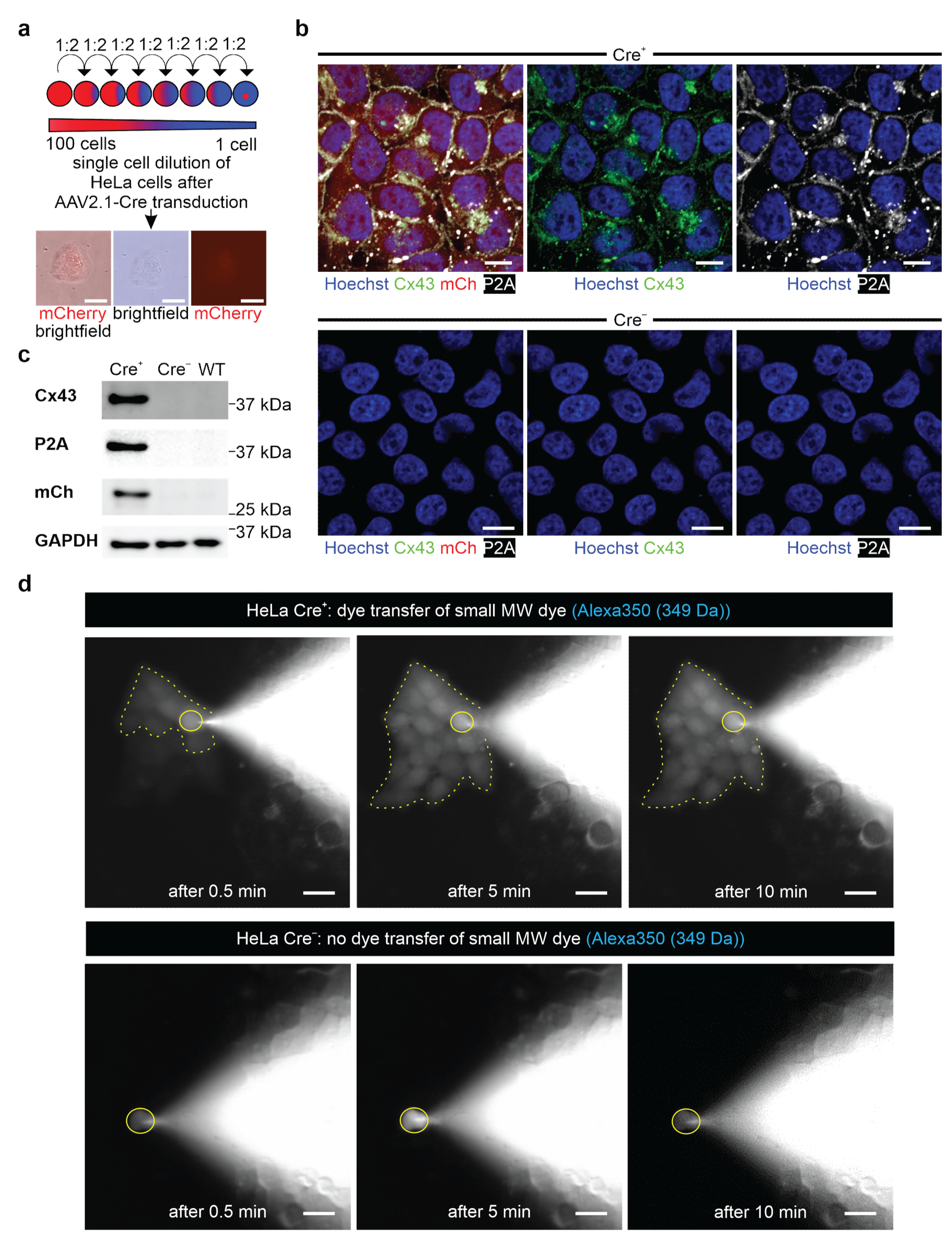
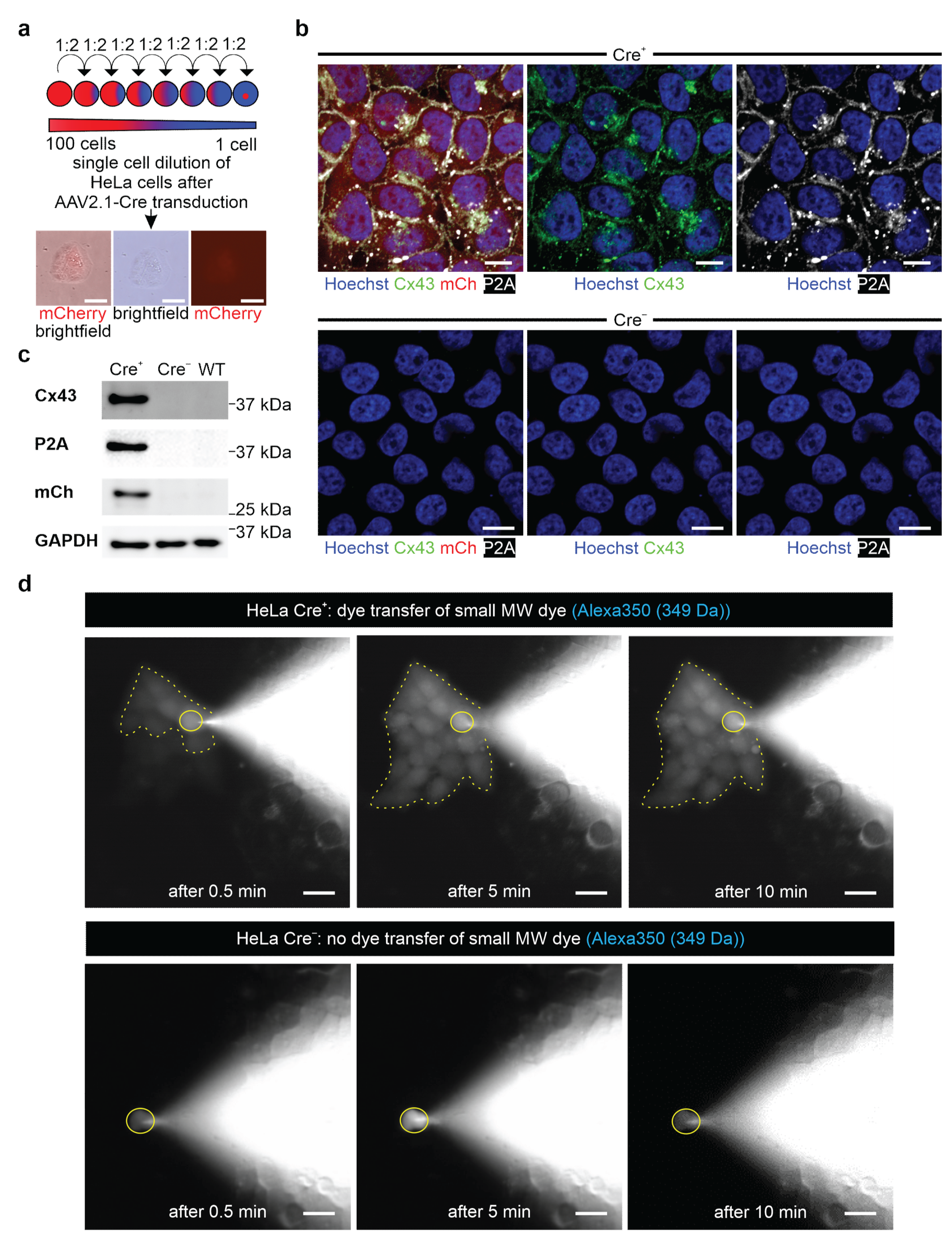
Resistant ES cells and HeLa colonies were isolated and expanded in 24-well plates; in case of the ES cells, these were coated with irradiated neomycin-resistant fibroblasts. Colonies of resistant transgenic 3T3 cells were pooled, and a single cell dilution of 3T3-fibroblasts was performed, as depicted in Figure 3a.
Neomycin-resistant colonies were analyzed for the presence of the expression cassette by PCR (see 2.9) and mCherry fluorescence after AAV2.1-Cre transduction.
