
Application and Design of Switches Used in CAR


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Nobel Prizes in Cell-Mediated Immunity Research and CAR
1.2. Dark Side of CAR Therapy
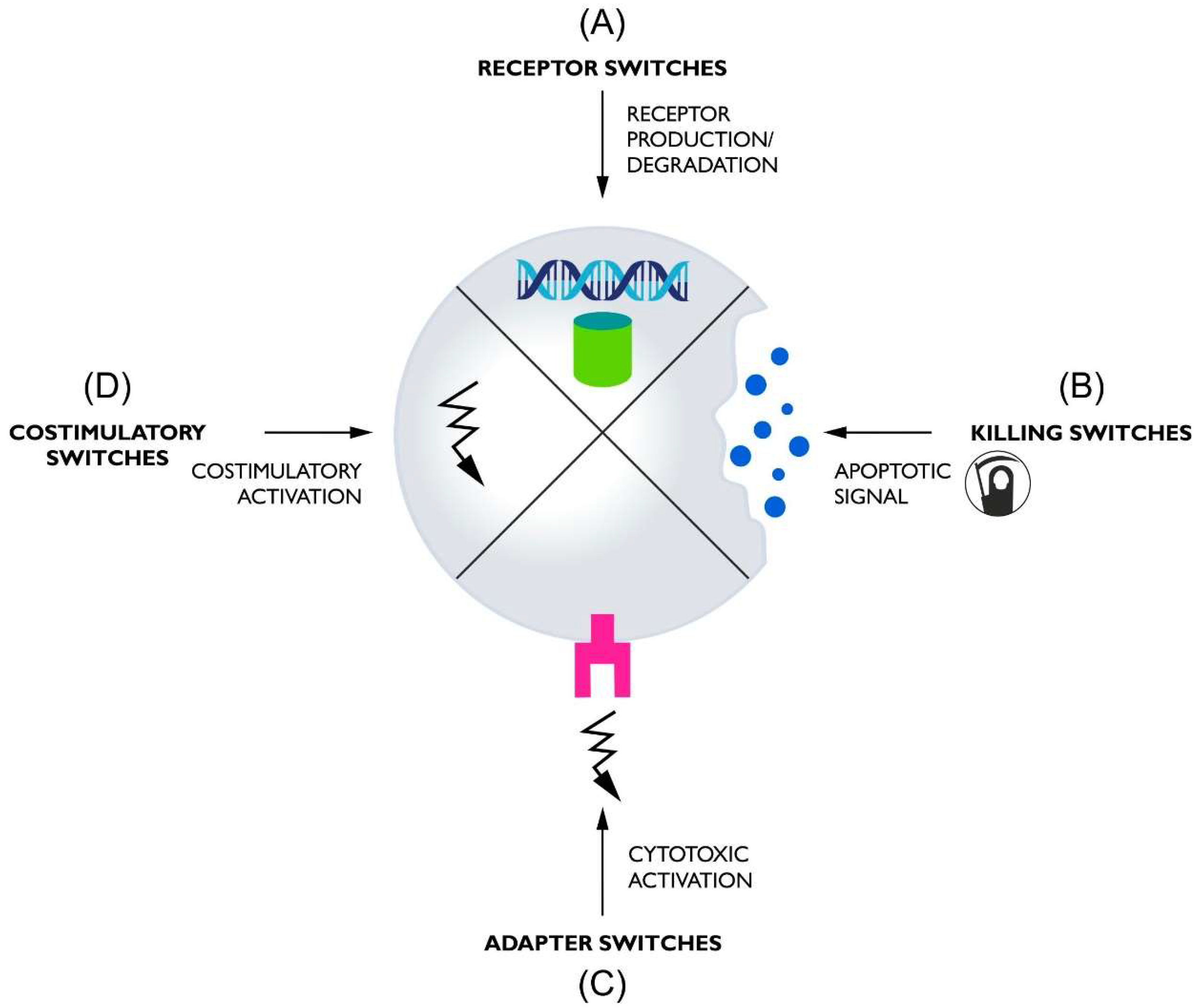
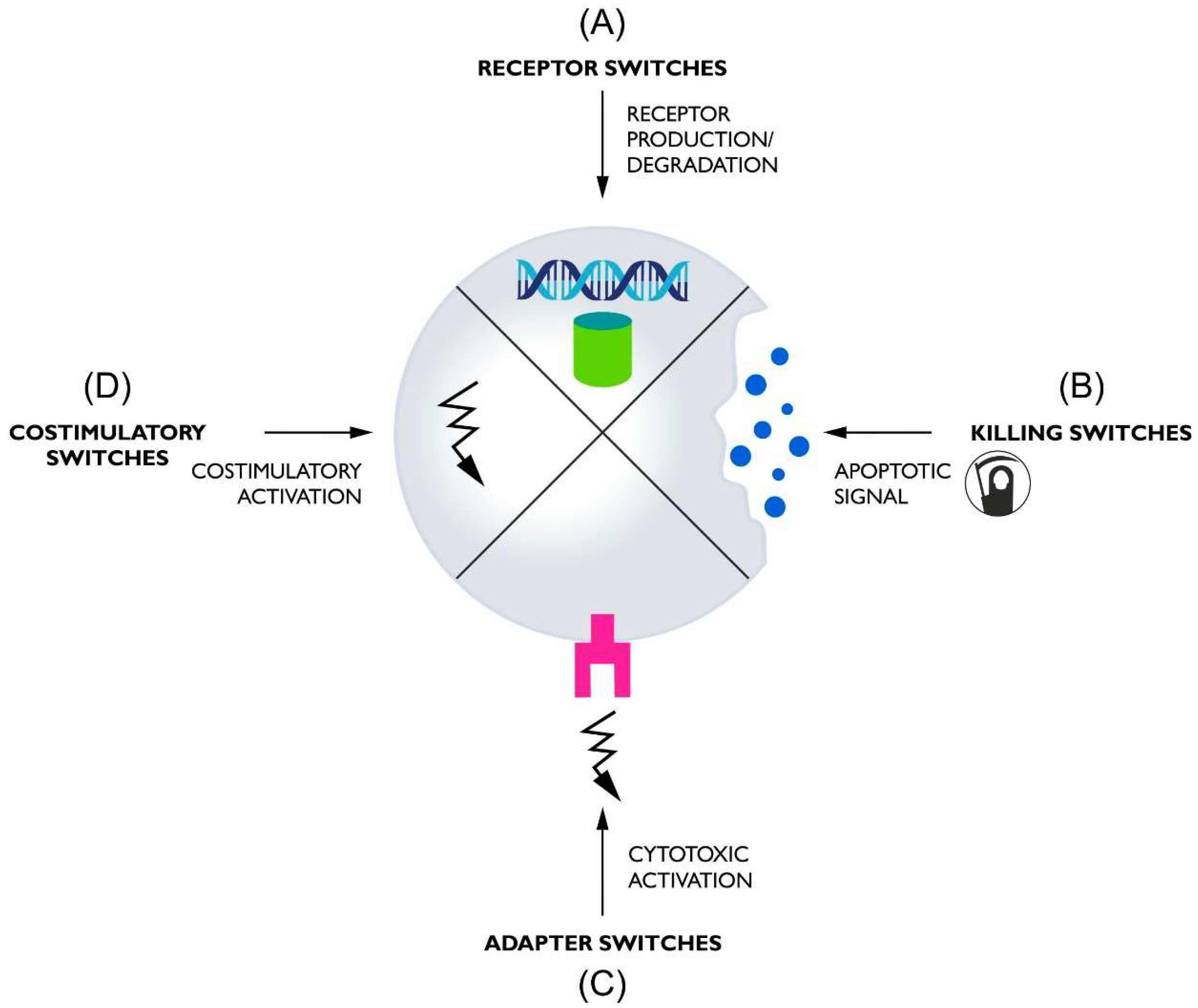
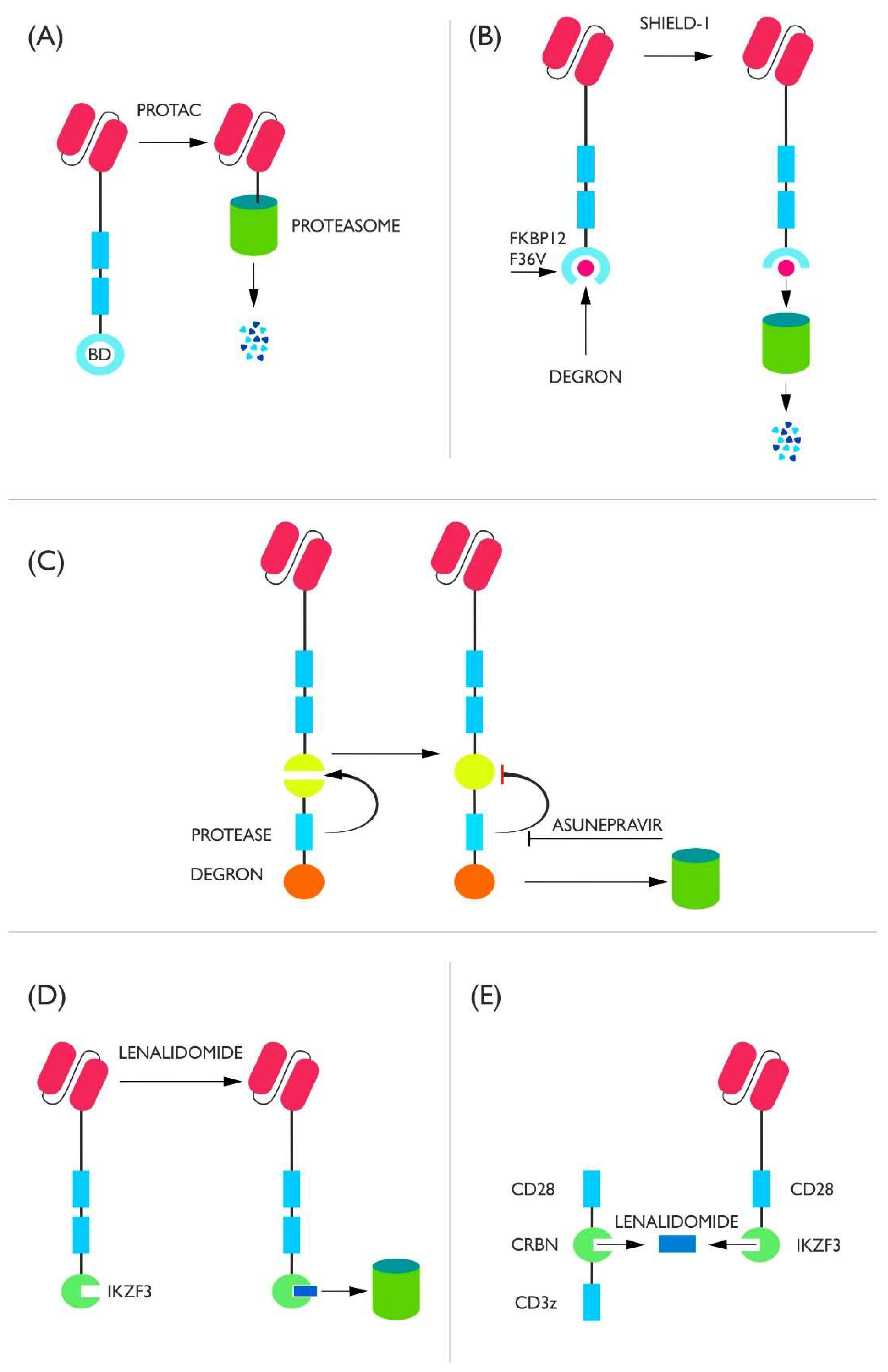
2. Receptor Switches
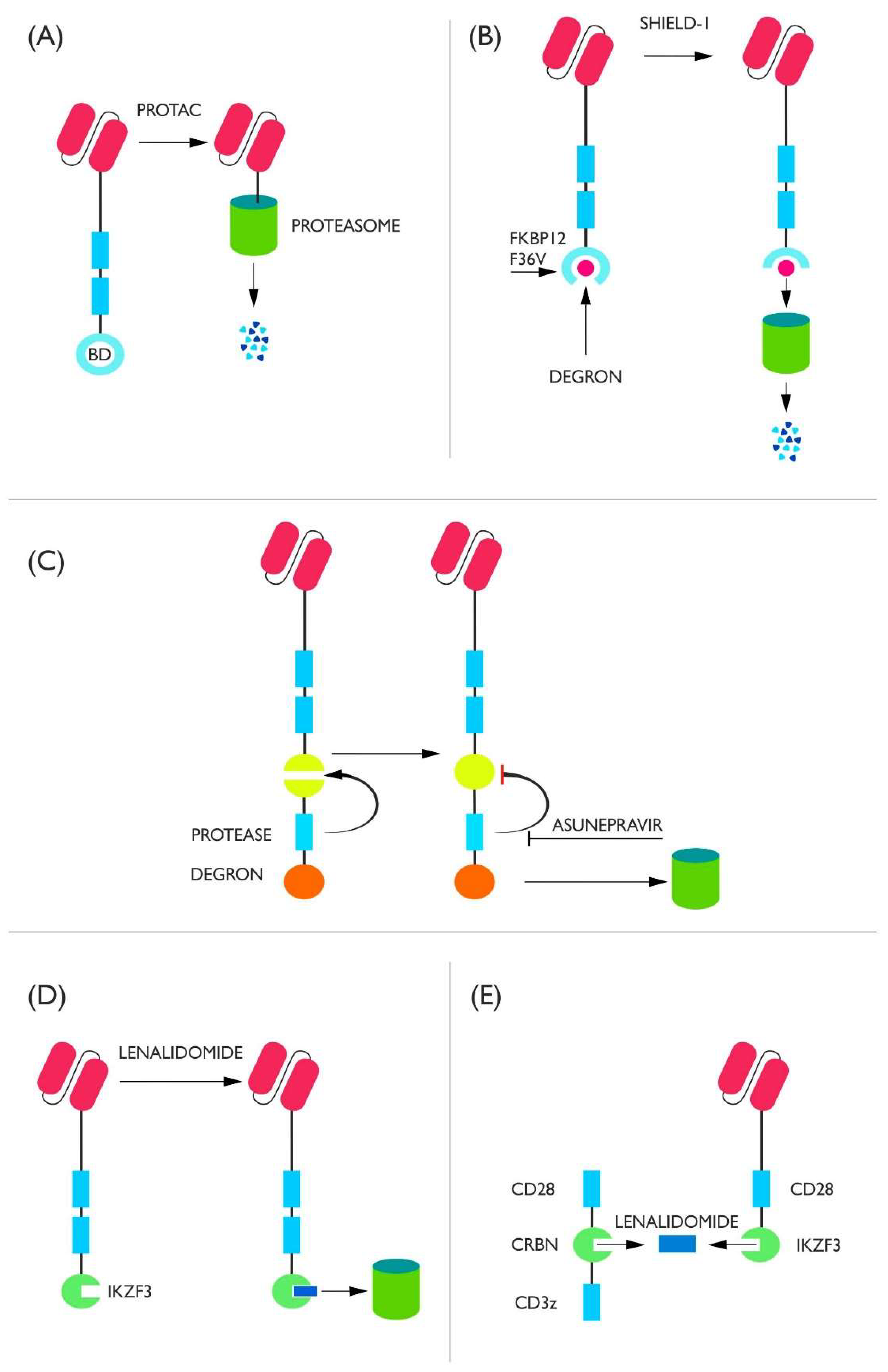
2.1. Degron Switches
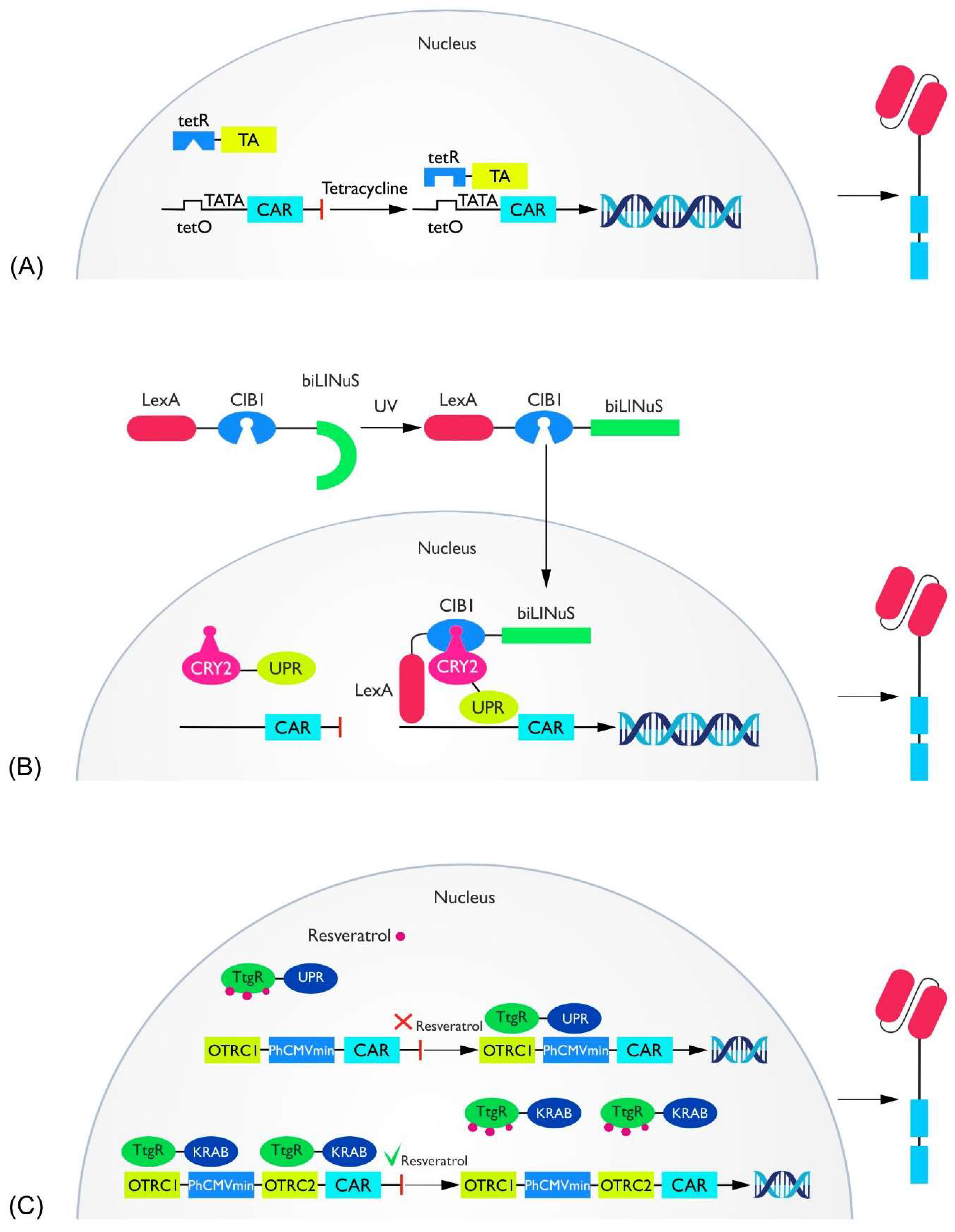
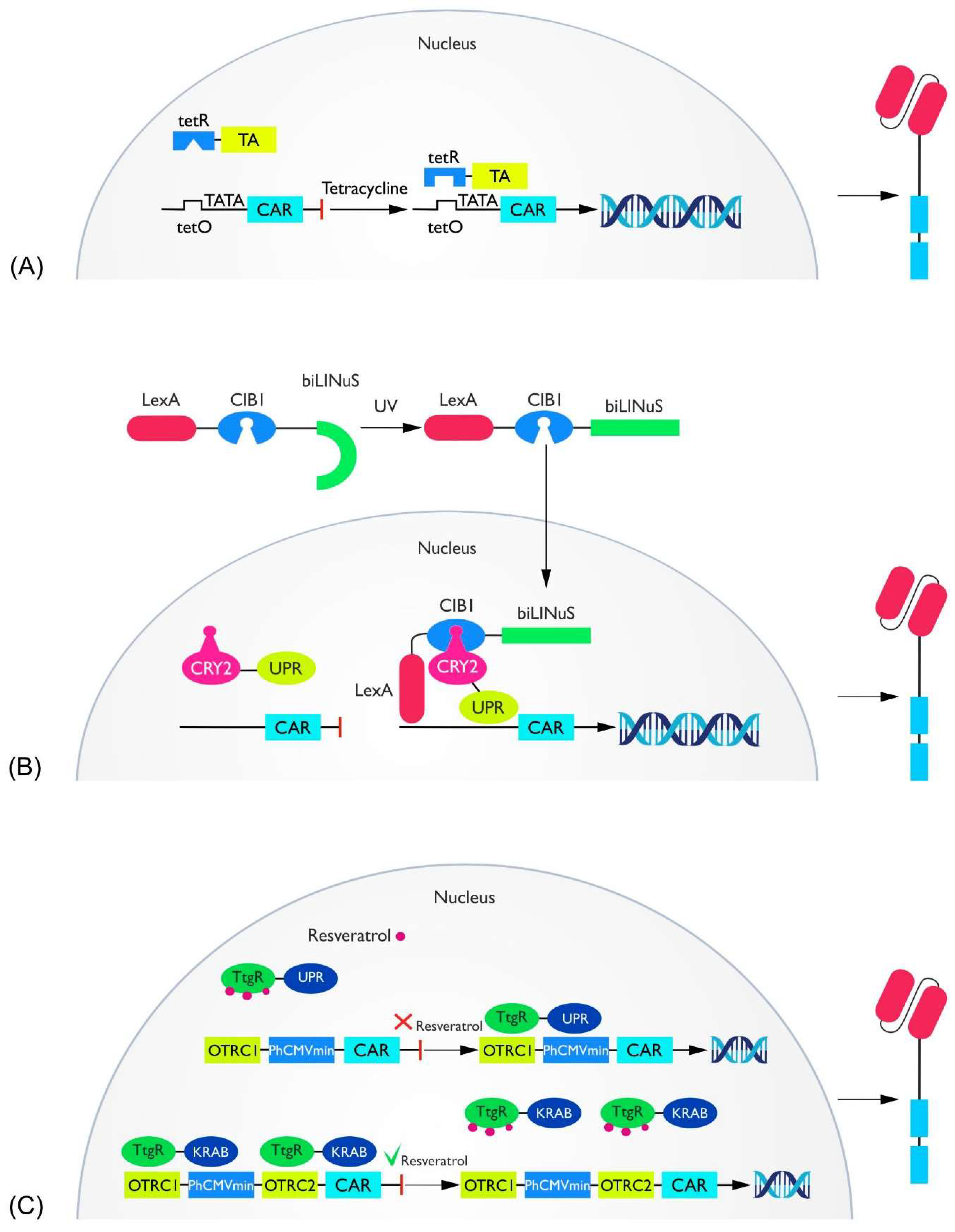
2.2. Transcriptional Switches
3. Killing Switches
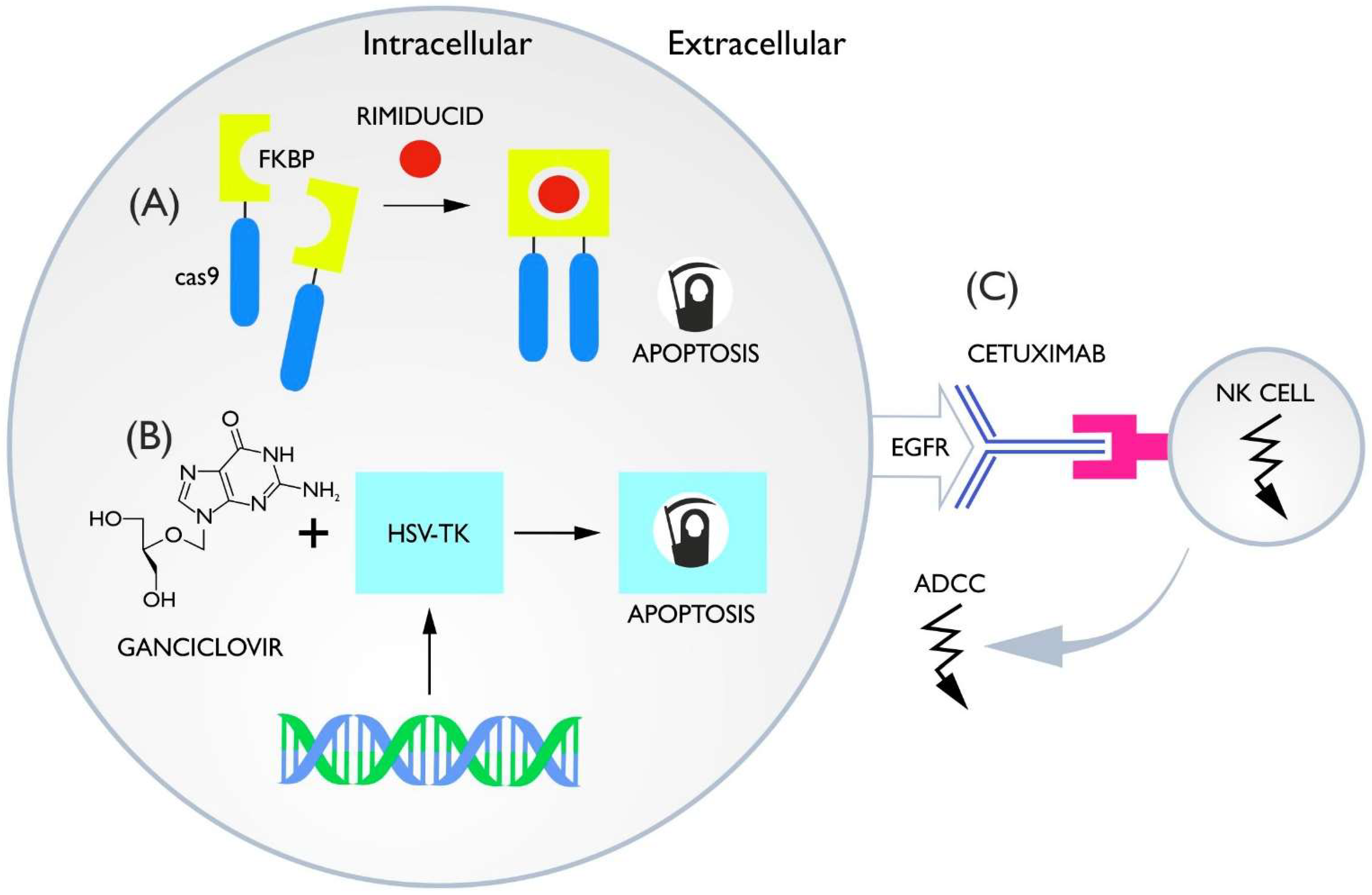
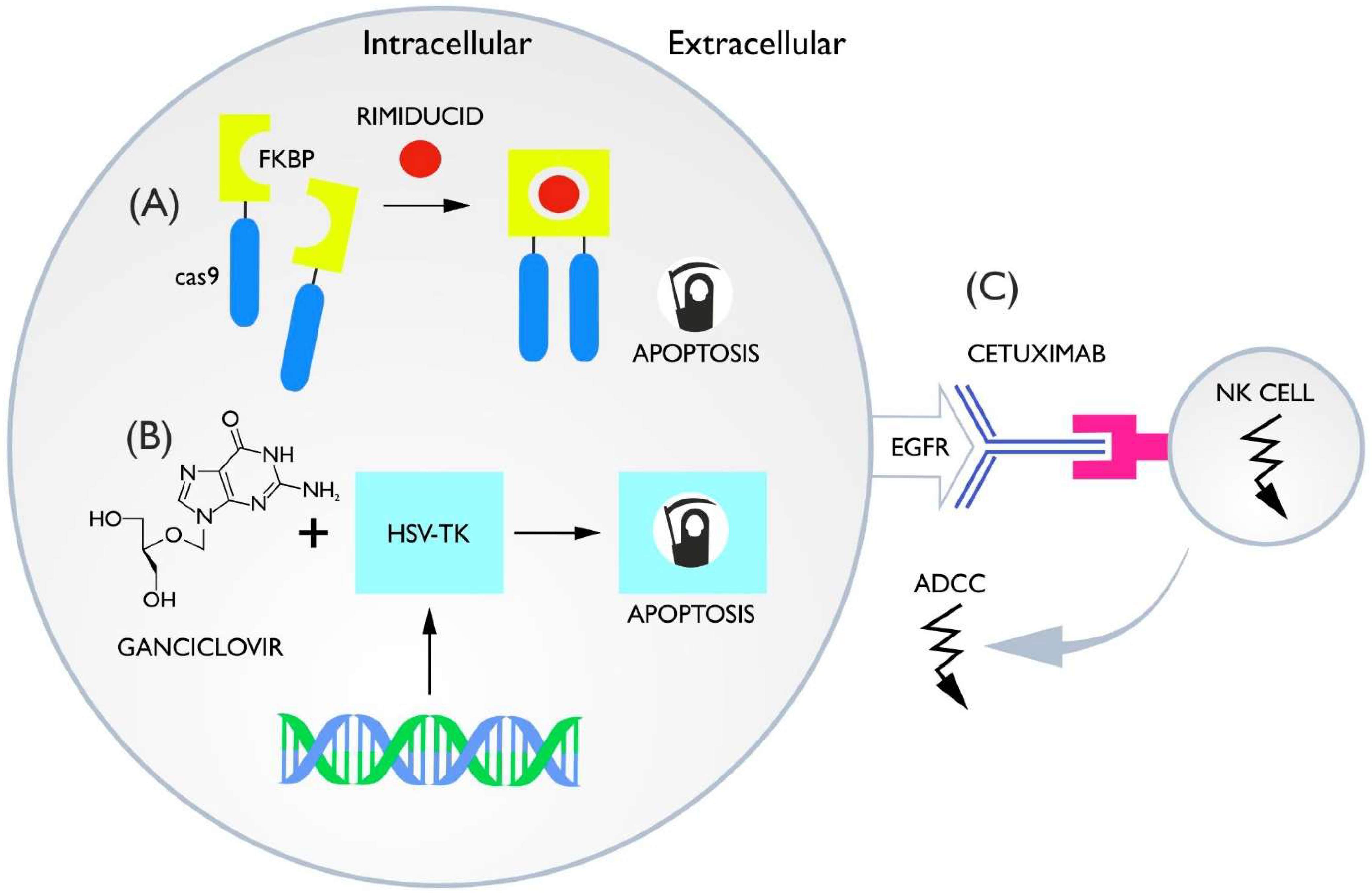
3.1. Inducible Caspase-9
3.2. HSV-TK Ganciclovir
3.3. ADCC Switches
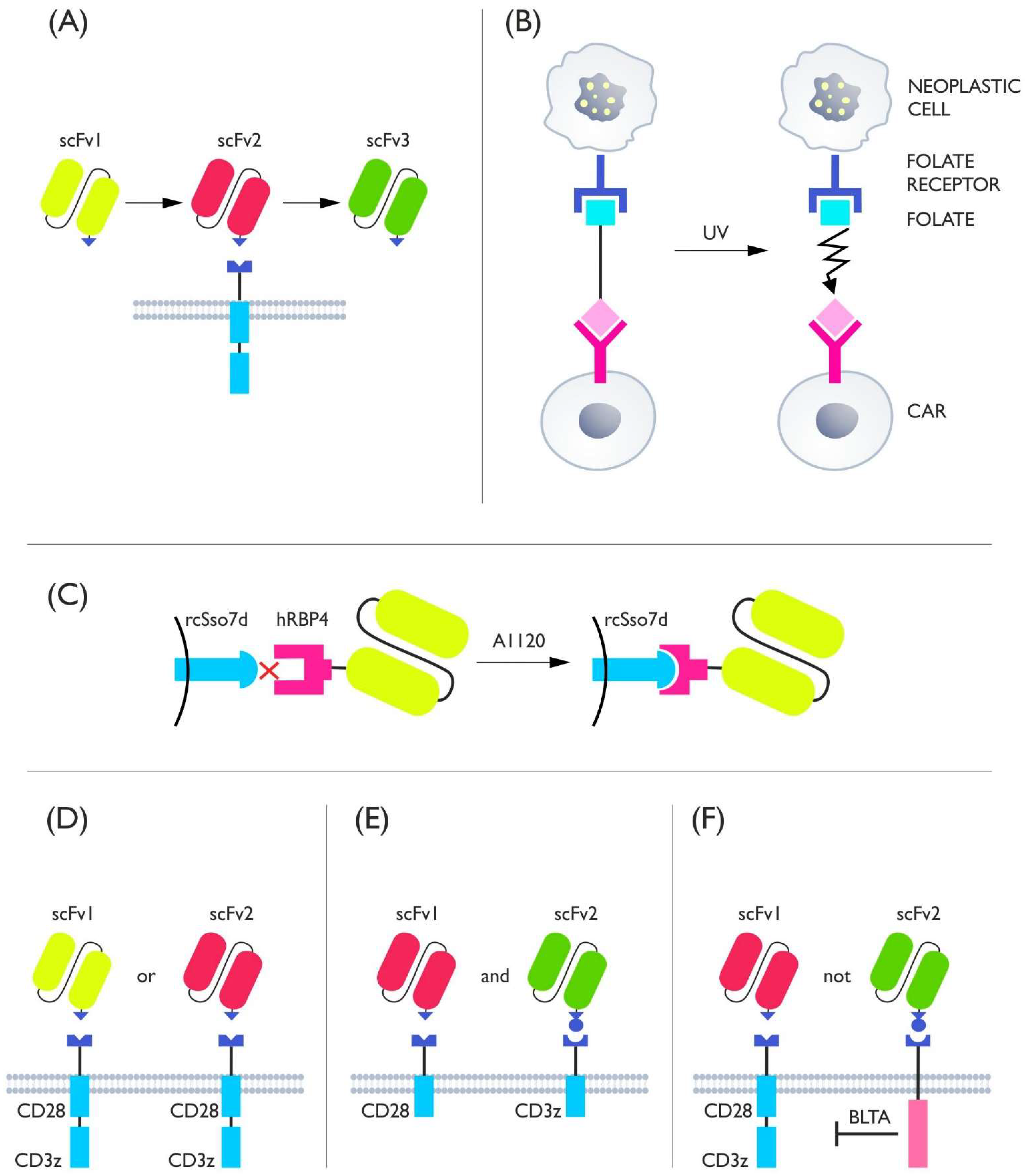
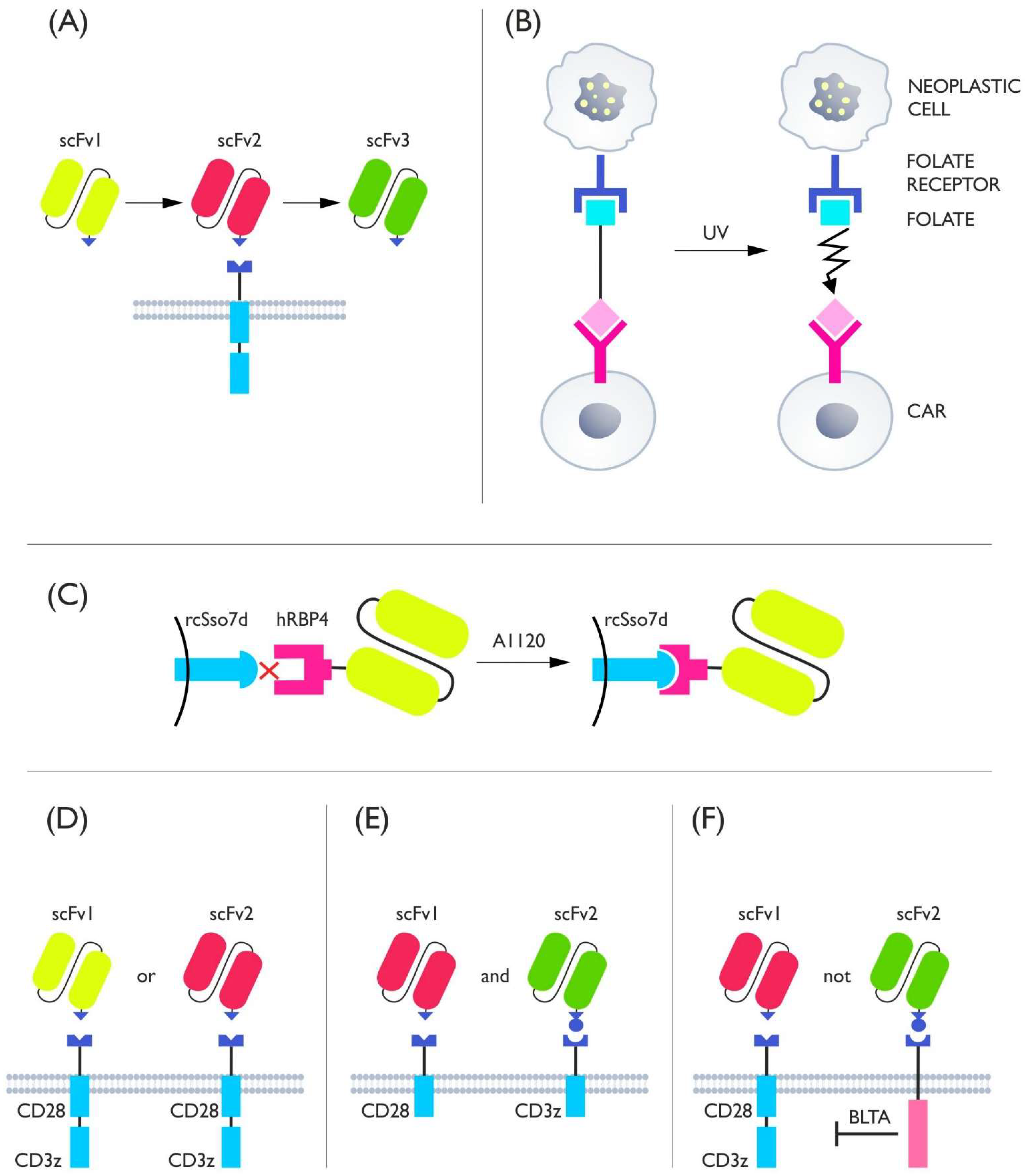
4. Adapter Switches
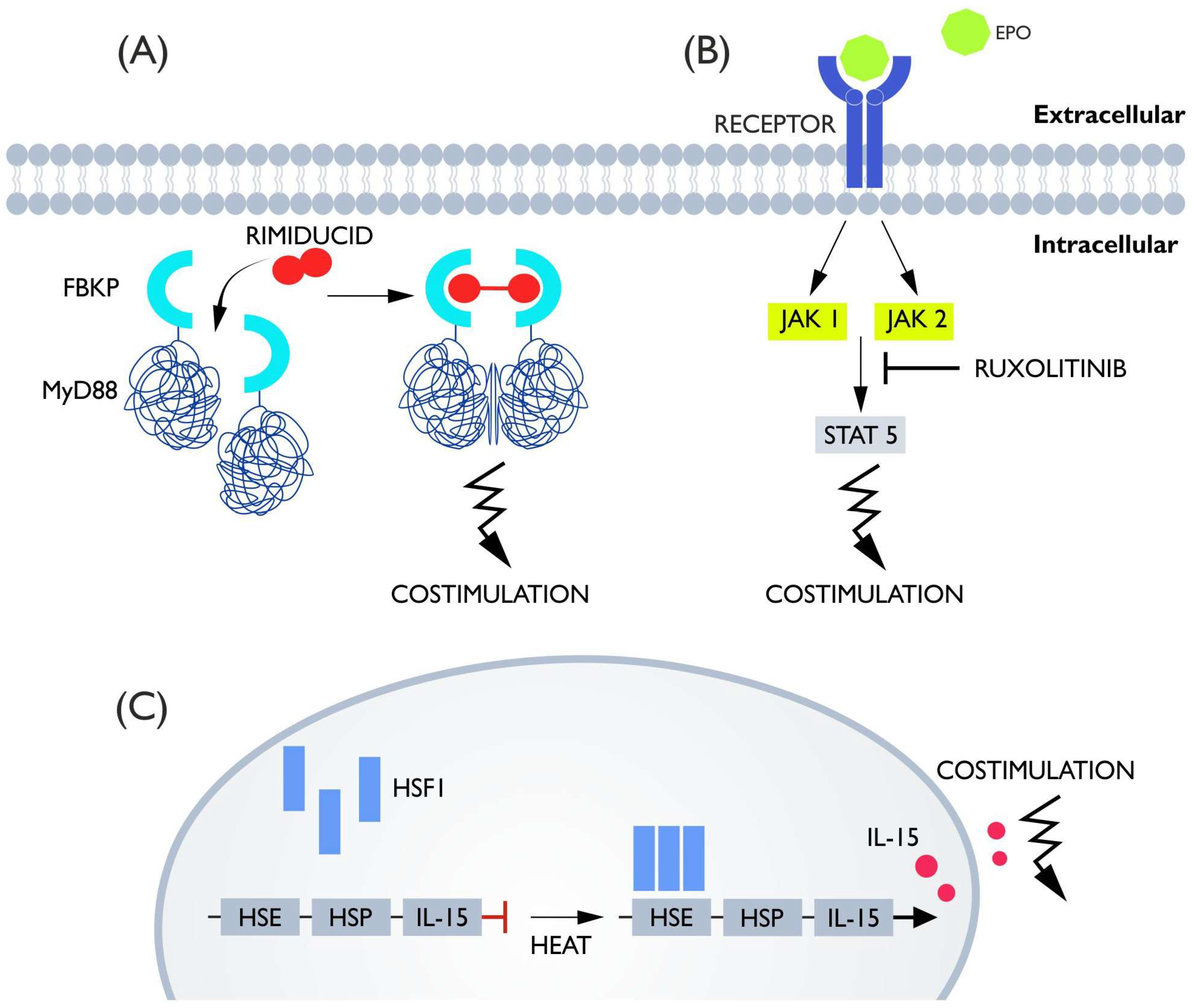
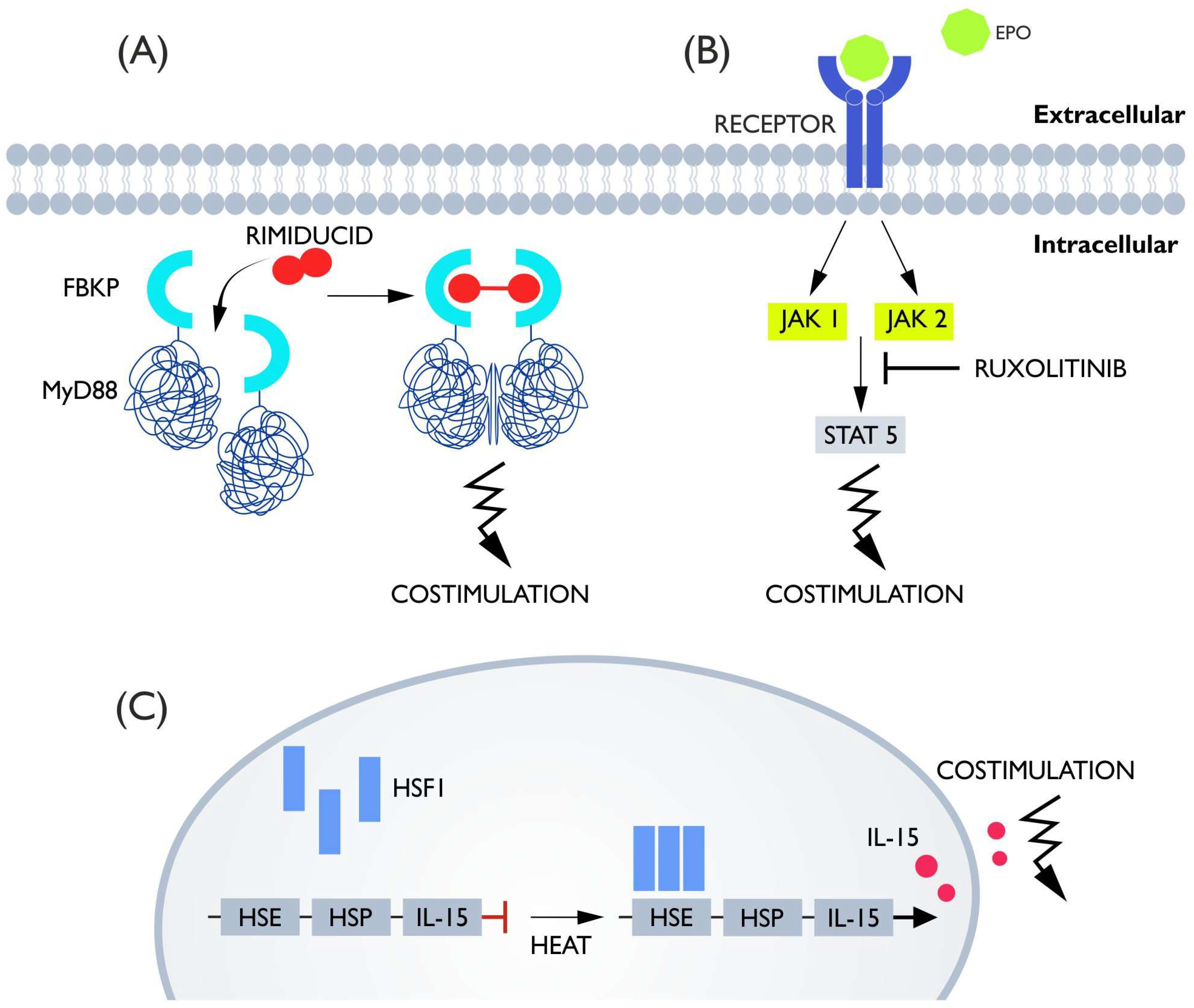
5. Costimulatory Switches
6. Allogeneic Adoptive Therapies
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Wiemann, B.; Starnes, C.O. Coley’s Toxins, Tumor Necrosis Factor and Cancer Research: A Historical Perspective. Pharmacol. Ther. 1994, 64, 529–564. [Google Scholar] [CrossRef]
- Guedan, S.; Posey, A.D.; Shaw, C.; Wing, A.; Da, T.; Patel, P.R.; McGettigan, S.E.; Casado-Medrano, V.; Kawalekar, O.U.; Uribe-Herranz, M.; et al. Enhancing CAR T Cell Persistence through ICOS and 4-1BB Costimulation. JCI Insight 2018, 3, e96976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowley, D.A.; Fitch, F.W. The Road to the Discovery of Dendritic Cells, a Tribute to Ralph Steinman. Cell. Immunol. 2012, 273, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Beutler, B.A. TLRs and Innate Immunity. Blood 2009, 113, 1399–1407. [Google Scholar] [CrossRef] [Green Version]
- Foster, A.E.; Mahendravada, A.; Shinners, N.P.; Chang, W.-C.; Crisostomo, J.; Lu, A.; Khalil, M.; Morschl, E.; Shaw, J.L.; Saha, S.; et al. Regulated Expansion and Survival of Chimeric Antigen Receptor-Modified T Cells Using Small Molecule-Dependent Inducible MyD88/CD40. Mol. Ther. 2017, 25, 2176–2188. [Google Scholar] [CrossRef] [Green Version]
