

Reprogramming T-Cell Metabolism for Better Anti-Tumor Immunity


{kind=link}
{kind=link}
Abstract
1. Introduction
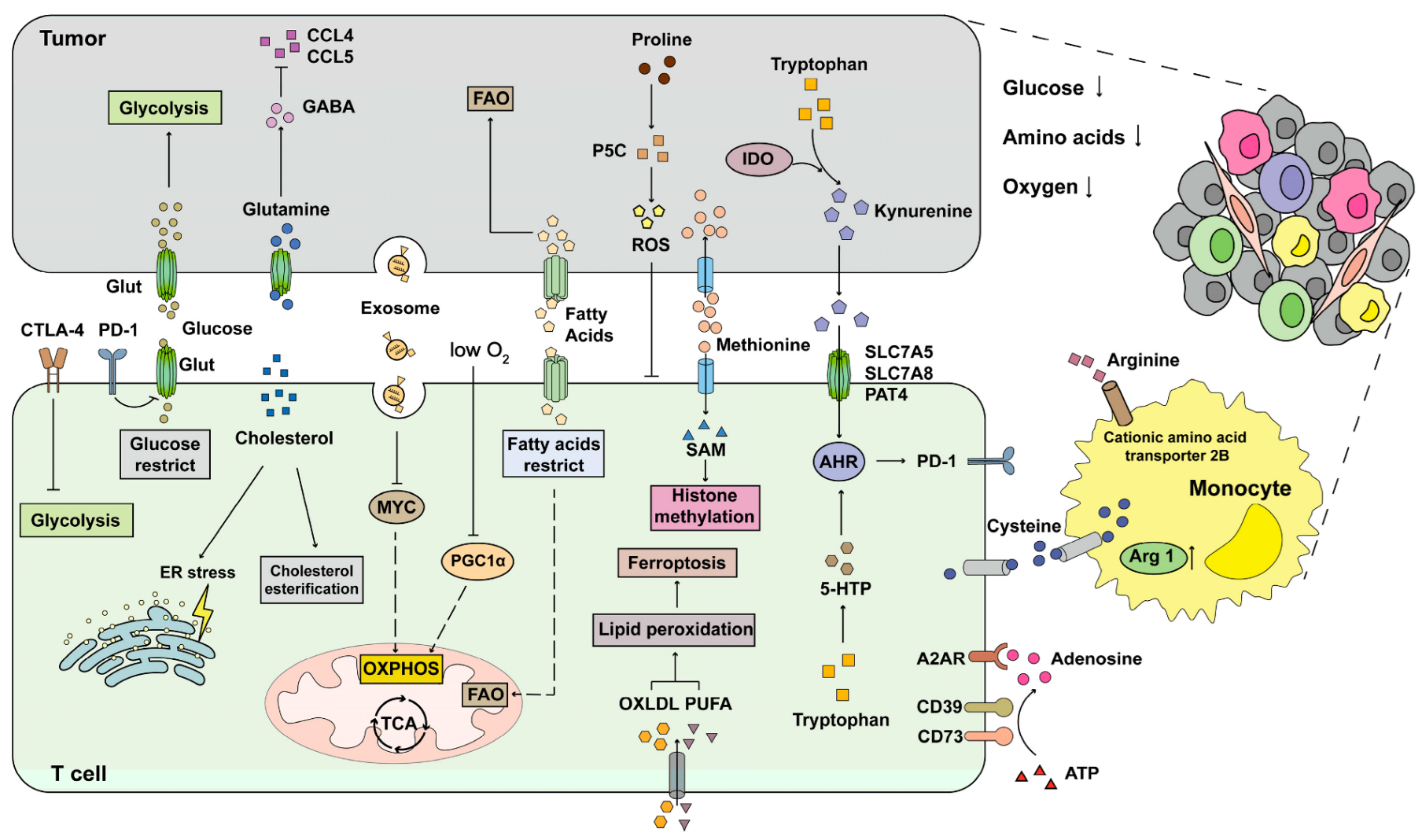
2. Metabolic Characteristics of T Cells during Anti-Tumor Response
2.1. Glucose Metabolism
2.2. Lipid Metabolism
2.3. Amino Acid Metabolism
2.4. Nucleotide Metabolism
2.5. Mitochondrial Metabolism
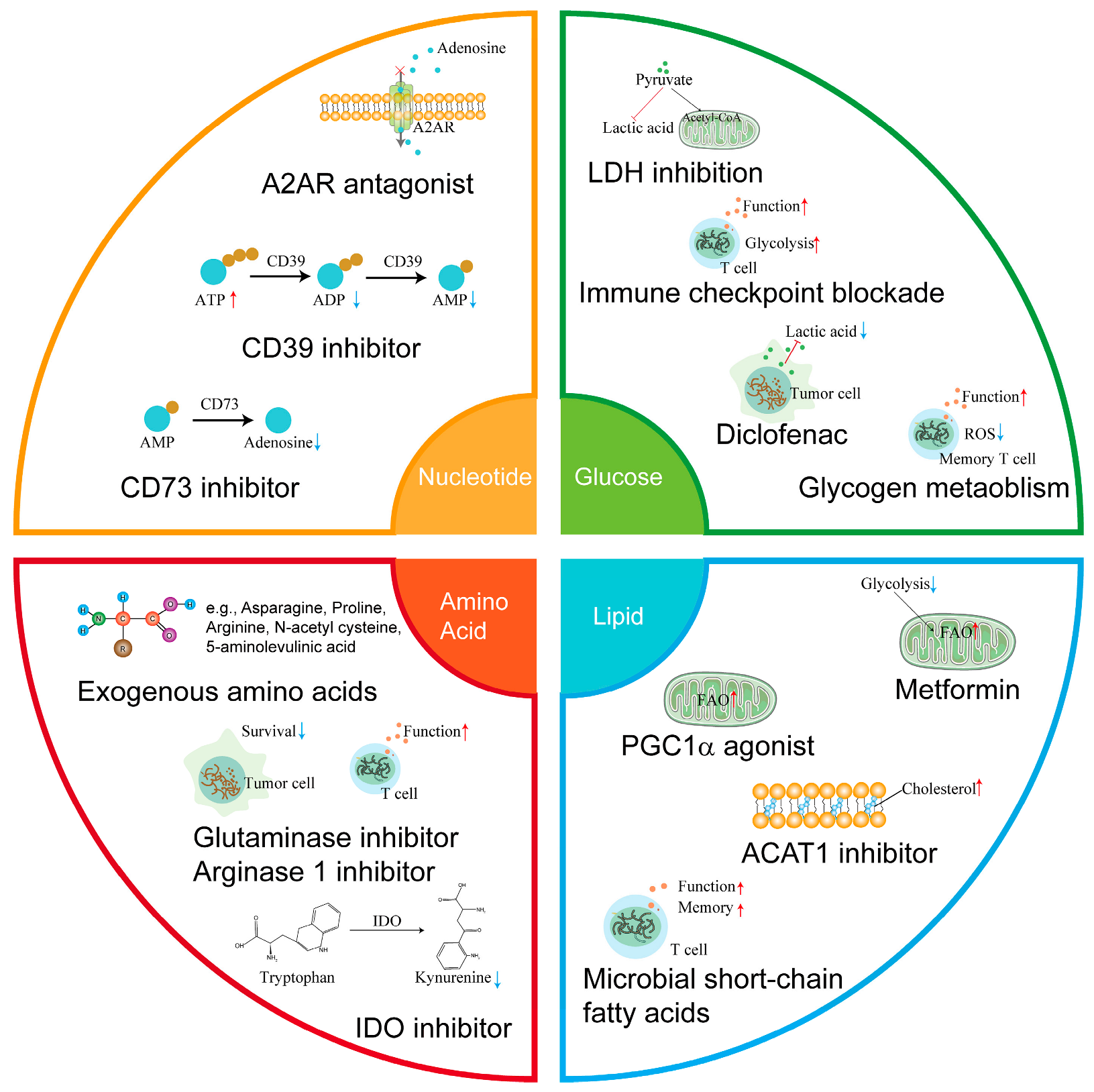
3. Targeting Metabolism to Improve T Cell Mediated Anti-Tumor Efficiency
3.1. Targeting Glucose Metabolism in the TME
3.2. Targeting Fatty Acid Metabolism
3.3. Targeting Amino Acid Metabolism
3.4. Targeting Adenosine Metabolism
4. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Smyth, M.J.; Teng, M.W. 2018 Nobel Prize in physiology or medicine. Clin. Transl. Immunol. 2018, 7, e1041. [Google Scholar] [CrossRef] [PubMed]
- Korman, A.J.; Garrett-Thomson, S.C.; Lonberg, N. The foundations of immune checkpoint blockade and the ipilimumab approval decennial. Nat. Rev. Drug Discov. 2021, 21, 509–528. [Google Scholar] [CrossRef]
- Dammeijer, F.; van Gulijk, M.; Mulder, E.E.; Lukkes, M.; Klaase, L.; van den Bosch, T.; van Nimwegen, M.; Lau, S.P.; Latupeirissa, K.; Schetters, S.; et al. The PD-1/PD-L1-Checkpoint Restrains T cell Immunity in Tumor-Draining Lymph Nodes. Cancer Cell 2020, 38, 685–700.e8. [Google Scholar] [CrossRef] [PubMed]
- Lozano, A.X.; Chaudhuri, A.A.; Nene, A.; Bacchiocchi, A.; Earland, N.; Vesely, M.D.; Usmani, A.; Turner, B.E.; Steen, C.B.; Luca, B.A.; et al. T cell characteristics associated with toxicity to immune checkpoint blockade in patients with melanoma. Nat. Med. 2022, 28, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Watson, R.A.; Tong, O.; Cooper, R.; Taylor, C.A.; Sharma, P.K.; de Los Aires, A.V.; Mahé, E.A.; Ruffieux, H.; Nassiri, I.; Middleton, M.R.; et al. Immune checkpoint blockade sensitivity and progression-free survival associates with baseline CD8 T cell clone size and cytotoxicity. Sci. Immunol. 2021, 6, eabj8825. [Google Scholar] [CrossRef] [PubMed]
- Budimir, N.; Thomas, G.D.; Dolina, J.S.; Salek-Ardakani, S. Reversing T-cell Exhaustion in Cancer: Lessons Learned from PD-1/PD-L1 Immune Checkpoint Blockade. Cancer Immunol. Res. 2022, 10, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef] [PubMed]
- McLane, L.M.; Abdel-Hakeem, M.S.; Wherry, E.J. CD8 T Cell Exhaustion during Chronic Viral Infection and Cancer. Annu. Rev. Immunol. 2019, 37, 457–495. [Google Scholar] [CrossRef] [PubMed]
- Reina-Campos, M.; Scharping, N.E.; Goldrath, A.W. CD8 T cell metabolism in infection and cancer. Nat. Rev. Immunol. 2021, 21, 718–738. [Google Scholar] [CrossRef] [PubMed]
- Franco, F.; Jaccard, A.; Romero, P.; Yu, Y.-R.; Ho, P.-C. Metabolic and epigenetic regulation of T-cell exhaustion. Nat. Metab. 2020, 2, 1001–1012. [Google Scholar] [CrossRef]
- Liu, X.; Hartman, C.L.; Li, L.; Albert, C.J.; Si, F.; Gao, A.; Huang, L.; Zhao, Y.; Lin, W.; Hsueh, E.C.; et al. Reprogramming lipid metabolism prevents effector T cell senescence and enhances tumor immunotherapy. Sci. Transl. Med. 2021, 13, eaaz6314. [Google Scholar] [CrossRef]
- Best, S.A.; Gubser, P.M.; Sethumadhavan, S.; Kersbergen, A.; Negrón Abril, Y.L.; Goldford, J.; Sellers, K.; Abeysekera, W.; Garnham, A.L.; McDonald, J.A.; et al. Glutaminase inhibition impairs CD8 T cell activation in STK11-/Lkb1-deficient lung cancer. Cell Metab. 2022, 34, 874–887.e6. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Yang, W.; Zhou, X.; Zöphel, D.; Soriano-Baguet, L.; Dolgener, D.; Carlein, C.; Hof, C.; Zhao, R.; Ye, S.; et al. High Glucose Enhances Cytotoxic T Lymphocyte-Mediated Cytotoxicity. Front. Immunol. 2021, 12, 689337. [Google Scholar] [CrossRef] [PubMed]
- Beckermann, K.E.; Dudzinski, S.O.; Rathmell, J.C. Dysfunctional T cell metabolism in the tumor microenvironment. Cytokine Growth Factor Rev. 2017, 35, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Tang, Y.Q.; Miao, H. Metabolism in tumor microenvironment: Implications for cancer immunotherapy. MedComm (2020) 2020, 1, 47–68. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Lei, Q.; Yang, L.; Qin, G.; Liu, S.; Wang, D.; Ping, Y.; Zhang, Y. Contradictory roles of lipid metabolism in immune response within the tumor microenvironment. J. Hematol. Oncol. 2021, 14, 187. [Google Scholar] [CrossRef]
- Heintzman, D.R.; Fisher, E.L.; Rathmell, J.C. Microenvironmental influences on T cell immunity in cancer and inflammation. Cell. Mol. Immunol. 2022, 19, 316–326. [Google Scholar] [CrossRef]
- Menk, A.V.; Scharping, N.E.; Moreci, R.S.; Zeng, X.; Guy, C.; Salvatore, S.; Bae, H.; Xie, J.; Young, H.A.; Wendell, S.G.; et al. Early TCR Signaling Induces Rapid Aerobic Glycolysis Enabling Distinct Acute T Cell Effector Functions. Cell Rep. 2018, 22, 1509–1521. [Google Scholar] [CrossRef]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef]