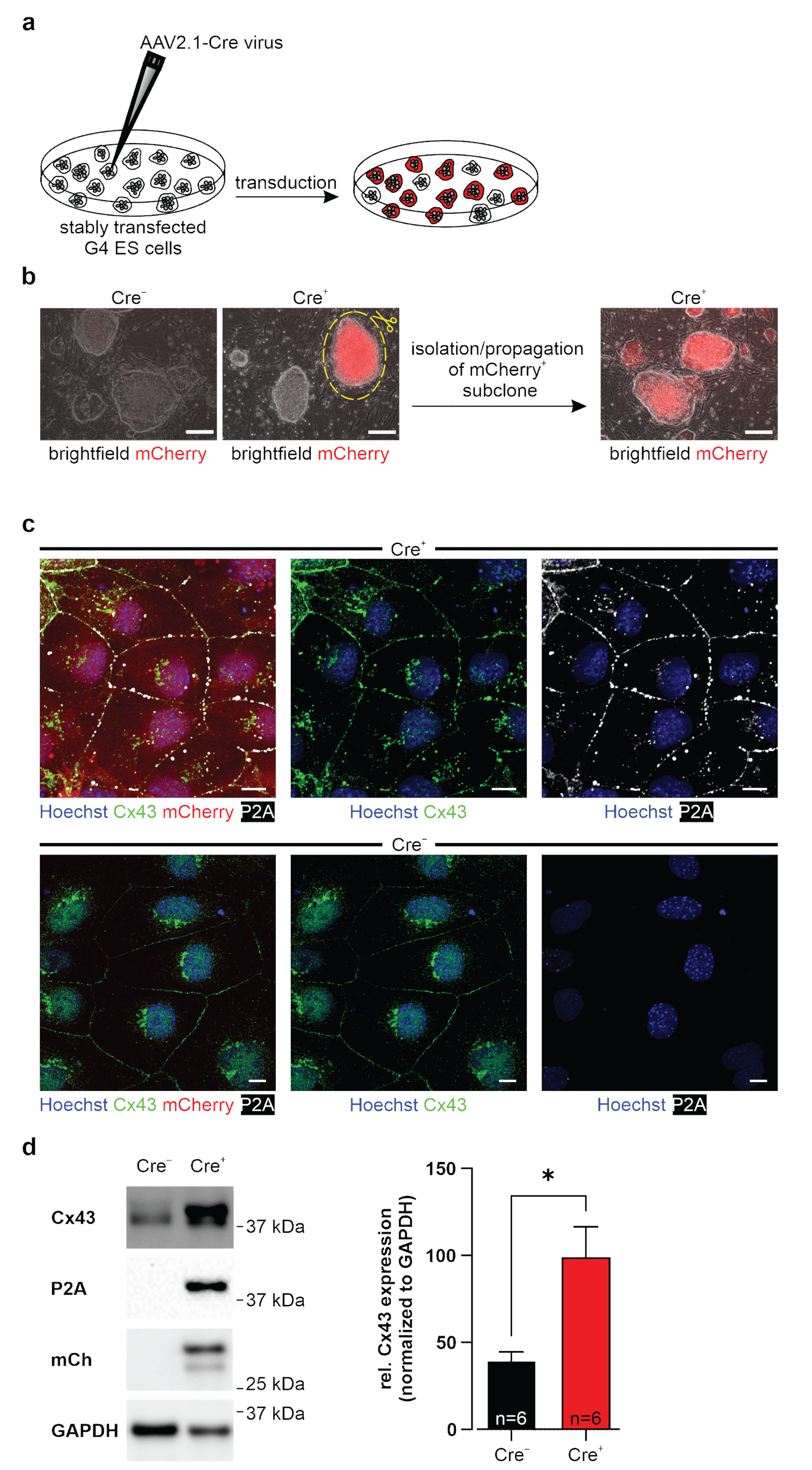
2.4. Generation of Transgene-Expressing Cre+ Cell Lines
The AAV2.1-Cre virus (pENN.AAV.CMVs.PI.Cre.rBG, Addgene, #105537-AAV1, 1.2 × 10
13 GC/mL) was titrated on a G4 ES cell line (BAG3P209L-eGFPflox3) [
34] expressing eGFP after Cre induction; 5.6 × 10
10 genome copies/mL per 7500 cells were used. Viral transduction was performed in cell culture medium of CAG-floxSTOP-Cx43-P2A-mCherry-Ai6 transfected (Cre
−) lines of ES, HeLa cells, and 3T3-fibroblasts. Two days post-transduction, the virus was washed off, and at day three post-transduction, mCherry-expressing colonies were observed. These were either picked, or single cell dilutions were performed.
For immunostainings, cells were cultured on fibronectin (0.5 µg/mL, Sigma-Aldrich, Merck) or 0.1% gelatin- (Merck) coated (I) 96 well glass-bottom plates (Greiner, Kremsmünster, Austria) or (II) cover glasses (VWR, Radnor, PA, USA), fixed with 4% Formaldehyde (FA) (PanReac AppliChem, Darmstadt, Germany) for 30 min.
2.5. Generation of AAV2.6-Cx43-P2A-mCherry Virus
The Cx43-P2A-mCherry insert was directly cloned under control of the CAG promoter of the pAAV.CAG.hChR2(H134R)-mCherry.WPRE.SV40 vector (Addgene, #100054; channelrhodopsin-2 cDNA was replaced). The CAG-floxSTOP-Cx43-P2A-mCherry-Ai6 plasmid was digested with FseI, blunted, and subsequently cut with BsrGI. The resulting 1935 bp fragment was isolated and inserted into the BamHI (blunted) and BsrGI sites of the AAV vector. The resulting pAAV-CAG-Cx43-P2A-mCherry vector was utilized for rAAV2.6-Cx43-P2A-mCherry virus production.
Plasmid DNAs were prepared using the EndoFree Plasmid Maxi Kit (Qiagen). All vectors were verified by sequencing (Eurofins Genomics, Ebersberg, Germany).
2.6. Isolation of Murine Neonatal Cardiomyocytes (NNCMs) and Transduction with the AAV2.6-Cx43-P2A-mCherry Virus
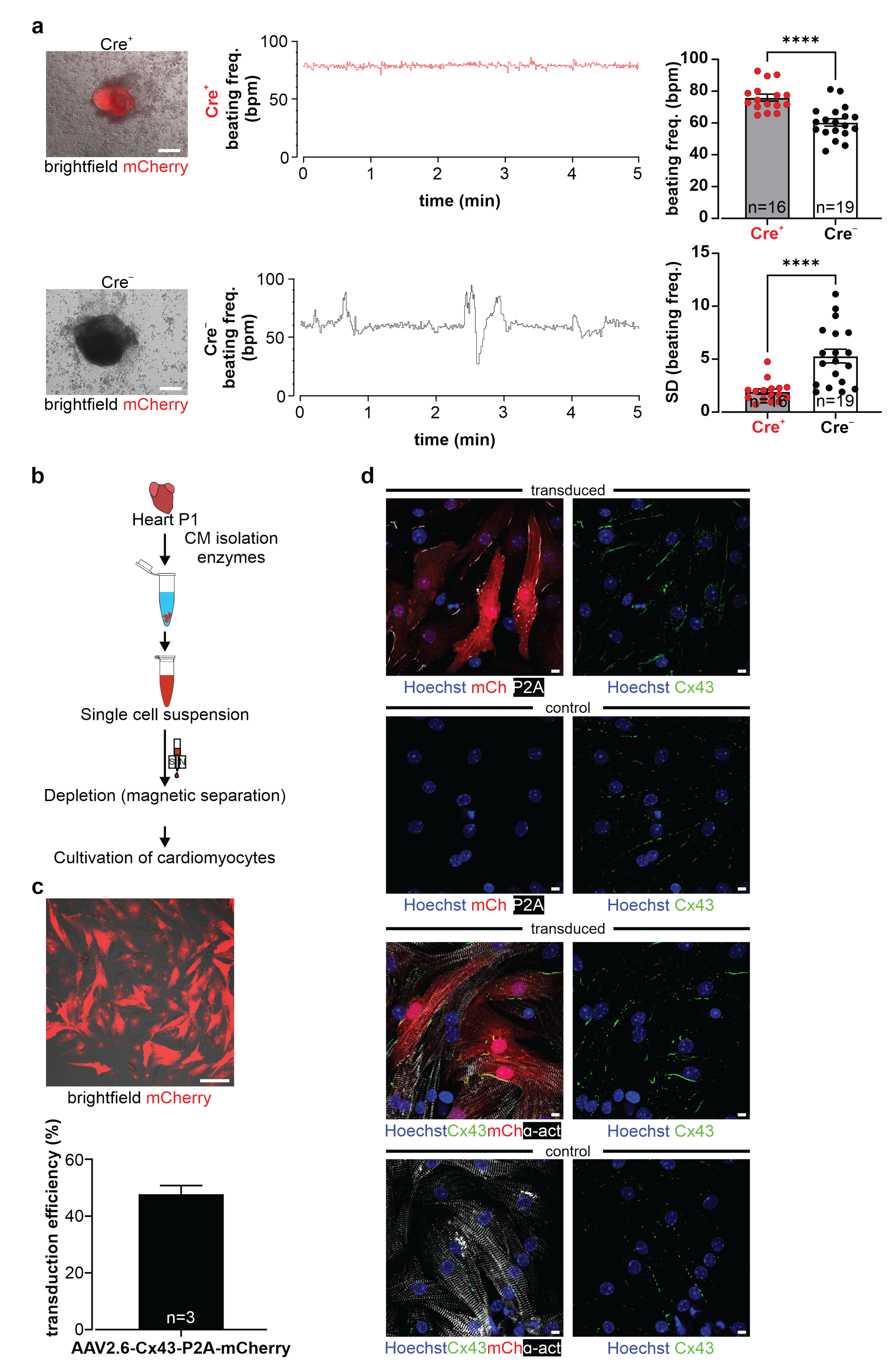
Neonatal cardiomyocytes (P1, CD1 background; breeding pairs were supplied by Charles River Laboratories, Sulzfeld, Germany) were isolated (Figure 7b) and enriched using the neonatal heart dissociation and isolation kits (Miltenyi) (for details, see: Raulf, Voeltz et al. [
35] and manufacturer’s instructions).