- McGonagle, D.; Georgouli, T. The Importance of Mechnikovs Thorn for an Improved Understanding of 21st Century Medicine and Immunology: A View from the Eye. Scand. J. Immunol. 2008, 68, 129–139. [Google Scholar] [CrossRef]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human Chimeric Antigen Receptor Macrophages for Cancer Immunotherapy. Nat. Biotechnol. 2020, 38, 947–953. [Google Scholar] [CrossRef]
- Kedzierska, K.; Koutsakos, M. The ABC of Major Histocompatibility Complexes and T Cell Receptors in Health and Disease. Viral Immunol. 2020, 33, 160–178. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Brzostek, J.; Sankaran, S.; Wei, Q.; Yap, J.; Tan, T.Y.Y.; Lai, J.; MacAry, P.A.; Gascoigne, N.R.J. Targeting CAR to the Peptide-MHC Complex Reveals Distinct Signaling Compared to That of TCR in a Jurkat T Cell Model. Cancers 2021, 13, 867. [Google Scholar] [CrossRef]
- Nicholls, M. Sir Frank Macfarlane Burnet. Eur. Heart J. 2020, 41, 1148–1150. [Google Scholar] [CrossRef]
- Ribatti, D. Peter Brian Medawar and the Discovery of Acquired Immunological Tolerance. Immunol. Lett. 2015, 167, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Barry, J.M.; Murray, J.E. The First Human Renal Transplants. J. Urol. 2006, 176, 888–890. [Google Scholar] [CrossRef]
- Thomas, E.D. Bone Marrow Transplantation from the Personal Viewpoint. Int. J. Hematol. 2005, 81, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, R.; Graham, C.; Yallop, D.; Jozwik, A.; Mirci-Danicar, O.C.; Lucchini, G.; Pinner, D.; Jain, N.; Kantarjian, H.; Boissel, N.; et al. Genome-Edited, Donor-Derived Allogeneic Anti-CD19 Chimeric Antigen Receptor T Cells in Paediatric and Adult B-Cell Acute Lymphoblastic Leukaemia: Results of Two Phase 1 Studies. Lancet 2020, 396, 1885–1894. [Google Scholar] [CrossRef]
- Liu, W.; Zang, X. Structures of immune checkpoints: An overview on the CD28-B7 family. In Structural Immunology; Jin, T., Yin, Q., Eds.; Advances in Experimental Medicine and Biology; Springer: Singapore, 2019; Volume 1172, pp. 63–78. ISBN 9789811393662. [Google Scholar]
- Cha, J.-H.; Chan, L.-C.; Li, C.-W.; Hsu, J.L.; Hung, M.-C. Mechanisms Controlling PD-L1 Expression in Cancer. Mol. Cell 2019, 76, 359–370. [Google Scholar] [CrossRef]
- Choi, B.D.; Yu, X.; Castano, A.P.; Darr, H.; Henderson, D.B.; Bouffard, A.A.; Larson, R.C.; Scarfò, I.; Bailey, S.R.; Gerhard, G.M.; et al. CRISPR-Cas9 Disruption of PD-1 Enhances Activity of Universal EGFRvIII CAR T Cells in a Preclinical Model of Human Glioblastoma. J. Immunother. Cancer 2019, 7, 304. [Google Scholar] [CrossRef] [PubMed]