- Zhao, E.; Maj, T.; Kryczek, I.; Li, W.; Wu, K.; Zhao, L.; Wei, S.; Crespo, J.; Wan, S.; Vatan, L.; et al. Cancer mediates effector T cell dysfunction by targeting microRNAs and EZH2 via glycolysis restriction. Nat. Immunol. 2016, 17, 95–103. [Google Scholar] [CrossRef]
- Ho, P.C.; Bihuniak, J.D.; Macintyre, A.N.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.C.; Cui, G.; Micevic, G.; Perales, J.C.; et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 2015, 162, 1217–1228. [Google Scholar] [CrossRef] [PubMed]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015, 6, 6692. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.K.; Lee, D.Y.; Lee, D.G.; Kim, Y.H.; Kim, S.H.; Oh, H.S.; Han, C.; Kwon, B.S. 4-1BB signaling activates glucose and fatty acid metabolism to enhance CD8(+) T cell proliferation. Cell. Mol. Immunol. 2017, 14, 748–757. [Google Scholar] [CrossRef]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef]
- Ringel, A.E.; Drijvers, J.M.; Baker, G.J.; Catozzi, A.; Garcia-Canaveras, J.C.; Gassaway, B.M.; Miller, B.C.; Juneja, V.R.; Nguyen, T.H.; Joshi, S.; et al. Obesity Shapes Metabolism in the Tumor Microenvironment to Suppress Anti-Tumor Immunity. Cell 2020, 183, 1848–1866 e1826. [Google Scholar] [CrossRef]
- Zhang, Y.; Kurupati, R.; Liu, L.; Zhou, X.Y.; Zhang, G.; Hudaihed, A.; Filisio, F.; Giles-Davis, W.; Xu, X.; Karakousis, G.C.; et al. Enhancing CD8 T Cell Fatty Acid Catabolism within a Metabolically Challenging Tumor Microenvironment Increases the Efficacy of Melanoma Immunotherapy. Cancer Cell 2017, 32, 377–391.e9. [Google Scholar] [CrossRef]
- Herber, D.L.; Cao, W.; Nefedova, Y.; Novitskiy, S.V.; Nagaraj, S.; Tyurin, V.A.; Corzo, A.; Cho, H.-I.; Celis, E.; Lennox, B.; et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat. Med. 2010, 16, 880–886. [Google Scholar] [CrossRef]
- Ma, X.; Xiao, L.; Liu, L.; Ye, L.; Su, P.; Bi, E.; Wang, Q.; Yang, M.; Qian, J.; Yi, Q. CD36-mediated ferroptosis dampens intratumoral CD8(+) T cell effector function and impairs their antitumor ability. Cell Metab. 2021, 33, 1001–1012.e1005. [Google Scholar] [CrossRef]
- Xu, S.; Chaudhary, O.; Rodriguez-Morales, P.; Sun, X.; Chen, D.; Zappasodi, R.; Xu, Z.; Pinto, A.F.M.; Williams, A.; Schulze, I.; et al. Uptake of oxidized lipids by the scavenger receptor CD36 promotes lipid peroxidation and dysfunction in CD8(+) T cells in tumors. Immunity 2021, 54, 1561–1577.e1567. [Google Scholar] [CrossRef]
- Chowdhury, P.S.; Chamoto, K.; Kumar, A.; Honjo, T. PPAR-Induced Fatty Acid Oxidation in T Cells Increases the Number of Tumor-Reactive CD8(+) T Cells and Facilitates Anti-PD-1 Therapy. Cancer Immunol. Res. 2018, 6, 1375–1387. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yue, C.; Herrmann, A.; Song, J.; Egelston, C.; Wang, T.; Zhang, Z.; Li, W.; Lee, H.; Aftabizadeh, M.; et al. STAT3 Activation-Induced Fatty Acid Oxidation in CD8(+) T Effector Cells Is Critical for Obesity-Promoted Breast Tumor Growth. Cell Metab. 2020, 31, 148–161.e145. [Google Scholar] [CrossRef] [PubMed]
- Buck, M.D.; O’Sullivan, D.; Klein Geltink, R.I.; Curtis, J.D.; Chang, C.H.; Sanin, D.E.; Qiu, J.; Kretz, O.; Braas, D.; van der Windt, G.J.; et al. Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming. Cell 2016, 166, 63–76. [Google Scholar] [CrossRef]
- Lin, R.; Zhang, H.; Yuan, Y.; He, Q.; Zhou, J.; Li, S.; Sun, Y.; Li, D.Y.; Qiu, H.B.; Wang, W.; et al. Fatty Acid Oxidation Controls CD8(+) Tissue-Resident Memory T-cell Survival in Gastric Adenocarcinoma. Cancer Immunol. Res. 2020, 8, 479–492. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Bai, Y.; Xiong, Y.; Zhang, J.; Chen, S.; Zheng, X.; Meng, X.; Li, L.; Wang, J.; Xu, C.; et al. Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism. Nature 2016, 531, 651–655. [Google Scholar] [CrossRef]
- Ma, X.; Bi, E.; Huang, C.; Lu, Y.; Xue, G.; Guo, X.; Wang, A.; Yang, M.; Qian, J.; Dong, C.; et al. Cholesterol negatively regulates IL-9-producing CD8(+) T cell differentiation and antitumor activity. J. Exp. Med. 2018, 215, 1555–1569. [Google Scholar] [CrossRef]
- Ma, X.; Bi, E.; Lu, Y.; Su, P.; Huang, C.; Liu, L.; Wang, Q.; Yang, M.; Kalady, M.F.; Qian, J.; et al. Cholesterol Induces CD8(+) T Cell Exhaustion in the Tumor Microenvironment. Cell Metab. 2019, 30, 143–156.e145. [Google Scholar] [CrossRef]
- Wu, D.; Zhu, Y. Role of kynurenine in promoting the generation of exhausted CD8(+) T cells in colorectal cancer. Am. J. Transl. Res. 2021, 13, 1535–1547. [Google Scholar]
- Amobi-McCloud, A.; Muthuswamy, R.; Battaglia, S.; Yu, H.; Liu, T.; Wang, J.; Putluri, V.; Singh, P.K.; Qian, F.; Huang, R.Y.; et al. IDO1 Expression in Ovarian Cancer Induces PD-1 in T Cells via Aryl Hydrocarbon Receptor Activation. Front. Immunol. 2021, 12, 678999. [Google Scholar] [CrossRef]