Then, NNCMS were seeded on fibronectin- (0.5 µg/mL, Sigma-Aldrich, Merck, Darmstadt, Germany) coated 96 well glass-bottom plates (Greiner) and cultured in a 20% IMDM medium (30,000 NNCMs/well). After 24 h, FCS was reduced to a 2% IMDM medium (IMDM, 2% v/v FCS, 100 U/mL/100 µg/mL P/S, 0.1 mM NEAA, and 0.1 mM β-ME), and the virus was added to the medium (0.55 Virus Genomes (VG)/cell). Two days post-transduction, the medium was changed (2% IMDM), and three days post-transduction, mCherry+ cells were observed. Transduction efficiency was calculated as a ratio of double positive NNCMs (α-actinin+ and mCherry+)/all NNCMs (α-actinin+) in four fields of view/well (Nikon Eclipse Ti and Nikon A1R MP system, Nikon, Tokyo, Japan) with DAPI, Cy3, and Cy5 filters; 20× objective; and Lasers (LU-N4 laser unit, 405 nm, 488 nm, 561 nm, 640 nm) using NIS-Elements AR 5.11.01 software (Nikon).
2.7. Karyotyping of Transgenic G4 ES Cell Clones
Suitable G4 ES cell clones were selected based on karyotyping. Cells were incubated with 0.1 µg/mL demecolcine solution (Sigma-Aldrich, Merck) in a G4 ES cell medium for 3 h, trypsinized (Thermo Fisher Scientific) and treated for 20 min with 75 mM KCl solution (Sigma-Aldrich, Merck); a fixation solution containing methanol (Carl Roth, Karlsruhe, Germany) and acetic acid (Carl Roth) (3:1) was added to preserve chromatin morphology. The suspension was dripped on glass slides (Marienfeld, Lauda-Königshofen, Germany). A Hoechst solution (1 µg/mL in PBS, Sigma-Aldrich, Merck) was used (15 min, RT) for chromosome staining, and microscopic pictures were taken (Axio Observer Microscope, Zen Software 2.6, Carl Zeiss MicroImaging, Inc., Oberkochen, Germany).
2.8. Analysis of Transgene Integration into the Rosa26 Locus by Southern Blot
2.8.1. Extraction of Genomic DNA
Total high-molecular-weight DNA of transgenic and WT ES cells was isolated by standard DNA isolation protocol. This consists of lysis in 17.5 mM sodium dodecyl sulfate (SDS), 50 mM Tris (pH 8.0), 10 mM ethylenediaminetetraacetic acid (EDTA), proteinase K (200 µg/mL, Carl Roth); phenol-chloroform-isoamyl alcohol extraction (Sigma-Aldrich, Merck, 25:24:1); chloroform-isoamyl extraction (Sigma-Aldrich, Merck, 24:1); precipitation and resuspension of DNA in 10 mM Tris and 1 mM EDTA, pH 8.0 (TE); RNase treatment (1 mg/mL, Qiagen); further extraction; and precipitation and resuspension of DNA in TE buffer.
2.8.2. Preparation of DNA Probes
The 391 bp Cx43 DNA probe was obtained by a PCR reaction using the CAG-floxSTOP-Cx43-P2A-mCherry-Ai6 construct DNA as a template and the following primer pair: fw: GAGCTCAAAGTGGCGCAGAC and rev: CCTTCACGCGAT CCTTAACG (PCR program: (I) 95 °C for 15 min; 30 cycles of (II-IV): (II) 95 °C for 20 s, (III) 60 °C for 60 s and (IV) 72 °C for 2 min; (V) 72 °C for 10 min; HOT FIREPol Blend Master Mix Ready to Load (Solis Biodyne, Tartu, Estonia)), followed by agarose gel electrophoresis (1% gel) and extraction of the 391 bp DNA band.
For the generation of the Rs26-specific DNA probe (pRs26-5′, see Soriano et al. [
36]), the plasmid pDonor MCS Rosa26 vector (Addgene, #37200) was digested with EcoRI, and the resulting 450 bp fragment was isolated. pRs26-5′probe sequence:
CAGGGAAAACGACAAAATCTGGCTCAATTCCAGGCTAGAACCCTACAAATTCAACAGGGATATCGCAAGGATACTGGGGCATACGCCACAGGGAGTCCAAGAATGTGAGGTGGGGGTGGCGAAGGTAATGTCTTTGGTGTGGGAAAAGCAGCAGCCATCTGAGATAGGAACTGGAAAACCAGAGGAGAGGCGTTCAGGAAGATTATGGAGGGGAGGACTGGGCCCCCACGAGCGACCAGAGTTGTCACAAGGCCGCAAGAACAGGGGAGGTGGGGGGCTCAGGGACAGAAAAAAAAGTATGTGTATTTTGAGAGCAGGGTTGGGAGGCCTCTCCTGAAAAGGGTATAAACGTGGAGTAGGCAATACCCAGGCAAAAAGGGGAGACCAGAGTAGGGGGAGGGGAAGAGTCCTGACCCAGGGAAGACATTAAAAAGGTAGTGGGGTCGACTAGATGAAGGAGAGCCTTTCTCTCTGGGCAAGAGCGGTGCAATGGTGTGTAAAGGTAGCTGAGAA
2.8.3. Blotting
For Southern blot analysis, 10 µg genomic DNA were digested with AflIII or double digested with EcoRV and HindIII and the addition of 2.5 mM of spermidine at 37 °C overnight. Enzymes were inactivated by incubation for 20 min at 60 °C and 80 °C, respectively, and digested DNAs were separated overnight (30 V) in 0.8% agarose gels containing 0.01% ethidium bromide. A Lambda HindIII digested DNA marker (Promega, Madison, WI, USA) was used to determine DNA fragment sizes. After 15 min in 0.25 N HCl, 5 min in distilled water, and two 15 min periods in 0.5 M NaOH, the DNA was transferred to a positively charged nylon membrane treated with distilled water and 0.5 M NaOH (BrightStar-Plus, Ambion, Thermo Fisher Scientific) via capillary blotting method (transfer buffer: 1M NaCl and 400 mM NaOH) for 3 h at room temperature. Following this, the membrane was neutralized for 15 min in 2× SSC buffer (0.3 M NaCl and 0.03 M sodium citrate tribasic dihydrate) and dried for 15 min at 80 °C (Heratherm incubator, Thermo Fisher Scientific) to immobilize the DNA. The membrane was then prehybridized for 3 h in a glass hybridization tube (Analytik Jena GmbH, Jena, Germany) placed in a hybridization oven (Analytik Jena GmbH) at 60 °C in an ULTRAhyb ultrasensitive hybridization buffer (Invitrogen, Thermo Fisher Scientific). DNA probes were labelled with α32P-dCTP (PerkinElmer 6000 Ci/mmol 20 mCi/mL Lead, 500 µCi, Waltham, MA, USA) using the Prime-It II random primer labeling kit (Agilent Technologies, Santa Clara, CA, USA). Labelled probes were purified with Bio-Spin P30 Tris Chromatography Columns (Bio-Rad) added to the prehybridized membrane, and hybridization was performed under stringent conditions (68 °C) overnight. Filters were washed four times (each 30 min, 55 °C) in 2× SSC containing 0.1% SDS and were exposed to hyperfilm MP8 films (GE Healthcare GmbH, Solingen, Germany) using intensifier screens (Amersham, GE Healthcare). The hyperfilms were exposed for 6–8 days before they were developed and fixed (Kodak GBX Fixer and Replenisher, Sigma-Aldrich, Merck; Kodak GBX Developer and Replenisher, Sigma-Aldrich, Merck; both 1:5 diluted with distilled water).