- Chan, L.Y.; Dass, S.A.; Tye, G.J.; Imran, S.A.M.; Wan Kamarul Zaman, W.S.; Nordin, F. CAR-T Cells/-NK Cells in Cancer Immunotherapy and the Potential of MSC to Enhance Its Efficacy: A Review. Biomedicines 2022, 10, 804. [Google Scholar] [CrossRef]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef]
- Tchou, J.; Zhao, Y.; Levine, B.L.; Zhang, P.J.; Davis, M.M.; Melenhorst, J.J.; Kulikovskaya, I.; Brennan, A.L.; Liu, X.; Lacey, S.F.; et al. Safety and Efficacy of Intratumoral Injections of Chimeric Antigen Receptor (CAR) T Cells in Metastatic Breast Cancer. Cancer Immunol. Res. 2017, 5, 1152–1161. [Google Scholar] [CrossRef] [Green Version]
- Kloss, C.C.; Lee, J.; Zhang, A.; Chen, F.; Melenhorst, J.J.; Lacey, S.F.; Maus, M.V.; Fraietta, J.A.; Zhao, Y.; June, C.H. Dominant-Negative TGF-β Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation And Augments Prostate Cancer Eradication. Mol. Ther. 2018, 26, 1855–1866. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Wang, Y.; Yuan, Y.; Sun, J.; Liu, L.; Huang, D.; Hu, J.; Wang, M.; Li, S.; Song, W.; et al. In Vitro Elimination of Autoreactive B Cells from Rheumatoid Arthritis Patients by Universal Chimeric Antigen Receptor T Cells. Ann. Rheum. Dis. 2021, 80, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Kansal, R.; Richardson, N.; Neeli, I.; Khawaja, S.; Chamberlain, D.; Ghani, M.; Ghani, Q.; Balazs, L.; Beranova-Giorgianni, S.; Giorgianni, F.; et al. Sustained B Cell Depletion by CD19-Targeted CAR T Cells Is a Highly Effective Treatment for Murine Lupus. Sci. Transl. Med. 2019, 11, eaav1648. [Google Scholar] [CrossRef]
- Maldini, C.R.; Claiborne, D.T.; Okawa, K.; Chen, T.; Dopkin, D.L.; Shan, X.; Power, K.A.; Trifonova, R.T.; Krupp, K.; Phelps, M.; et al. Dual CD4-Based CAR T Cells with Distinct Costimulatory Domains Mitigate HIV Pathogenesis In Vivo. Nat. Med. 2020, 26, 1776–1787. [Google Scholar] [CrossRef]