- Greene, L.I.; Bruno, T.C.; Christenson, J.L.; D’Alessandro, A.; Culp-Hill, R.; Torkko, K.; Borges, V.F.; Slansky, J.E.; Richer, J.K. A Role for Tryptophan-2,3-dioxygenase in CD8 T-cell Suppression and Evidence of Tryptophan Catabolism in Breast Cancer Patient Plasma. Mol. Cancer Res. 2019, 17, 131–139. [Google Scholar] [CrossRef]
- Sinclair, L.V.; Neyens, D.; Ramsay, G.; Taylor, P.M.; Cantrell, D.A. Single cell analysis of kynurenine and System L amino acid transport in T cells. Nat. Commun. 2018, 9, 1981. [Google Scholar] [CrossRef] [PubMed]
- Ninomiya, S.; Narala, N.; Huye, L.; Yagyu, S.; Savoldo, B.; Dotti, G.; Heslop, H.E.; Brenner, M.K.; Rooney, C.M.; Ramos, C.A. Tumor indoleamine 2,3-dioxygenase (IDO) inhibits CD19-CAR T cells and is downregulated by lymphodepleting drugs. Blood 2015, 125, 3905–3916. [Google Scholar] [CrossRef]
- Liu, Y.; Liang, X.; Dong, W.; Fang, Y.; Lv, J.; Zhang, T.; Fiskesund, R.; Xie, J.; Liu, J.; Yin, X.; et al. Tumor-Repopulating Cells Induce PD-1 Expression in CD8(+) T Cells by Transferring Kynurenine and AhR Activation. Cancer Cell 2018, 33, 480–494.e487. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, N.; Zhou, L.; Wang, J.; Zhou, Y.; Zhang, T.; Fang, Y.; Deng, J.; Gao, Y.; Liang, X.; et al. IL-2 regulates tumor-reactive CD8(+) T cell exhaustion by activating the aryl hydrocarbon receptor. Nat. Immunol. 2021, 22, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Dillon, C.P.; Shi, L.Z.; Milasta, S.; Carter, R.; Finkelstein, D.; McCormick, L.L.; Fitzgerald, P.; Chi, H.; Munger, J.; et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 2011, 35, 871–882. [Google Scholar] [CrossRef]
- Toriyama, K.; Kuwahara, M.; Kondoh, H.; Mikawa, T.; Takemori, N.; Konishi, A.; Yorozuya, T.; Yamada, T.; Soga, T.; Shiraishi, A.; et al. T cell-specific deletion of Pgam1 reveals a critical role for glycolysis in T cell responses. Commun. Biol. 2020, 3, 394. [Google Scholar] [CrossRef] [PubMed]
- Bailis, W.; Shyer, J.A.; Zhao, J.; Canaveras, J.C.G.; Al Khazal, F.J.; Qu, R.; Steach, H.R.; Bielecki, P.; Khan, O.; Jackson, R.; et al. Distinct modes of mitochondrial metabolism uncouple T cell differentiation and function. Nature 2019, 571, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Ma, E.H.; Verway, M.J.; Johnson, R.M.; Roy, D.G.; Steadman, M.; Hayes, S.; Williams, K.S.; Sheldon, R.D.; Samborska, B.; Kosinski, P.A.; et al. Metabolic Profiling Using Stable Isotope Tracing Reveals Distinct Patterns of Glucose Utilization by Physiologically Activated CD8(+) T Cells. Immunity 2019, 51, 856–870.e855. [Google Scholar] [CrossRef] [PubMed]
- Edwards, D.N.; Ngwa, V.M.; Raybuck, A.L.; Wang, S.; Hwang, Y.; Kim, L.C.; Cho, S.H.; Paik, Y.; Wang, Q.; Zhang, S.; et al. Selective glutamine metabolism inhibition in tumor cells improves antitumor T lymphocyte activity in triple-negative breast cancer. J. Clin. Investig. 2021, 131, e140100. [Google Scholar] [CrossRef] [PubMed]
- Leone, R.D.; Zhao, L.; Englert, J.M.; Sun, I.M.; Oh, M.H.; Sun, I.H.; Arwood, M.L.; Bettencourt, I.A.; Patel, C.H.; Wen, J.; et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 2019, 366, 1013–1021. [Google Scholar] [CrossRef]
- Swamy, M.; Pathak, S.; Grzes, K.M.; Damerow, S.; Sinclair, L.V.; van Aalten, D.M.; Cantrell, D.A. Glucose and glutamine fuel protein O-GlcNAcylation to control T cell self-renewal and malignancy. Nat. Immunol. 2016, 17, 712–720. [Google Scholar] [CrossRef]
- Huang, D.; Wang, Y.; Thompson, J.W.; Yin, T.; Alexander, P.B.; Qin, D.; Mudgal, P.; Wu, H.; Liang, Y.; Tan, L.; et al. Cancer-cell-derived GABA promotes beta-catenin-mediated tumour growth and immunosuppression. Nat. Cell Biol. 2022, 24, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Sosnowska, A.; Chlebowska-Tuz, J.; Matryba, P.; Pilch, Z.; Greig, A.; Wolny, A.; Grzywa, T.M.; Rydzynska, Z.; Sokolowska, O.; Rygiel, T.P.; et al. Inhibition of arginase modulates T-cell response in the tumor microenvironment of lung carcinoma. Oncoimmunology 2021, 10, 1956143. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, P.C.; Quiceno, D.G.; Zabaleta, J.; Ortiz, B.; Zea, A.H.; Piazuelo, M.B.; Delgado, A.; Correa, P.; Brayer, J.; Sotomayor, E.M.; et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004, 64, 5839–5849. [Google Scholar] [CrossRef]
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M.; et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 2016, 167, 829–842.e813. [Google Scholar] [CrossRef] [PubMed]
- Fultang, L.; Booth, S.; Yogev, O.; Martins da Costa, B.; Tubb, V.; Panetti, S.; Stavrou, V.; Scarpa, U.; Jankevics, A.; Lloyd, G.; et al. Metabolic engineering against the arginine microenvironment enhances CAR-T cell proliferation and therapeutic activity. Blood 2020, 136, 1155–1160. [Google Scholar] [CrossRef] [PubMed]