2.9. Selection of Suitable G4 ES and 3T3 Cell Clones Using PCR Analysis
Cells of the different G4 ES and 3T3 clones were lysed, and DNA was extracted using the Gentra Puregene Cell Kit (Qiagen). DNA was amplified applying 5× HOT FIREPol Blend Master Mix Ready to Load (Solis Biodyne) or GoTaqG2 Hot Start Green Master Mix (Promega) and following primer pairs for G4 ES cell clones: (I) integration into the 5′ end of Rs26 locus: fw (endogenous Rs26 sequence): GCCGCCTAAAGAAGAGGCTG, rev (CAG promoter of the insert): GGCGGGCCATTTACCGTAAG; (II) integration into the 3′ end of Rs26 locus: fw (plasmid specific sequence): AGCCTCTGTTCCACATACAC, rev (endogenous Rs26 sequence): GTGCAGTGTTGAGGGCAATC; (III) WT Rs26 allele: fw: AAGGGAGCTGCAGTGGAGTA, rev: CCGAAAATCTGTGGGAAGTC; (IV) detection of Cx43: fw: TGTGGCTGTCGGTGCTCTTC, rev: GAGCCAAGTACAGGA GTGTG; (V) mCherry: fw: AGACCGCCAAGCTGAAGGTG, rev: TCAGCTTCAGCCT CTGCTTGATCT and for 3T3-fibroblasts: (IV) Cx43: fw: TGTGGCTGTCGGTGCTCTTC, rev: GAGCCAAGTACAGGAGTGTG; (VI) Cx43-mCherry: fw: TGTGGCTGTCGG TGCTCTTC, rev: TCAGCTTCAGCCTCTGCTTGATCT.
To test the functionality of the Cre-loxP system RNA of Cre− as well as Cre+ G4 ES cell clones 29 and 31 were isolated using the RNeasy Mini Kit (Qiagen), and cDNA was synthesized with the SuperScript VILO cDNA Synthesis Kit (Invitrogen, Thermo Fisher Scientific). CAG-driven expression of the transgene using a forward primer that binds to the first exon of the CAG promoter in combination with a reverse primer binding to Cx43 sequence demonstrated functionality of the Cre-loxP system: fw: CTGACTGACCGCGTTACTC, rev: AGCTGACTCAACCGCTGTCC.
Agarose gel electrophoreses were run to separate PCR products, which were visualized by ethidium bromide (Carl Roth) using an UV light imager Intas GelSTICK Touch (Intas Science Imaging Instruments GmbH, Göttingen, Germany) in combination with Intas GDS Touch 2 software 1.1.5.4 (Intas Science Imaging Instruments GmbH).
2.10. Copy Number Determination by qPCR
To analyze actual transgene copy numbers of G4 ES cells, the genomic DNA of three Cre− clones was isolated (EchoLUTION CellCulture DNA kit, BioEchoLife Sciences GmbH, Cologne, Germany) according to manufacturer’s instructions. Then, qPCR was performed as triple determination with 200 ng of genomic DNA, TaqMan Gene Expression Master-Mix (Applied Biosystems, Thermo Fisher Scientific), a Tfrc-VIC housekeeper TaqMan probe (TaqMan Copy Number Reference Assay, mouse, Tfrc, Applied Biosystems, Thermo Fisher Scientific, #4458366), and a custom-made mCherry binding TaqMan probe (Thermo Fisher Scientific). A CFX 96 Real-Time System (Bio-Rad) and 96 well PCR plates (Bio-Rad) with seals (Bio-Rad) (PCR program: (I) 95 °C for 10 min, 35 cycles of (II–IV): (II) 95 °C for 15 s, (III) 60 °C for 30 s, (IV) 72 °C for 30 s) were used. CT values for the target (mCherry) and reference (Tfrc, two copy numbers) gene were computed with Bio-Rad CFX Manager 3.1 software (Bio-Rad). The ∆CTTR values (CTtarget − CTreference) and copy numbers (2−ΔCTTR × 2) of the mCherry-containing transgene were calculated.
2.11. RT qPCR
To test for Cx43 (endogenous and endogenous + exogenous), Cx45, Cx40, and Cx30.2 expression, the RNA of G4 ES cells and G4 ES cell-derived EBs (Cre− and Cre+) was extracted using the RNeasy Micro Kit (Qiagen), and cDNA was synthesized as described in 2.10. Then, qPCR was performed as triple determination with 25 ng of cDNA, TaqMan probes (Thermo Fisher Scientific) for endogenous Cx43 (exon 1, Mm00439105, Gja1), total (endogenous + exogenous) Cx43 (exon 2 cDNA coding sequence, custom-made), Cx45 (Mm01253027, Gjc1), Cx40 (Mm07294003, Gja5), Cx30.2 (Mm01204089, Gjc3), and the TaqMan Gene Expression Master-Mix (Applied Biosystems, Thermo Fisher Scientific) were used. As internal control GAPDH (Thermo Fisher Scientific, #4352339E) was applied. Then, qPCR was performed on a CFX 96 cycler (Bio-Rad) as described in 2.10 (39 cycles of (II–IV)). CT values for the Cx isotypes and the reference (GAPDH) cDNAs were computed with Bio-Rad CFX Manager 3.1 software (Bio-Rad) to calculate the ∆CTTR values (CTtarget − CTreference).
2.12. Immunofluorescence Staining and Microscopy
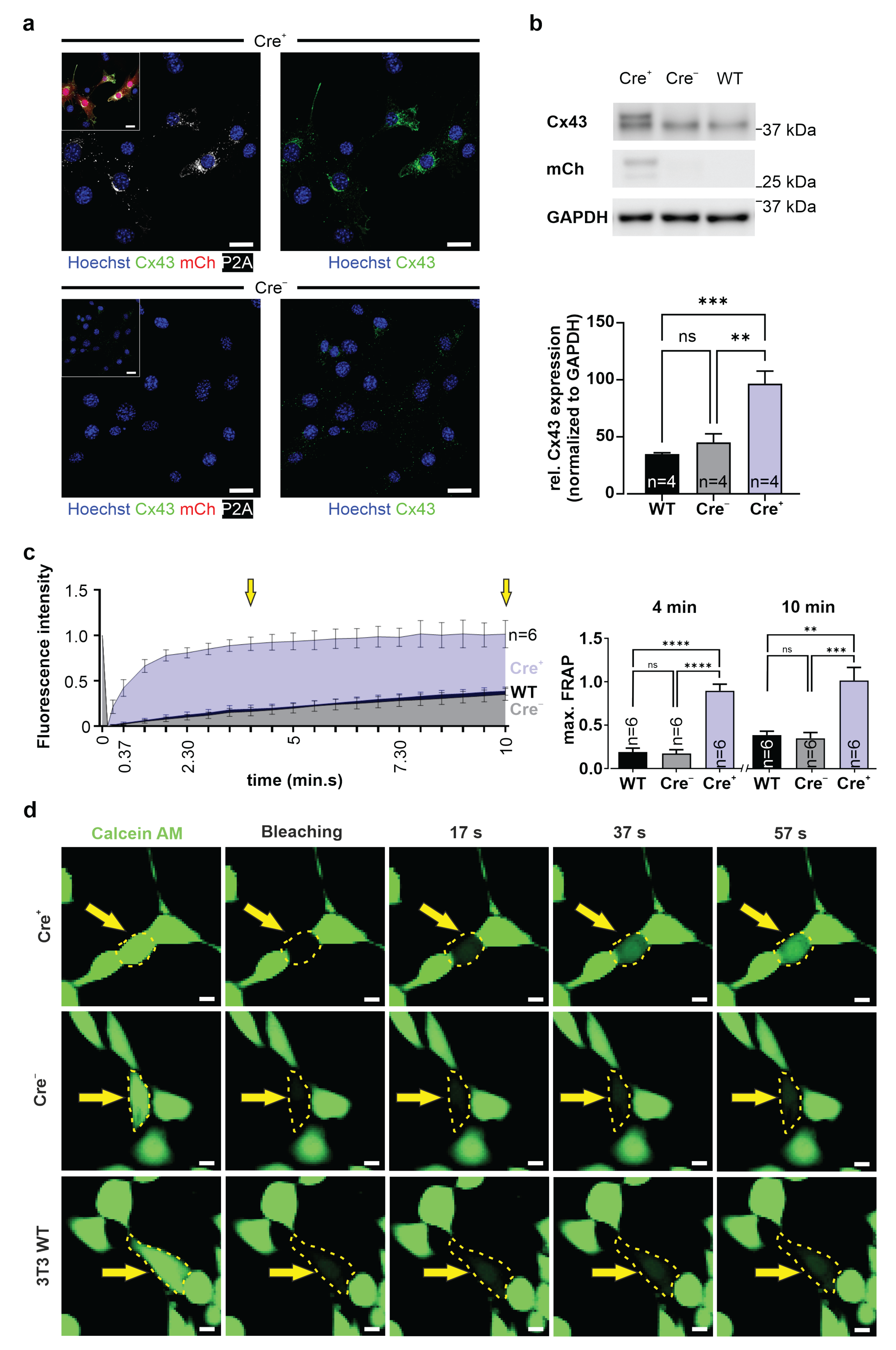
Cells on coverslips and EBs were fixed for 30 min with 4% FA (PanReac AppliChem). EBs were embedded in Tissue-Tek O.C.T. Compound (Sakura Finetek, Torrance, CA, USA) and cryosectioned (10 µm, Cryostat (Leica, CM 3050S, Wetzlar, Germany)). Prior to staining, cells and EB slices were permeabilized with 0.2% Triton X (Sigma-Aldrich, Merck) in PBS (PBS Tablets, Gibco, Thermo Fisher) for 10 min. For immunostainings, the following antibodies (diluted in 5% donkey serum (Jackson ImmunoResearch, West Grove, PA, USA), 2 h at RT or overnight at 4 °C) were used: Cx43 (1:3000, custom-produced rabbit polyclonal antibody, PSL GmbH, Heidelberg, Germany; immunizing peptide sequence: CDQRPSSRASSRASSRPRPDDLEI [