- Al-Utaibi, K.A.; Nutini, A.; Sohail, A.; Arif, R.; Tunc, S.; Sait, S.M. Forecasting the Action of CAR-T Cells against SARS-Corona Virus-II Infection with Branching Process. Model. Earth Syst. Environ. 2021, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wu, Z.; Zhao, N. Individual Patient Data Meta-Analysis from 16 Trials for Safety Factors in Cytokine Release Syndrome After CAR-T Therapy in Patients with Non-Hodgkin Lymphoma (NHL) and Acute Lymphoblastic Leukemia. Adv. Ther. 2019, 36, 2881–2894. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, J.C.; Weiss, S.L.; Maude, S.L.; Barrett, D.M.; Lacey, S.F.; Melenhorst, J.J.; Shaw, P.; Berg, R.A.; June, C.H.; Porter, D.L.; et al. Cytokine Release Syndrome After Chimeric Antigen Receptor T Cell Therapy for Acute Lymphoblastic Leukemia. Crit. Care Med. 2017, 45, e124–e131. [Google Scholar] [CrossRef] [PubMed]
- Santomasso, B.D.; Park, J.H.; Salloum, D.; Riviere, I.; Flynn, J.; Mead, E.; Halton, E.; Wang, X.; Senechal, B.; Purdon, T.; et al. Clinical and Biological Correlates of Neurotoxicity Associated with CAR T-Cell Therapy in Patients with B-Cell Acute Lymphoblastic Leukemia. Cancer Discov. 2018, 8, 958–971. [Google Scholar] [CrossRef] [Green Version]
- Rejeski, K.; Kunz, W.G.; Rudelius, M.; Bücklein, V.; Blumenberg, V.; Schmidt, C.; Karschnia, P.; Schöberl, F.; Dimitriadis, K.; von Baumgarten, L.; et al. Severe Candida Glabrata Pancolitis and Fatal Aspergillus Fumigatus Pulmonary Infection in the Setting of Bone Marrow Aplasia after CD19-Directed CAR T-Cell Therapy–a Case Report. BMC Infect. Dis. 2021, 21, 121. [Google Scholar] [CrossRef]
- Qasrawi, A.; Arora, R.; Ramlal, R.; Munker, R.; Hildebrandt, G.C. Allogenic Hematopoietic Stem Cell Transplantation for Prolonged Bone Marrow Aplasia after Chimeric Antigen Receptor (CAR) T-cell Therapy for Relapsed Diffuse Large B-cell Lymphoma. Am. J. Hematol. 2020, 95, E89–E91. [Google Scholar] [CrossRef] [Green Version]
- Goto, H.; Makita, S.; Kato, K.; Tokushige, K.; Fujita, T.; Akashi, K.; Izutsu, K.; Teshima, T. Efficacy and Safety of Tisagenlecleucel in Japanese Adult Patients with Relapsed/Refractory Diffuse Large B-Cell Lymphoma. Int. J. Clin. Oncol. 2020, 25, 1736–1743. [Google Scholar] [CrossRef]