- Crump, N.T.; Hadjinicolaou, A.V.; Xia, M.; Walsby-Tickle, J.; Gileadi, U.; Chen, J.L.; Setshedi, M.; Olsen, L.R.; Lau, I.J.; Godfrey, L.; et al. Chromatin accessibility governs the differential response of cancer and T cells to arginine starvation. Cell Rep. 2021, 35, 109101. [Google Scholar] [CrossRef]
- Marti i Lindez, A.A.; Dunand-Sauthier, I.; Conti, M.; Gobet, F.; Nunez, N.; Hannich, J.T.; Riezman, H.; Geiger, R.; Piersigilli, A.; Hahn, K.; et al. Mitochondrial arginase-2 is a cellautonomous regulator of CD8+ T cell function and antitumor efficacy. JCI Insight 2019, 4, e132975. [Google Scholar] [CrossRef]
- Srivastava, M.K.; Sinha, P.; Clements, V.K.; Rodriguez, P.; Ostrand-Rosenberg, S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 2010, 70, 68–77. [Google Scholar] [CrossRef]
- Yan, Y.; Chang, L.; Tian, H.; Wang, L.; Zhang, Y.; Yang, T.; Li, G.; Hu, W.; Shah, K.; Chen, G.; et al. 1-Pyrroline-5-carboxylate released by prostate Cancer cell inhibit T cell proliferation and function by targeting SHP1/cytochrome c oxidoreductase/ROS Axis. J. Immunother. Cancer 2018, 6, 148. [Google Scholar] [CrossRef] [PubMed]
- Lian, G.; Gnanaprakasam, J.R.; Wang, T.; Wu, R.; Chen, X.; Liu, L.; Shen, Y.; Yang, M.; Yang, J.; Chen, Y.; et al. Glutathione de novo synthesis but not recycling process coordinates with glutamine catabolism to control redox homeostasis and directs murine T cell differentiation. elife 2018, 7, e36158. [Google Scholar] [CrossRef]
- Siska, P.J.; Kim, B.; Ji, X.; Hoeksema, M.D.; Massion, P.P.; Beckermann, K.E.; Wu, J.; Chi, J.T.; Hong, J.; Rathmell, J.C. Fluorescence-based measurement of cystine uptake through xCT shows requirement for ROS detoxification in activated lymphocytes. J. Immunol. Methods 2016, 438, 51–58. [Google Scholar] [CrossRef]
- Bian, Y.; Li, W.; Kremer, D.M.; Sajjakulnukit, P.; Li, S.; Crespo, J.; Nwosu, Z.C.; Zhang, L.; Czerwonka, A.; Pawlowska, A.; et al. Cancer SLC43A2 alters T cell methionine metabolism and histone methylation. Nature 2020, 585, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Ron-Harel, N.; Santos, D.; Ghergurovich, J.M.; Sage, P.T.; Reddy, A.; Lovitch, S.B.; Dephoure, N.; Satterstrom, F.K.; Sheffer, M.; Spinelli, J.B.; et al. Mitochondrial Biogenesis and Proteome Remodeling Promote One-Carbon Metabolism for T Cell Activation. Cell Metab. 2016, 24, 104–117. [Google Scholar] [CrossRef]
- Ma, E.H.; Bantug, G.; Griss, T.; Condotta, S.; Johnson, R.M.; Samborska, B.; Mainolfi, N.; Suri, V.; Guak, H.; Balmer, M.L.; et al. Serine Is an Essential Metabolite for Effector T Cell Expansion. Cell Metab. 2017, 25, 345–357. [Google Scholar] [CrossRef]
- Peeters, M.J.W.; Aehnlich, P.; Pizzella, A.; Molgaard, K.; Seremet, T.; Met, O.; Rasmussen, L.J.; Thor Straten, P.; Desler, C. Mitochondrial-Linked De Novo Pyrimidine Biosynthesis Dictates Human T-Cell Proliferation but Not Expression of Effector Molecules. Front. Immunol. 2021, 12, 718863. [Google Scholar] [CrossRef]
- Ledderose, C.; Liu, K.; Kondo, Y.; Slubowski, C.J.; Dertnig, T.; Denicolo, S.; Arbab, M.; Hubner, J.; Konrad, K.; Fakhari, M.; et al. Purinergic P2X4 receptors and mitochondrial ATP production regulate T cell migration. J. Clin. Investig. 2018, 128, 3583–3594. [Google Scholar] [CrossRef]
- Borges da Silva, H.; Peng, C.; Wang, H.; Wanhainen, K.M.; Ma, C.; Lopez, S.; Khoruts, A.; Zhang, N.; Jameson, S.C. Sensing of ATP via the Purinergic Receptor P2RX7 Promotes CD8(+) Trm Cell Generation by Enhancing Their Sensitivity to the Cytokine TGF-beta. Immunity 2020, 53, 158–171.e156. [Google Scholar] [CrossRef] [PubMed]
- Borges da Silva, H.; Beura, L.K.; Wang, H.; Hanse, E.A.; Gore, R.; Scott, M.C.; Walsh, D.A.; Block, K.E.; Fonseca, R.; Yan, Y.; et al. The purinergic receptor P2RX7 directs metabolic fitness of long-lived memory CD8(+) T cells. Nature 2018, 559, 264–268. [Google Scholar] [CrossRef]
- Vardam-Kaur, T.; van Dijk, S.; Peng, C.; Wanhainen, K.M.; Jameson, S.C.; Borges da Silva, H. The Extracellular ATP Receptor P2RX7 Imprints a Promemory Transcriptional Signature in Effector CD8(+) T Cells. J. Immunol. 2022, 208, 1686–1699. [Google Scholar] [CrossRef]
- Stark, R.; Wesselink, T.H.; Behr, F.M.; Kragten, N.A.M.; Arens, R.; Koch-Nolte, F.; van Gisbergen, K.; van Lier, R.A.W. TRM maintenance is regulated by tissue damage via P2RX7. Sci. Immunol. 2018, 3, eaau1022. [Google Scholar] [CrossRef]