15,
37,
38]), P2A (1:700, mouse monoclonal IgG1 kappa antibody, Novus Biologicals, Littleton, CO, USA, #NBP2-59627), mCherry (1:5000, chicken polyclonal IgY antibody, Novus Biologicals, #NBP2-25158), α-actinin (1:200, Sigma-Aldrich, Merck, #A7811), and cCasp3 (1:50, Cell Signaling Technology, Danvers, MA, USA, #9661). After several washing steps, secondary antibody incubation was performed (diluted in 1 μg/mL Hoechst/PBS (Sigma-Aldrich, Merck) for 1 h at RT): donkey anti-rabbit IgG (H + L) Alexa Fluor 488/Cy2-conjugated (1:400, AffiniPure, Jackson ImmunoResearch, #711-545-152/#711-225-152); donkey anti-chicken IgY (IgG) (H + L) Cy3 conjugated antibody (1:3000, AffiniPure, Jackson ImmunoResearch, #703-165-155); donkey anti-mouse (IgG) (H+L) Alexa Fluor 647 conjugated antibody (1:3000, AffiniPure, Jackson ImmunoResearch, #715-605-151); and donkey anti-rabbit (IgG) (H + L) Alexa Fluor 647 conjugated antibody (1:3000, AffiniPure, Jackson ImmunoResearch, #711-605-152). After washing, EB slices and cells were mounted with Aqua-Poly/Mount (Polysciences, Warrington, PA, USA). Cells seeded on 96 well glass plates were mounted with PBS. Imaging was performed (I) prior fixation with a fluorescence microscope (Axio Vert A1, Carl Zeiss) using a Cy3 filter, 10× and 20× air objectives, and (II) after fixation and staining, pictures were taken with a confocal microscope (Nikon Eclipse Ti and Nikon A1R MP system, Nikon) with DAPI, Cy2, Cy3, and Cy5 filters, 20×, 40×, and 60× water objectives, Lasers (LU-N4 laser unit (405 nm, 488 nm, 561 nm, 640 nm)) using NIS-Elements AR 5.11.01 software (Nikon).
2.13. Western Blot Analysis of Cx43, P2A, and mCherry Protein Expression of Transgenic Cell Lines
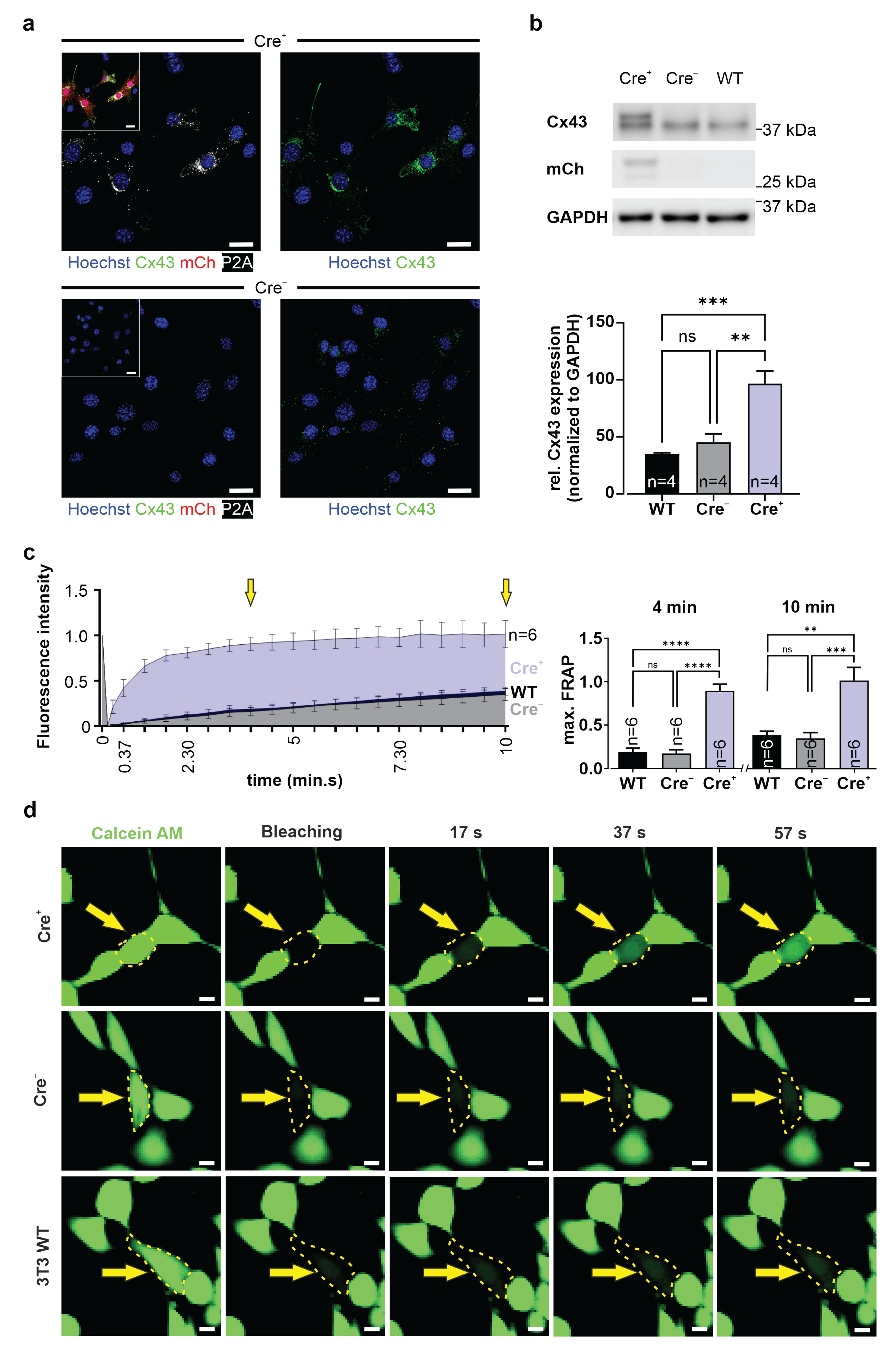
Cells were lysed on ice in freshly prepared RIPA buffer (1 mM EDTA (Applichem); 50 mM Tris HCl (pH 7.5) (Carl Roth); 150 mM NaCl (Carl Roth); 0.25% sodium deoxycholate (Sigma-Aldrich, Merck); 1% IGEPAL CA-630 (Sigma-Aldrich, Merck); 1 mM phenylmethanesulfonyl fluoride (Sigma-Aldrich, Merck); Protease and Phosphatase Inhibitor (Thermo Fisher)). SDS mini gels were cast (10% separating gel: 10% acrylamide (Bio-Rad) in 375 mM Tris HCl (pH 8.8), 0.1% SDS (Carl Roth), 0.05% APS (Bio-Rad), 0.05% TEMED (Sigma-Aldrich, Merck); 5% stacking gel: 5% acrylamide in 125 mM Tris HCl (pH 6.8), 0.1% SDS, 0.05% APS, 0.1% TEMED). Protein lysates were mixed 1:1 with 2× Laemmli Buffer (Bio-Rad) supplemented with β-ME (Sigma-Aldrich, Merck) and denatured for 10 min at 100 °C. Protein samples and Precision Plus Protein WesternC Standards (Bio-Rad) separated by gel electrophoresis (ProSieve EX Running Buffer, Lonza, Basel, Switzerland) were transferred onto a methanol-activated (Carl Roth) 0.22 µm PVDF-FL membrane (Biozym) by tankblot (ProSieve EX Western Blot Transfer Buffer, Lonza). Non-specific binding sites of membranes were blocked for 1 h with 5% skim-milk powder (VWR) solved in TBST (20 mM Tris (Sigma-Aldrich, Merck), 150 mM NaCl, 0.1% Tween-20 (Applichem)). Membranes were incubated with Cx43 (1:3000, custom-produced rabbit polyclonal antibody, PSL GmbH, Heidelberg, Germany; 1:8000, rabbit polyclonal antibody, Sigma-Aldrich, Merck, #C6219), P2A (1:700, mouse monoclonal IgG1 kappa antibody, Novus Biologicals, #NBP2-59627), or mCherry (1:5000, chicken polyclonal IgY antibody, Novus Biologicals, #NBP2-25158) antibodies over night at 4 °C. After washing with TBST (3 × 5 min, RT), they were incubated for 1 h at RT with a donkey anti-rabbit IgG (H+L) Alexa Fluor 488 conjugated antibody (1:3000, AffiniPure, Jackson ImmunoResearch, #711-545-152), a goat anti-mouse IgG peroxidase conjugated antibody (1:3000, Sigma-Aldrich, Merck, #A2304), a donkey anti-chicken IgG (H+L) Alexa Fluor 647 conjugated antibody (1:3000, AffiniPure, Jackson ImmunoResearch, #703-175-155), and an Alexa 555 conjugated-GAPDH loading control monoclonal antibody (1:5000, Thermo Fisher, #MA5-15738). Blots with peroxidase-conjugated secondary antibodies were incubated with Pierce ECL Western Blotting Substrate (Thermo Fisher). First and secondary antibodies were diluted in 5% skim milk powder solved in TBST. All blots were detected with the ChemiDoc MP Imaging System (Bio-Rad). Image Lab 5.2.1 software (Bio-Rad) was used for semi-quantitative analysis of Cx43 protein expression, which was normalized to GAPDH as housekeeper.