- Cai, C.; Tang, D.; Han, Y.; Shen, E.; Ahmed, O.A.; Guo, C.; Shen, H.; Zeng, S. A Comprehensive Analysis of the Fatal Toxic Effects Associated with CD19 CAR-T Cell Therapy. Aging 2020, 12, 18741–18753. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Teachey, D.T.; Pequignot, E.; Frey, N.; Porter, D.; Maude, S.L.; Grupp, S.A.; June, C.H.; Melenhorst, J.J.; Lacey, S.F. Measuring IL-6 and SIL-6R in Serum from Patients Treated with Tocilizumab and/or Siltuximab Following CAR T Cell Therapy. J. Immunol. Methods 2016, 434, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wehrli, M.; Gallagher, K.; Chen, Y.-B.; Leick, M.B.; McAfee, S.L.; El-Jawahri, A.R.; DeFilipp, Z.; Horick, N.; O’Donnell, P.; Spitzer, T.; et al. Single-Center Experience Using Anakinra for Steroid-Refractory Immune Effector Cell-Associated Neurotoxicity Syndrome (ICANS). J. Immunother. Cancer 2022, 10, e003847. [Google Scholar] [CrossRef]
- Liu, D.; Xu, X.; Dai, Y.; Zhao, X.; Bao, S.; Ma, W.; Zha, L.; Liu, S.; Liu, Y.; Zheng, J.; et al. Blockade of AIM2 Inflammasome or A1-AR Ameliorates IL-1β Release and Macrophage-Mediated Immunosuppression Induced by CAR-T Treatment. J. Immunother. Cancer 2021, 9, e001466. [Google Scholar] [CrossRef] [PubMed]
- Ruella, M.; Barrett, D.M.; Shestova, O.; Perazzelli, J.; Posey, A.D.; Hong, S.J.; Kozlowski, M.; Lacey, S.F.; Melenhorst, J.J.; June, C.H.; et al. A Cellular Antidote to Specifically Deplete Anti-CD19 Chimeric Antigen Receptor–Positive Cells. Blood 2020, 135, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Han, W. Biomarkers of Cytokine Release Syndrome and Neurotoxicity Related to CAR-T Cell Therapy. Biomark Res. 2018, 6, 4. [Google Scholar] [CrossRef] [Green Version]
- Weber, E.W.; Parker, K.R.; Sotillo, E.; Lynn, R.C.; Anbunathan, H.; Lattin, J.; Good, Z.; Belk, J.A.; Daniel, B.; Klysz, D.; et al. Transient Rest Restores Functionality in Exhausted CAR-T Cells through Epigenetic Remodeling. Science 2021, 372, eaba1786. [Google Scholar] [CrossRef] [PubMed]
- Guedan, S.; Madar, A.; Casado-Medrano, V.; Shaw, C.; Wing, A.; Liu, F.; Young, R.M.; June, C.H.; Posey, A.D. Single Residue in CD28-Costimulated CAR-T Cells Limits Long-Term Persistence and Antitumor Durability. J. Clin. Investig. 2020, 130, 3087–3097. [Google Scholar] [CrossRef] [Green Version]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB Costimulation Ameliorates T Cell Exhaustion Induced by Tonic Signaling of Chimeric Antigen Receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.M.; Kang, C.H.; Choi, S.U.; Kim, Y.; Hwang, J.Y.; Jeong, H.G.; Park, C.H. A Chemical Switch System to Modulate Chimeric Antigen Receptor T Cell Activity through Proteolysis-Targeting Chimaera Technology. ACS Synth. Biol. 2020, 9, 987–992. [Google Scholar] [CrossRef]