- Canale, F.P.; Ramello, M.C.; Nunez, N.; Araujo Furlan, C.L.; Bossio, S.N.; Gorosito Serran, M.; Tosello Boari, J.; Del Castillo, A.; Ledesma, M.; Sedlik, C.; et al. CD39 Expression Defines Cell Exhaustion in Tumor-Infiltrating CD8(+) T Cells. Cancer Res. 2018, 78, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Schneider, E.; Winzer, R.; Rissiek, A.; Ricklefs, I.; Meyer-Schwesinger, C.; Ricklefs, F.L.; Bauche, A.; Behrends, J.; Reimer, R.; Brenna, S.; et al. CD73-mediated adenosine production by CD8 T cell-derived extracellular vesicles constitutes an intrinsic mechanism of immune suppression. Nat. Commun. 2021, 12, 5911. [Google Scholar] [CrossRef]
- Vigano, S.; Alatzoglou, D.; Irving, M.; Menetrier-Caux, C.; Caux, C.; Romero, P.; Coukos, G. Targeting Adenosine in Cancer Immunotherapy to Enhance T-Cell Function. Front. Immunol. 2019, 10, 925. [Google Scholar] [CrossRef]
- Mastelic-Gavillet, B.; Navarro Rodrigo, B.; Decombaz, L.; Wang, H.; Ercolano, G.; Ahmed, R.; Lozano, L.E.; Ianaro, A.; Derre, L.; Valerio, M.; et al. Adenosine mediates functional and metabolic suppression of peripheral and tumor-infiltrating CD8(+) T cells. J. Immunother. Cancer 2019, 7, 257. [Google Scholar] [CrossRef]
- Shinohara, Y.; Tsukimoto, M. Guanine and inosine nucleotides/nucleosides suppress murine T cell activation. Biochem. Biophys. Res. Commun. 2018, 498, 764–768. [Google Scholar] [CrossRef]
- Weiler, M.; Schmetzer, H.; Braeu, M.; Buhmann, R. Inhibitory effect of extracellular purine nucleotide and nucleoside concentrations on T cell proliferation. Exp. Cell Res. 2016, 349, 1–14. [Google Scholar] [CrossRef] [PubMed]
- van der Windt, G.J.; Everts, B.; Chang, C.H.; Curtis, J.D.; Freitas, T.C.; Amiel, E.; Pearce, E.J.; Pearce, E.L. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity 2012, 36, 68–78. [Google Scholar] [CrossRef] [PubMed]
- van der Windt, G.J.; O’Sullivan, D.; Everts, B.; Huang, S.C.; Buck, M.D.; Curtis, J.D.; Chang, C.H.; Smith, A.M.; Ai, T.; Faubert, B.; et al. CD8 memory T cells have a bioenergetic advantage that underlies their rapid recall ability. Proc. Natl. Acad. Sci. USA 2013, 110, 14336–14341. [Google Scholar] [CrossRef]
- Wenes, M.; Jaccard, A.; Wyss, T.; Maldonado-Perez, N.; Teoh, S.T.; Lepez, A.; Renaud, F.; Franco, F.; Waridel, P.; Yacoub Maroun, C.; et al. The mitochondrial pyruvate carrier regulates memory T cell differentiation and antitumor function. Cell Metab. 2022, 34, 731–746.e739. [Google Scholar] [CrossRef]
- Siska, P.J.; Beckermann, K.E.; Mason, F.M.; Andrejeva, G.; Greenplate, A.R.; Sendor, A.B.; Chiang, Y.J.; Corona, A.L.; Gemta, L.F.; Vincent, B.G.; et al. Mitochondrial dysregulation and glycolytic insufficiency functionally impair CD8 T cells infiltrating human renal cell carcinoma. JCI Insight 2017, 2, e93411. [Google Scholar] [CrossRef]
- Vardhana, S.A.; Hwee, M.A.; Berisa, M.; Wells, D.K.; Yost, K.E.; King, B.; Smith, M.; Herrera, P.S.; Chang, H.Y.; Satpathy, A.T.; et al. Impaired mitochondrial oxidative phosphorylation limits the self-renewal of T cells exposed to persistent antigen. Nat. Immunol. 2020, 21, 1022–1033. [Google Scholar] [CrossRef]
- Yu, Y.R.; Imrichova, H.; Wang, H.; Chao, T.; Xiao, Z.; Gao, M.; Rincon-Restrepo, M.; Franco, F.; Genolet, R.; Cheng, W.C.; et al. Disturbed mitochondrial dynamics in CD8(+) TILs reinforce T cell exhaustion. Nat. Immunol. 2020, 21, 1540–1551. [Google Scholar] [CrossRef]
- Scharping, N.E.; Menk, A.V.; Moreci, R.S.; Whetstone, R.D.; Dadey, R.E.; Watkins, S.C.; Ferris, R.L.; Delgoffe, G.M. The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction. Immunity 2016, 45, 374–388. [Google Scholar] [CrossRef]
- Scharping, N.E.; Rivadeneira, D.B.; Menk, A.V.; Vignali, P.D.A.; Ford, B.R.; Rittenhouse, N.L.; Peralta, R.; Wang, Y.; Wang, Y.; DePeaux, K.; et al. Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat. Immunol. 2021, 22, 205–215. [Google Scholar] [CrossRef]
- Liu, Y.N.; Yang, J.F.; Huang, D.J.; Ni, H.H.; Zhang, C.X.; Zhang, L.; He, J.; Gu, J.M.; Chen, H.X.; Mai, H.Q.; et al. Hypoxia Induces Mitochondrial Defect That Promotes T Cell Exhaustion in Tumor Microenvironment Through MYC-Regulated Pathways. Front. Immunol. 2020, 11, 1906. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, F.; Wang, L.; Qiu, S.; Yao, Y.; Yan, C.; Xiong, X.; Chen, X.; Ji, Q.; Cao, J.; et al. NAD(+) supplement potentiates tumor-killing function by rescuing defective TUB-mediated NAMPT transcription in tumor-infiltrated T cells. Cell Rep. 2021, 36, 109516. [Google Scholar] [CrossRef]
- Najjar, Y.G.; Menk, A.V.; Sander, C.; Rao, U.; Karunamurthy, A.; Bhatia, R.; Zhai, S.; Kirkwood, J.M.; Delgoffe, G.M. Tumor cell oxidative metabolism as a barrier to PD-1 blockade immunotherapy in melanoma. JCI Insight 2019, 4, e124989. [Google Scholar] [CrossRef]
- Sukumar, M.; Liu, J.; Ji, Y.; Subramanian, M.; Crompton, J.G.; Yu, Z.; Roychoudhuri, R.; Palmer, D.C.; Muranski, P.; Karoly, E.D.; et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J. Clin. Investig. 2013, 123, 4479–4488. [Google Scholar] [CrossRef]