2.14. Dye Transfer Experiments in HeLa Cells
Cells were seeded on 0.1% gelatin- (Merck) coated cover glasses (VWR) placed in 24-well plates containing 20% DMEM medium plus additives, as described above. A small blue fluorescent Alexa Fluor 350 Hydrazide dye (349 Da, Invitrogen, Thermo Fisher Scientific, #A10439) and a large red fluorescent dextran-coupled Alexa Fluor 647 dye (10 kDa, Invitrogen, Thermo Fisher Scientific, #D22914) were diluted to a concentration of 0.5 µg/µL in intracellular solution (40 mM KCl (Sigma-Aldrich, Merck), 90 mM KAsp (Sigma-Aldrich, Merck), 10 mM NaCl (Carl Roth), 3 mM Mg-ATP (Sigma-Aldrich, Merck), 1 mM MgCl (Sigma-Aldrich, Merck) and 10 mM HEPES (Sigma-Aldrich, Merck) and adjusted to pH 7.4 with KOH (Sigma-Aldrich, Merck)). HeLa cells were dialyzed with a patch clamp pipette (BF 150-86-10, Science Products, Hofheim, Germany) and filled with the above-mentioned solutions under microscopic control. When the patch pipette touched the cell, a bright field and a fluorescence picture were taken (a DAPI filter (F36 500 HC, Nikon) for Alexa Fluor 350 or a Cy5 filter (F 46-009 ET, Nikon) for Alexa Fluor 647 (20× Objective Fluor, Nikon)). The membrane resistance was monitored through an EPC10 amplifier (HEKA Elektronik GmbH, Reutlingen, Germany) until reaching whole cell configuration in the voltage clamp mode (holding potential: −40 mV). Fluorescence pictures were taken after obtaining whole-cell configuration at 0.5 min, 5 min, and 10 min. Pictures were taken with the Grasshopper 3 Camera (GS3-U3-23S6C-C, Teledyne Flir LCC, Wilsonville, OR, USA) controlled by the Micro-Manager 2.0 beta software (Image J, Bethesda, CA, USA).
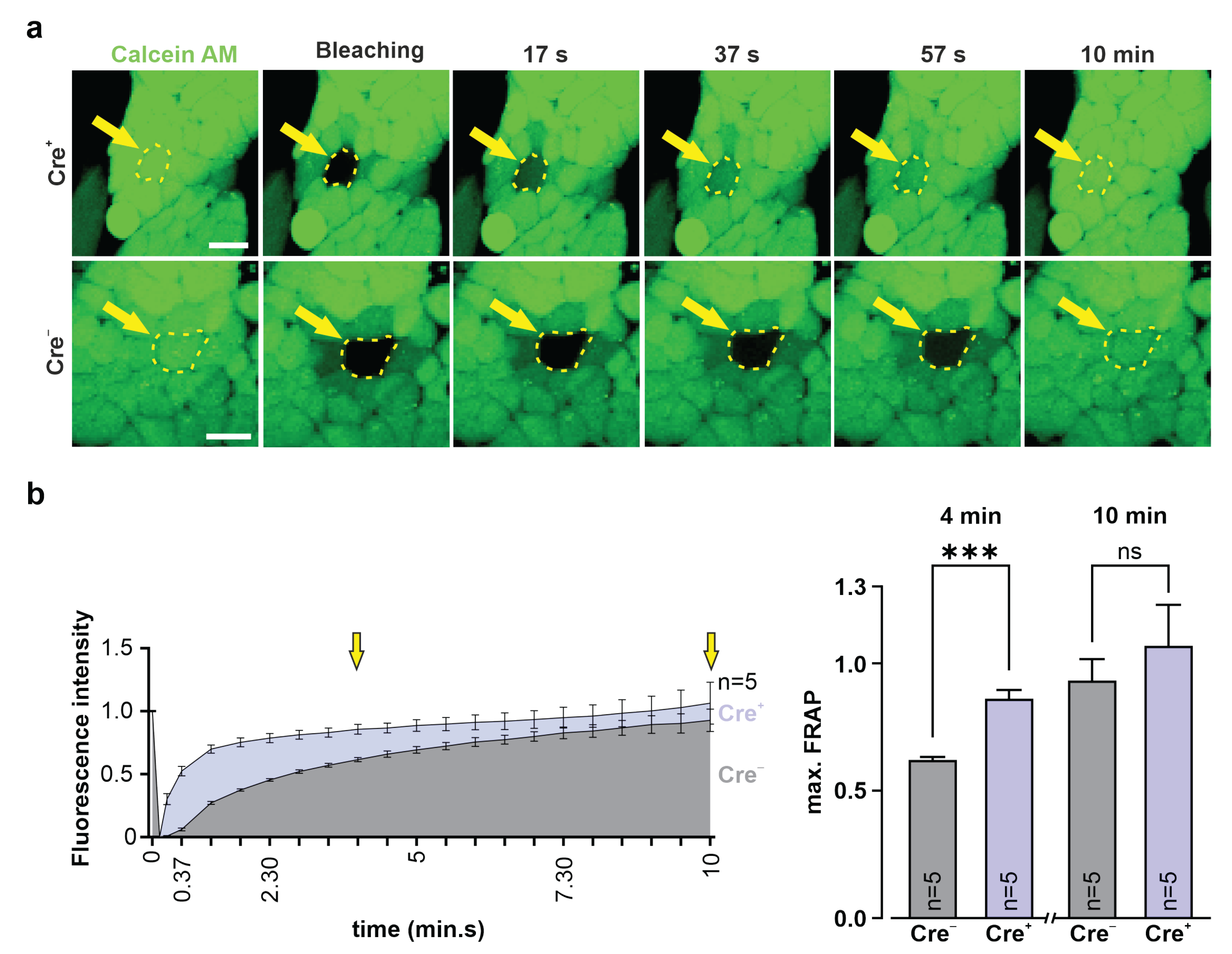
2.15. Fluorescence Recovery after Photobleaching (FRAP)
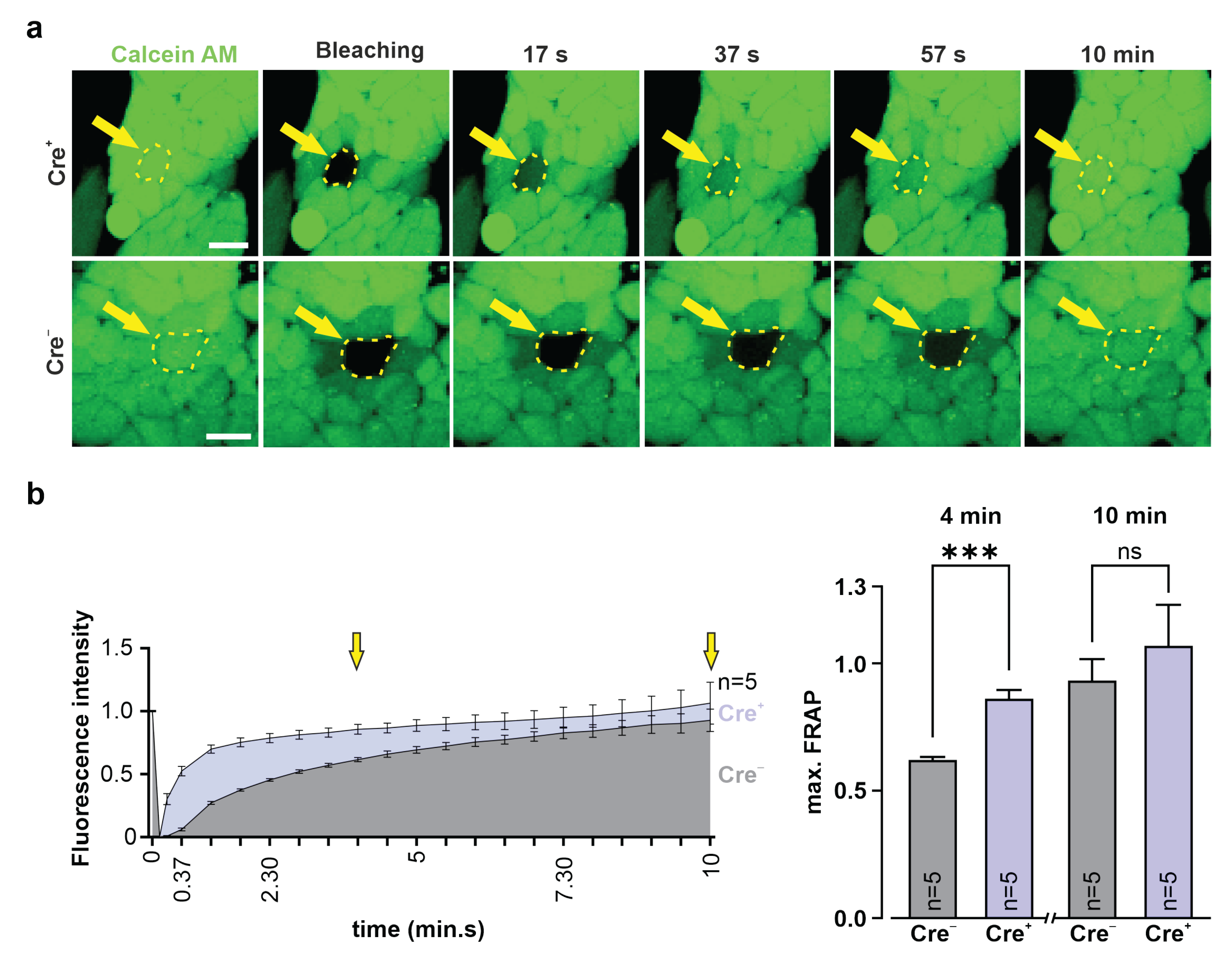
3T3-fibroblasts and G4 ES cells were incubated for 20 min at 37 °C with calcein AM (0.38 µM, Invitrogen, Thermo Fisher Scientific, #C3100MP) diluted in 10% DMEM medium (3T3, see above) or 15% KnockOut-DMEM (G4 ES cells, see above). Fluorescence intensity of a cluster of calcein+ 3T3-fibroblasts or G4 ES cells was measured using a confocal microscope (Nikon Eclipse Ti, 40× objective; CFI Apo Lambda S LWD 40XWI, NA 1.25, Nikon). Next, a single fibroblast or G4 ES cell in the cluster was bleached with a 561 nm laser pulse (5 s, 2.5 mW), fluorescence intensity was measured, and pictures were taken over 10 min at 15 s intervals. Fluorescence intensities were converted into values between 1 (fluorescence intensity before bleaching) and 0 (fluorescence intensity immediately after bleaching).
2.16. Video Microscopy of Cell Clusters of Dissociated Beating EBs
Cre
+ and Cre