- Richman, S.A.; Wang, L.-C.; Moon, E.K.; Khire, U.R.; Albelda, S.M.; Milone, M.C. Ligand-Induced Degradation of a CAR Permits Reversible Remote Control of CAR T Cell Activity In Vitro and In Vivo. Mol. Ther. 2020, 28, 1600–1613. [Google Scholar] [CrossRef] [PubMed]
- Maynard-Smith, L.A.; Chen, L.; Banaszynski, L.A.; Ooi, A.G.L.; Wandless, T.J. A Directed Approach for Engineering Conditional Protein Stability Using Biologically Silent Small Molecules. J. Biol. Chem. 2007, 282, 24866–24872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mestermann, K.; Giavridis, T.; Weber, J.; Rydzek, J.; Frenz, S.; Nerreter, T.; Mades, A.; Sadelain, M.; Einsele, H.; Hudecek, M. The Tyrosine Kinase Inhibitor Dasatinib Acts as a Pharmacologic on/off Switch for CAR T Cells. Sci. Transl. Med. 2019, 11, eaau5907. [Google Scholar] [CrossRef] [PubMed]
- Juillerat, A.; Tkach, D.; Busser, B.W.; Temburni, S.; Valton, J.; Duclert, A.; Poirot, L.; Depil, S.; Duchateau, P. Modulation of Chimeric Antigen Receptor Surface Expression by a Small Molecule Switch. BMC Biotechnol. 2019, 19, 44. [Google Scholar] [CrossRef] [Green Version]
- Carbonneau, S.; Sharma, S.; Peng, L.; Rajan, V.; Hainzl, D.; Henault, M.; Yang, C.; Hale, J.; Shulok, J.; Tallarico, J.; et al. An IMiD-Inducible Degron Provides Reversible Regulation for Chimeric Antigen Receptor Expression and Activity. Cell Chem. Biol. 2021, 28, 802812.e6. [Google Scholar] [CrossRef]
- Jan, M.; Scarfò, I.; Larson, R.C.; Walker, A.; Schmidts, A.; Guirguis, A.A.; Gasser, J.A.; Słabicki, M.; Bouffard, A.A.; Castano, A.P.; et al. Reversible ON- and OFF-Switch Chimeric Antigen Receptors Controlled by Lenalidomide. Sci. Transl. Med. 2021, 13, eabb6295. [Google Scholar] [CrossRef]
- Corbel, S.Y.; Rossi, F.M.V. Latest Developments and in Vivo Use of the Tet System: Ex Vivo and in Vivo Delivery of Tetracycline-Regulated Genes. Curr. Opin. Biotechnol. 2002, 13, 448–452. [Google Scholar] [CrossRef]
- T Das, A.; Tenenbaum, L.; Berkhout, B. Tet-On Systems for Doxycycline-Inducible Gene Expression. CGT 2016, 16, 156–167. [Google Scholar] [CrossRef] [Green Version]