- Wang, Z.-H.; Peng, W.-B.; Zhang, P.; Yang, X.-P.; Zhou, Q. Lactate in the tumour microenvironment: From immune modulation to therapy. eBioMedicine 2021, 73, 103627. [Google Scholar] [CrossRef]
- Hermans, D.; Gautam, S.; Garcia-Canaveras, J.C.; Gromer, D.; Mitra, S.; Spolski, R.; Li, P.; Christensen, S.; Nguyen, R.; Lin, J.X.; et al. Lactate dehydrogenase inhibition synergizes with IL-21 to promote CD8(+) T cell stemness and antitumor immunity. Proc. Natl. Acad. Sci. USA 2020, 117, 6047–6055. [Google Scholar] [CrossRef]
- Renner, K.; Bruss, C.; Schnell, A.; Koehl, G.; Becker, H.M.; Fante, M.; Menevse, A.N.; Kauer, N.; Blazquez, R.; Hacker, L.; et al. Restricting Glycolysis Preserves T Cell Effector Functions and Augments Checkpoint Therapy. Cell Rep. 2019, 29, 135–150.e139. [Google Scholar] [CrossRef]
- Zhang, H.; Ma, J.; Tang, K.; Huang, B. Beyond energy storage: Roles of glycogen metabolism in health and disease. FEBS J. 2021, 288, 3772–3783. [Google Scholar] [CrossRef]
- Ma, J.; Wei, K.; Liu, J.; Tang, K.; Zhang, H.; Zhu, L.; Chen, J.; Li, F.; Xu, P.; Chen, J.; et al. Glycogen metabolism regulates macrophage-mediated acute inflammatory responses. Nat. Commun. 2020, 11, 1769. [Google Scholar] [CrossRef]
- Ma, R.; Ji, T.; Zhang, H.; Dong, W.; Chen, X.; Xu, P.; Chen, D.; Liang, X.; Yin, X.; Liu, Y.; et al. A Pck1-directed glycogen metabolic program regulates formation and maintenance of memory CD8(+) T cells. Nat. Cell Biol. 2018, 20, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, J.; Yang, Z.; Zeng, L.; Wei, K.; Zhu, L.; Tang, L.; Wang, D.; Zhou, Y.; Lv, J.; et al. TCR activation directly stimulates PYGB-dependent glycogenolysis to fuel the early recall response in CD8(+) memory T cells. Mol. Cell 2022, 82, 3077–3088.e6. [Google Scholar] [CrossRef]
- Zhang, H.; Tang, K.; Ma, J.; Zhou, L.; Liu, J.; Zeng, L.; Zhu, L.; Xu, P.; Chen, J.; Wei, K.; et al. Ketogenesis-generated beta-hydroxybutyrate is an epigenetic regulator of CD8(+) T-cell memory development. Nat. Cell Biol. 2020, 22, 18–25. [Google Scholar] [CrossRef]
- Luo, L.; Li, X.; Zhang, J.; Zhu, C.; Jiang, M.; Luo, Z.; Qin, B.; Wang, Y.; Chen, B.; Du, Y.; et al. Enhanced immune memory through a constant photothermal-metabolism regulation for cancer prevention and treatment. Biomaterials 2021, 270, 120678. [Google Scholar] [CrossRef] [PubMed]
- Hao, M.; Hou, S.; Li, W.; Li, K.; Xue, L.; Hu, Q.; Zhu, L.; Chen, Y.; Sun, H.; Ju, C.; et al. Combination of metabolic intervention and T cell therapy enhances solid tumor immunotherapy. Sci. Transl. Med. 2020, 12, eaaz6667. [Google Scholar] [CrossRef]
- Li, M.; Yang, Y.; Wei, J.; Cun, X.; Lu, Z.; Qiu, Y.; Zhang, Z.; He, Q. Enhanced chemo-immunotherapy against melanoma by inhibition of cholesterol esterification in CD8(+) T cells. Nanomedicine 2018, 14, 2541–2550. [Google Scholar] [CrossRef]
- Luu, M.; Riester, Z.; Baldrich, A.; Reichardt, N.; Yuille, S.; Busetti, A.; Klein, M.; Wempe, A.; Leister, H.; Raifer, H.; et al. Microbial short-chain fatty acids modulate CD8(+) T cell responses and improve adoptive immunotherapy for cancer. Nat. Commun. 2021, 12, 4077. [Google Scholar] [CrossRef]
- Bachem, A.; Makhlouf, C.; Binger, K.J.; de Souza, D.P.; Tull, D.; Hochheiser, K.; Whitney, P.G.; Fernandez-Ruiz, D.; Dahling, S.; Kastenmuller, W.; et al. Microbiota-Derived Short-Chain Fatty Acids Promote the Memory Potential of Antigen-Activated CD8(+) T Cells. Immunity 2019, 51, 285–297.e285. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Que, W.; Hirano, H.; Wang, Z.; Nozawa, N.; Ishii, T.; Ishizuka, M.; Ito, H.; Takahashi, K.; Nakajima, M.; et al. 5-Aminolevulinic acid/sodium ferrous citrate enhanced the antitumor effects of programmed cell death-ligand 1 blockade by regulation of exhausted T cell metabolism in a melanoma model. Cancer Sci. 2021, 112, 2652–2663. [Google Scholar] [CrossRef]
- Scheffel, M.J.; Scurti, G.; Wyatt, M.M.; Garrett-Mayer, E.; Paulos, C.M.; Nishimura, M.I.; Voelkel-Johnson, C. N-acetyl cysteine protects anti-melanoma cytotoxic T cells from exhaustion induced by rapid expansion via the downmodulation of Foxo1 in an Akt-dependent manner. Cancer Immunol. Immunother. 2018, 67, 691–702. [Google Scholar] [CrossRef]
- Wu, J.; Li, G.; Li, L.; Li, D.; Dong, Z.; Jiang, P. Asparagine enhances LCK signalling to potentiate CD8(+) T-cell activation and anti-tumour responses. Nat. Cell Biol. 2021, 23, 75–86. [Google Scholar] [CrossRef]
- Ye, L.; Park, J.J.; Peng, L.; Yang, Q.; Chow, R.D.; Dong, M.B.; Lam, S.Z.; Guo, J.; Tang, E.; Zhang, Y.; et al. A genome-scale gain-of-function CRISPR screen in CD8 T cells identifies proline metabolism as a means to enhance CAR-T therapy. Cell Metab. 2022, 34, 595–614.e514. [Google Scholar] [CrossRef] [PubMed]