− beating EBs were harvested at d12/d13 of differentiation. After partial dissociation with trypsin for 20 min at 37 °C, cell clusters were seeded on 0.1% gelatin- (Merck) coated 24-well plates filled with 20% IMDM medium. One day later, frequency measurements were performed via video microscopy of beating clusters on a Nikon Eclipse Ti2 inverted microscope system with a 4× objective (Fluor, Nikon) using a brightfield setting. EB clusters plated in the 24 wells were heated by an Ibidi temperature controller (Ibidi GmbH, Gräfelfing, Germany), and CO
2 was maintained at 5% through an Ibidi gas mixer (Ibidi GmbH). Spontaneous contractions were recorded with a Grasshopper 3 Camera (GS3-U3-23S6C-C, Teledyne Flir LCC) analyzed online and offline using a custom-designed software (LabView, National Instruments, Austin, TX, USA), as described earlier in Makowka, Bruegmann et al. [
39]. Time points of individual beats were recorded by a PowerLab system through the sound recorder of the PC (PowerLab 4/35 and Labchart 8 software, AD Instruments, Sydney, Australia) and used to calculate the frequency in offline analysis. For statistical analysis, the mean of beats per min (bpm) and its standard deviation (SD) of 5 min recording (sampling every second) was calculated. The mCherry expression of Cre
+ cell clusters was verified before the experiment with a Cy3 filter (F 46-004 ET, Nikon) using a prime BSI camera (Teledyne Photometrics, Tuscon, AZ, USA) controlled by the NIS-Elements AR 5.21.03 software (Nikon).
2.17. Statistical Analyses
Relative Cx43 expression data of Western blots were either compared using an unpaired two-tailed t-test (Cre+ vs. Cre− ES cells) or an ordinary one-way ANOVA test followed, in case of significance, by a Tukey’s multiple comparisons test (comparison of WT-, Cre−-, and Cre+ 3T3-fibroblasts). Ordinary one-way ANOVA and Tukey’s multiple comparisons tests (comparison of WT-, Cre−-, and Cre+ 3T3-fibroblasts) or an unpaired two-tailed t-test (Cre+ vs. Cre− ES cells) were determined to evaluate statistical differences of FRAP experiments. Beating frequencies and standard deviations of beating frequencies were compared using an unpaired two-tailed t-test. Cx isoform expression of RT qPCR analysis was compared using an unpaired two-tailed t-test (Cre+ vs. Cre− EBs). All error bars are provided as SEMs. Statistical significance was considered a p-value of ≤0.05. All statistical analyses were calculated using GraphPad Prism 9.3.0 (GraphPad Software Inc., San Diego, CA, USA).
4. Discussion
Herein we report the generation of a new genetic model that allows inducible overexpression of Cx43 in mouse ES cells. Cx43 is widely distributed in mammals, and we have chosen the ES cell system because in vitro differentiation of pluripotent cells allows us to assess the effects of Cx43 overexpression in different somatic cell types, such as cardiomyocytes and astrocytes. We have also established an AAV virus, which enables Cx43 overexpression of cells, tissues, and organs in vitro and in vivo.