- Sakemura, R.; Terakura, S.; Watanabe, K.; Julamanee, J.; Takagi, E.; Miyao, K.; Koyama, D.; Goto, T.; Hanajiri, R.; Nishida, T.; et al. A Tet-On Inducible System for Controlling CD19-Chimeric Antigen Receptor Expression upon Drug Administration. Cancer Immunol. Res. 2016, 4, 658–668. [Google Scholar] [CrossRef] [Green Version]
- Gu, X.; He, D.; Li, C.; Wang, H.; Yang, G. Development of Inducible CD19-CAR T Cells with a Tet-On System for Controlled Activity and Enhanced Clinical Safety. Int. J. Mol. Sci. 2018, 19, 3455. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.-Y.; Wei, D.; Liu, Z.-K.; Yong, Y.-L.; Wei, W.; Zhang, Z.-Y.; Lv, J.-J.; Zhang, Z.; Chen, Z.-N.; Bian, H. Doxycycline Inducible Chimeric Antigen Receptor T Cells Targeting CD147 for Hepatocellular Carcinoma Therapy. Front. Cell Dev. Biol. 2019, 7, 233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drent, E.; Poels, R.; Mulders, M.J.; van de Donk, N.W.C.J.; Themeli, M.; Lokhorst, H.M.; Mutis, T. Feasibility of Controlling CD38-CAR T Cell Activity with a Tet-on Inducible CAR Design. PLoS ONE 2018, 13, e0197349. [Google Scholar] [CrossRef] [PubMed]
- Ali Hosseini Rad, S.M.; Poudel, A.; Tan, G.M.Y.; McLellan, A.D. Optimisation of Tet-On Inducible Systems for Sleeping Beauty-Based Chimeric Antigen Receptor (CAR) Applications. Sci. Rep. 2020, 10, 13125. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M.; Suzuki, Y.; Nagasaki, S.C.; Okuno, H.; Imayoshi, I. Light Control of the Tet Gene Expression System in Mammalian Cells. Cell Rep. 2018, 25, 487–500.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.; Wu, Y.; Allen, M.E.; Pan, Y.; Kyriakakis, P.; Lu, S.; Chang, Y.-J.; Wang, X.; Chien, S.; Wang, Y. Engineering Light-Controllable CAR T Cells for Cancer Immunotherapy. Sci. Adv. 2020, 6, eaay9209. [Google Scholar] [CrossRef] [Green Version]
- Allen, M.E.; Zhou, W.; Thangaraj, J.; Kyriakakis, P.; Wu, Y.; Huang, Z.; Ho, P.; Pan, Y.; Limsakul, P.; Xu, X.; et al. An AND-Gated Drug and Photoactivatable Cre- LoxP System for Spatiotemporal Control in Cell-Based Therapeutics. ACS Synth. Biol. 2019, 8, 2359–2371. [Google Scholar] [CrossRef]