- Satoh, Y.; Kotani, H.; Iida, Y.; Taniura, T.; Notsu, Y.; Harada, M. Supplementation of l-arginine boosts the therapeutic efficacy of anticancer chemoimmunotherapy. Cancer Sci. 2020, 111, 2248–2258. [Google Scholar] [CrossRef]
- Varghese, S.; Pramanik, S.; Williams, L.J.; Hodges, H.R.; Hudgens, C.W.; Fischer, G.M.; Luo, C.K.; Knighton, B.; Tan, L.; Lorenzi, P.L.; et al. The Glutaminase Inhibitor CB-839 (Telaglenastat) Enhances the Antimelanoma Activity of T-Cell-Mediated Immunotherapies. Mol. Cancer Ther. 2021, 20, 500–511. [Google Scholar] [CrossRef]
- Zhao, Y.; Feng, X.; Chen, Y.; Selfridge, J.E.; Gorityala, S.; Du, Z.; Wang, J.M.; Hao, Y.; Cioffi, G.; Conlon, R.A.; et al. 5-Fluorouracil Enhances the Antitumor Activity of the Glutaminase Inhibitor CB-839 against PIK3CA-Mutant Colorectal Cancers. Cancer Res. 2020, 80, 4815–4827. [Google Scholar] [CrossRef]
- Riess, J.W.; Frankel, P.; Shackelford, D.; Dunphy, M.; Badawi, R.D.; Nardo, L.; Cherry, S.R.; Lanza, I.; Reid, J.; Gonsalves, W.I.; et al. Phase 1 Trial of MLN0128 (Sapanisertib) and CB-839 HCl (Telaglenastat) in Patients with Advanced NSCLC (NCI 10327): Rationale and Study Design. Clin. Lung Cancer 2021, 22, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Gross, M.I.; Demo, S.D.; Dennison, J.B.; Chen, L.; Chernov-Rogan, T.; Goyal, B.; Janes, J.R.; Laidig, G.J.; Lewis, E.R.; Li, J.; et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol. Cancer Ther. 2014, 13, 890–901. [Google Scholar] [CrossRef]
- Steggerda, S.M.; Bennett, M.K.; Chen, J.; Emberley, E.; Huang, T.; Janes, J.R.; Li, W.; MacKinnon, A.L.; Makkouk, A.; Marguier, G.; et al. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J. Immunother. Cancer 2017, 5, 101. [Google Scholar] [CrossRef]
- Chang, K.-Y.; Chiang, N.-J.; Wu, S.-Y.; Yen, C.-J.; Chen, S.-H.; Yeh, Y.-M.; Li, C.-F.; Feng, X.; Wu, K.; Johnston, A.; et al. Phase 1b study of pegylated arginine deiminase (ADI-PEG 20) plus Pembrolizumab in advanced solid cancers. Oncoimmunology 2021, 10, 1943253. [Google Scholar] [CrossRef]
- Harding, J.J.; Do, R.K.; Dika, I.E.; Hollywood, E.; Uhlitskykh, K.; Valentino, E.; Wan, P.; Hamilton, C.; Feng, X.; Johnston, A.; et al. A phase 1 study of ADI-PEG 20 and modified FOLFOX6 in patients with advanced hepatocellular carcinoma and other gastrointestinal malignancies. Cancer Chemother. Pharmacol. 2018, 82, 429–440. [Google Scholar] [CrossRef]
- Yao, S.; Janku, F.; Koenig, K.; Tsimberidou, A.M.; Piha-Paul, S.A.; Shi, N.; Stewart, J.; Johnston, A.; Bomalaski, J.; Meric-Bernstam, F.; et al. Phase 1 trial of ADI-PEG 20 and liposomal doxorubicin in patients with metastatic solid tumors. Cancer Med. 2022, 11, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Lowery, M.A.; Yu, K.H.; Kelsen, D.P.; Harding, J.J.; Bomalaski, J.S.; Glassman, D.C.; Covington, C.M.; Brenner, R.; Hollywood, E.; Barba, A.; et al. A phase 1/1B trial of ADI-PEG 20 plus nab-paclitaxel and gemcitabine in patients with advanced pancreatic adenocarcinoma. Cancer 2017, 123, 4556–4565. [Google Scholar] [CrossRef]
- Ott, P.A.; Carvajal, R.D.; Pandit-Taskar, N.; Jungbluth, A.A.; Hoffman, E.W.; Wu, B.-W.; Bomalaski, J.S.; Venhaus, R.; Pan, L.; Old, L.J.; et al. Phase I/II study of pegylated arginine deiminase (ADI-PEG 20) in patients with advanced melanoma. Investig. New Drugs 2013, 31, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Yen, M.-C.; Lin, C.-C.; Chen, Y.-L.; Huang, S.-S.; Yang, H.-J.; Chang, C.-P.; Lei, H.-Y.; Lai, M.-D. A novel cancer therapy by skin delivery of indoleamine 2,3-dioxygenase siRNA. Clin. Cancer Res. 2009, 15, 641–649. [Google Scholar] [CrossRef]
- Huang, T.-T.; Yen, M.-C.; Lin, C.-C.; Weng, T.-Y.; Chen, Y.-L.; Lin, C.-M.; Lai, M.-D. Skin delivery of short hairpin RNA of indoleamine 2,3 dioxygenase induces antitumor immunity against orthotopic and metastatic liver cancer. Cancer Sci. 2011, 102, 2214–2220. [Google Scholar] [CrossRef]
- Shao, J.; Hou, L.; Liu, J.; Liu, Y.; Ning, J.; Zhao, Q.; Zhang, Y. Indoleamine 2,3-Dioxygenase 1 Inhibitor-Loaded Nanosheets Enhance CAR-T Cell Function in Esophageal Squamous Cell Carcinoma. Front. Immunol. 2021, 12, 661357. [Google Scholar] [CrossRef] [PubMed]
- Yue, E.W.; Sparks, R.; Polam, P.; Modi, D.; Douty, B.; Wayland, B.; Glass, B.; Takvorian, A.; Glenn, J.; Zhu, W.; et al. INCB24360 (Epacadostat), a Highly Potent and Selective Indoleamine-2,3-dioxygenase 1 (IDO1) Inhibitor for Immuno-oncology. ACS Med. Chem. Lett. 2017, 8, 486–491. [Google Scholar] [CrossRef]
- Gibney, G.T.; Hamid, O.; Lutzky, J.; Olszanski, A.J.; Mitchell, T.C.; Gajewski, T.F.; Chmielowski, B.; Hanks, B.A.; Zhao, Y.; Newton, R.C.; et al. Phase 1/2 study of epacadostat in combination with ipilimumab in patients with unresectable or metastatic melanoma. J. Immunother. Cancer 2019, 7, 80. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.-J.; Kim, T.M.; et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): A phase 3, randomised, double-blind study. Lancet Oncol. 2019, 20, 1083–1097. [Google Scholar] [CrossRef]