We have opted for an inducible genetic system because CMV-driven ubiquitous Cx43 overexpression in mice was reported to cause morbidity and postnatal mortality due to neural tube and conotruncal heart defects [
29], a phenotype which was reminiscent of Cx43 KO models [
21,
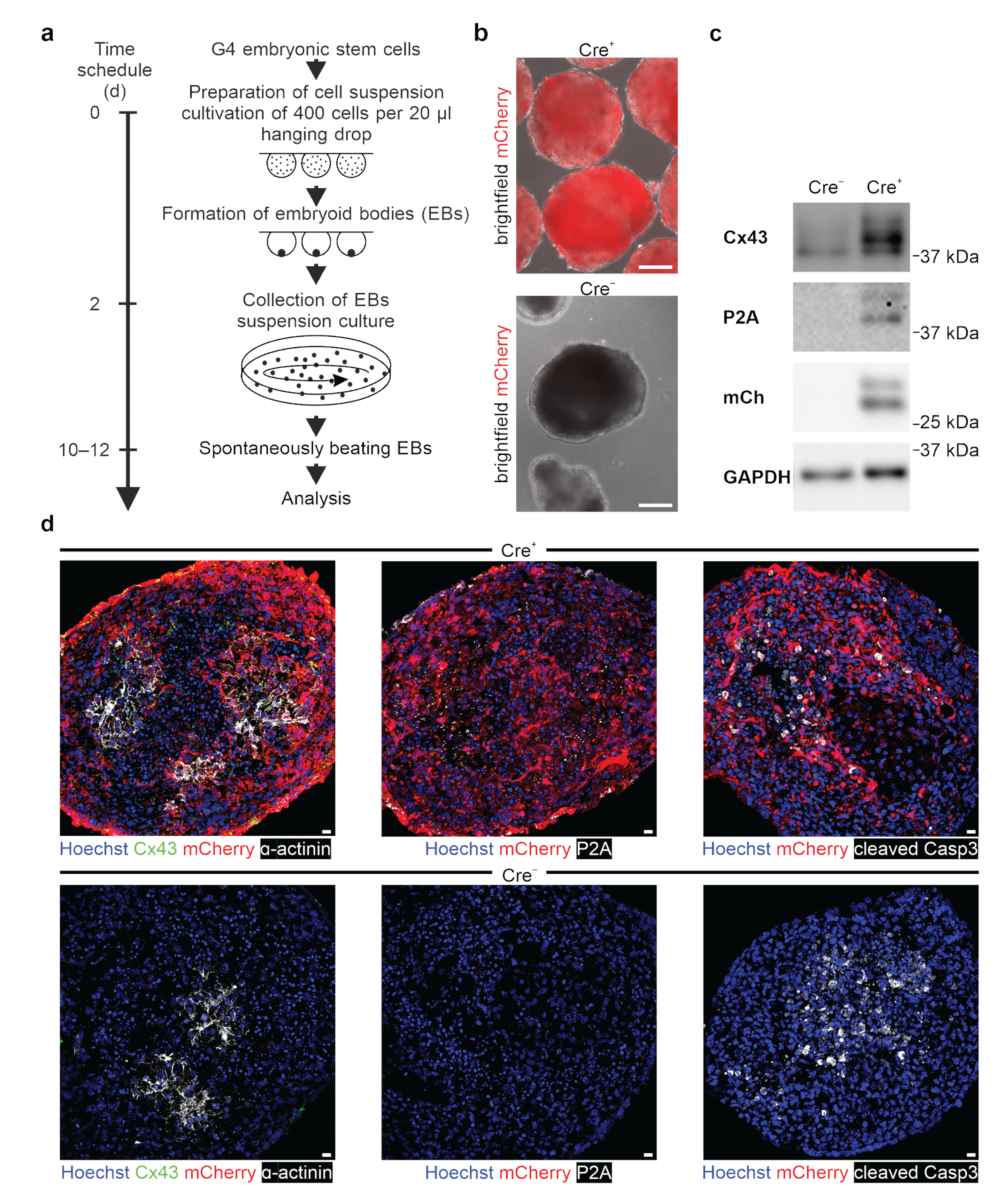
47]. We tested our genetic construct in ES cells, as well as HeLa cells and 3T3-fibroblasts, and observed no leakiness in the absence of Cre, whereas AAV-Cre-mediated excision of the floxed stop cassettes resulted in Cx43 overexpression in cells with and without endogenous Cx43 expression. A double reporter system was implemented to detect both Cx43 overexpressing cells based on mCherry fluorescence and the location and distribution pattern of exogenous Cx43 molecules based on their fusion to the small self-cleaving peptide P2A [
48], which is detectable by antibodies (
Figure 2c,d). P2A is a powerful tool, as it allows co-translational expression of P2A-linked genes in equal amounts in contrast to IRES sites [
49]. Earlier work reported that the tagging of Cx43 is challenging, as fusion of proteins to its N-terminus resulted in the expression of non-functional hemichannels and gap junctions [
43], fusion to its C-terminus in altered gating properties [
50], and altered regulation of connexons and gap junctions [
44,
51]. We therefore chose to fuse the small P2A peptide to the C-terminal site of Cx43, and our data clearly show that this did neither alter the expression pattern nor the biology of the exogenous protein. This was underscored by dye diffusion in HeLa cells and FRAP experiments in 3T3-fibroblasts and G4 ES cells, proving the formation of functional Cx43 gap junction channels. As perturbation of the C terminus of Cx43 could lead to alterations of interactions with proteins involved in plaque dynamics [
52], this aspect needs be explored in the future by performing specific biochemical assays [
53,
54]. Similarly, phosphorylation is known to be a key modulator of Cx43 gap junction function, and, therefore, the phosphorylation pattern of exogeneous Cx43 should be investigated using phospho-specific antibodies [
55]. When analyzing the exogeneous Cx43 distribution pattern with confocal microscopy, we also found that it overlapped with endogenous Cx43 (
Figure 2c, upper panel;
Figure 4a, upper panel). Super-resolution microscopy and biochemical approaches are required to determine whether exogenous P2A
+- and endogenous P2A
−-Cx43 molecules assemble to form connexons and functional gap junctions.
Because of the reported adverse effects of Cx43 overexpression in vivo, we have explored this aspect in our system and differentiated mouse ES cells into spontaneously beating EBs in vitro upon Cre-induction. We could not observe prominent cell biological differences, as neither cardiac α-actinin immunostainings (
Figure 6d, left panel), nor the degree of apoptosis (
Figure 6d, right panel), nor the time point of initiation of spontaneous beating differed strongly between Cx43 overexpression and control EBs. Interestingly, when quantifying beating rates in Cx43 overexpression and control cell clusters, we noticed that mutant clusters were beating at faster rates and in a clearly more rhythmic fashion. At this point, we can only speculate on the mechanism and propose that the ubiquitous Cx43 overexpression provides better overall conduction between cardiomyocytes and also with non-cardiomyocytes within EBs, as we did not detect changes in the expression of other cardiac Cx isoforms. Recent experiments in human-induced pluripotent cell-derived cardiac microtissues found that Cx43 gap junctions are critical for the maturation of cardiomyocytes in these constructs and that this depends on the diffusion of cAMP through Cx43 gap junction channels [
56]. It is possible that also, in our case, the observed higher beating rates are related to more mature cardiomyocytes, which is difficult to assess based on purely morphological criteria. As expected for cultured cardiomyocytes, endogenous and exogenous Cx43 was found to be distributed in ES cell-derived cardiomyocytes and in NNCMs diffusely on the cell surface, whereas in the mature heart, its location is primarily restricted to intercalated discs.
Taken together, we have generated and characterized an inducible Cx43 overexpression system in murine ES cells and an AAV-Cx43 overexpression system enabling the investigation of both the cell biological and functional consequences of overexpressing Cx43 in vitro and in vivo. We envision for both systems multiple applications. Our group could show that grafting of Cx43-expressing muscle cells into the infarct area or overexpression of Cx43 in resident myofibroblasts of the scar strongly reduced the incidence of post-infarction ventricular tachycardias in mice in vivo [
37,
57]. Despite the great translational potential of these strategies for specific patient cohorts, potential cell biological (e.g., cell differentiation, maturation, viability) and functional (e.g., expression of ion channels and transporters) drawbacks of Cx43 (over)expression in cardiomyocytes and non-cardiomyocytes need to be investigated. The in vitro ES cell differentiation system is ideally suited for this purpose. Similarly, it has been suggested that some forms of epilepsy could benefit from the increased opening of Cx43 gap junction channels in astrocytes [
58]. Therefore, novel Cx43 gap junction activators and openers could be studied in ES cell-derived astrocytes in regard to their effects on Cx43 gap junction channels. In addition, potential adverse effects of such Cx43 modulators could be investigated in terms of safety pharmacology in transgenic ES cell-derived cardiomyocytes. The transgenic G4 ES cells containing the CAG-floxSTOP-Cx43-P2A-mCherry construct can be used to generate transgenic mice. Such a mouse model would allow for the testing of the therapeutic potential of Cx43 overexpression in any cell type, depending on crossings with Cre-deleter mouse lines. For instance, for us and other groups working on cardiac arrhythmias, it would be of great interest to induce homogenous overexpression of Cx43 in border zone cardiomyocytes or myofibroblasts of the scar area and to study the effects on ventricular arrhythmias after myocardial infarction.



 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}