- Naing, A.; Powderly, J.D.; Nemunaitis, J.J.; Luke, J.J.; Mansfield, A.S.; Messersmith, W.A.; Sahebjam, S.; LoRusso, P.M.; Garrido-Laguna, I.; Leopold, L.; et al. Exploring the safety, effect on the tumor microenvironment, and efficacy of itacitinib in combination with epacadostat or parsaclisib in advanced solid tumors: A phase I study. J. Immunother. Cancer 2022, 10, e004223. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, D.; Young, A.; Teng, M.W.L.; Smyth, M.J. Targeting immunosuppressive adenosine in cancer. Nat. Rev. Cancer 2017, 17, 709–724. [Google Scholar] [CrossRef]
- Piovesan, D.; Tan, J.B.L.; Becker, A.; Banuelos, J.; Narasappa, N.; DiRenzo, D.; Zhang, K.; Chen, A.; Ginn, E.; Udyavar, A.R.; et al. Targeting CD73 with AB680 (Quemliclustat), a Novel and Potent Small-Molecule CD73 Inhibitor, Restores Immune Functionality and Facilitates Antitumor Immunity. Mol. Cancer Ther. 2022, 21, 948–959. [Google Scholar] [CrossRef]
- Ma, S.-R.; Deng, W.-W.; Liu, J.-F.; Mao, L.; Yu, G.-T.; Bu, L.-L.; Kulkarni, A.B.; Zhang, W.-F.; Sun, Z.-J. Blockade of adenosine A2A receptor enhances CD8 T cells response and decreases regulatory T cells in head and neck squamous cell carcinoma. Mol. Cancer 2017, 16, 99. [Google Scholar] [CrossRef] [PubMed]
- Beavis, P.A.; Henderson, M.A.; Giuffrida, L.; Mills, J.K.; Sek, K.; Cross, R.S.; Davenport, A.J.; John, L.B.; Mardiana, S.; Slaney, C.Y.; et al. Targeting the adenosine 2A receptor enhances chimeric antigen receptor T cell efficacy. J. Clin. Investig. 2017, 127, 929–941. [Google Scholar] [CrossRef]
- Deng, W.-W.; Li, Y.-C.; Ma, S.-R.; Mao, L.; Yu, G.-T.; Bu, L.-L.; Kulkarni, A.B.; Zhang, W.-F.; Sun, Z.-J. Specific blockade CD73 alters the “exhausted” phenotype of T cells in head and neck squamous cell carcinoma. Int. J. Cancer 2018, 143, 1494–1504. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-Y.; Moesta, A.K.; Xiao, C.; Nakamura, K.; Casey, M.; Zhang, H.; Madore, J.; Lepletier, A.; Aguilera, A.R.; Sundarrajan, A.; et al. Targeting CD39 in Cancer Reveals an Extracellular ATP- and Inflammasome-Driven Tumor Immunity. Cancer Discov. 2019, 9, 1754–1773. [Google Scholar] [CrossRef]
- Perrot, I.; Michaud, H.A.; Giraudon-Paoli, M.; Augier, S.; Docquier, A.; Gros, L.; Courtois, R.; Dejou, C.; Jecko, D.; Becquart, O.; et al. Blocking Antibodies Targeting the CD39/CD73 Immunosuppressive Pathway Unleash Immune Responses in Combination Cancer Therapies. Cell Rep. 2019, 27, 2411–2425 e2419. [Google Scholar] [CrossRef]
- Kashyap, A.S.; Thelemann, T.; Klar, R.; Kallert, S.M.; Festag, J.; Buchi, M.; Hinterwimmer, L.; Schell, M.; Michel, S.; Jaschinski, F.; et al. Antisense oligonucleotide targeting CD39 improves anti-tumor T cell immunity. J. Immunother. Cancer 2019, 7, 67. [Google Scholar] [CrossRef]
- Willingham, S.B.; Ho, P.Y.; Hotson, A.; Hill, C.; Piccione, E.C.; Hsieh, J.; Liu, L.; Buggy, J.J.; McCaffery, I.; Miller, R.A. A2AR Antagonism with CPI-444 Induces Antitumor Responses and Augments Efficacy to Anti-PD-(L)1 and Anti-CTLA-4 in Preclinical Models. Cancer Immunol. Res. 2018, 6, 1136–1149. [Google Scholar] [CrossRef]
- Herbst, R.S.; Majem, M.; Barlesi, F.; Carcereny, E.; Chu, Q.; Monnet, I.; Sanchez-Hernandez, A.; Dakhil, S.; Camidge, D.R.; Winzer, L.; et al. COAST: An Open-Label, Phase II, Multidrug Platform Study of Durvalumab Alone or in Combination with Oleclumab or Monalizumab in Patients with Unresectable, Stage III Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2022, 40, 3383–3393. [Google Scholar] [CrossRef]
- Fong, L.; Hotson, A.; Powderly, J.D.; Sznol, M.; Heist, R.S.; Choueiri, T.K.; George, S.; Hughes, B.G.M.; Hellmann, M.D.; Shepard, D.R.; et al. Adenosine 2A Receptor Blockade as an Immunotherapy for Treatment-Refractory Renal Cell Cancer. Cancer Discov. 2020, 10, 40–53. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ping, Y.; Shen, C.; Huang, B.; Zhang, Y. Reprogramming T-Cell Metabolism for Better Anti-Tumor Immunity. Cells 2022, 11, 3103. https://doi.org/10.3390/cells11193103
Ping Y, Shen C, Huang B, Zhang Y. Reprogramming T-Cell Metabolism for Better Anti-Tumor Immunity. Cells. 2022; 11(19):3103. https://doi.org/10.3390/cells11193103
Chicago/Turabian StylePing, Yu, Chunyi Shen, Bo Huang, and Yi Zhang. 2022. "Reprogramming T-Cell Metabolism for Better Anti-Tumor Immunity" Cells 11, no. 19: 3103. https://doi.org/10.3390/cells11193103
APA StylePing, Y., Shen, C., Huang, B., & Zhang, Y. (2022). Reprogramming T-Cell Metabolism for Better Anti-Tumor Immunity. Cells, 11(19), 3103. https://doi.org/10.3390/cells11193103