- Karschnia, P.; Strübing, F.; Teske, N.; Blumenberg, V.; Bücklein, V.L.; Schmidt, C.; Schöberl, F.; Dimitriadis, K.; Forbrig, R.; Stemmler, H.-J.; et al. Clinicopathologic Findings in Fatal Neurotoxicity After Adoptive Immunotherapy With CD19-Directed CAR T-Cells. HemaSphere 2021, 5, e533. [Google Scholar] [CrossRef]
- Salehpour, F.; Cassano, P.; Rouhi, N.; Hamblin, M.R.; De Taboada, L.; Farajdokht, F.; Mahmoudi, J. Penetration Profiles of Visible and Near-Infrared Lasers and Light-Emitting Diode Light Through the Head Tissues in Animal and Human Species: A Review of Literature. Photobiomodul. Photomed. Laser Surg. 2019, 37, 581–595. [Google Scholar] [CrossRef]
- Hong, A.-R.; Han, J.S.; Kang, G.; Ko, H.; Jang, H.S. Bright Blue, Green, and Red Luminescence from Dye-Sensitized Core@Shell Upconversion Nanophosphors under 800 Nm Near-Infrared Light. Materials 2020, 13, 5338. [Google Scholar] [CrossRef]
- Ovais, M.; Mukherjee, S.; Pramanik, A.; Das, D.; Mukherjee, A.; Raza, A.; Chen, C. Designing Stimuli-Responsive Upconversion Nanoparticles That Exploit the Tumor Microenvironment. Adv. Mater. 2020, 32, 2000055. [Google Scholar] [CrossRef]
- Kotter, B.; Engert, F.; Krueger, W.; Roy, A.; Rawashdeh, W.A.; Cordes, N.; Drees, B.; Webster, B.; Werchau, N.; Lock, D.; et al. Titratable Pharmacological Regulation of CAR T Cells Using Zinc Finger-Based Transcription Factors. Cancers 2021, 13, 4741. [Google Scholar] [CrossRef]
- Yang, L.; Yin, J.; Wu, J.; Qiao, L.; Zhao, E.M.; Cai, F.; Ye, H. Engineering Genetic Devices for in Vivo Control of Therapeutic T Cell Activity Triggered by the Dietary Molecule Resveratrol. Proc. Natl. Acad. Sci. USA 2021, 118, e2106612118. [Google Scholar] [CrossRef]
- Foster, M.C.; Savoldo, B.; Lau, W.; Rubinos, C.; Grover, N.; Armistead, P.; Coghill, J.; Hagan, R.S.; Morrison, K.; Buchanan, F.B.; et al. Utility of a Safety Switch to Abrogate CD19.CAR T-Cell–Associated Neurotoxicity. Blood 2021, 137, 3306–3309. [Google Scholar] [CrossRef]
- Ruella, M.; Xu, J.; Barrett, D.M.; Fraietta, J.A.; Reich, T.J.; Ambrose, D.E.; Klichinsky, M.; Shestova, O.; Patel, P.R.; Kulikovskaya, I.; et al. Induction of Resistance to Chimeric Antigen Receptor T Cell Therapy by Transduction of a Single Leukemic B Cell. Nat. Med. 2018, 24, 1499–1503. [Google Scholar] [CrossRef]
- Warda, W.; Da Rocha, M.N.; Trad, R.; Haderbache, R.; Salma, Y.; Bouquet, L.; Roussel, X.; Nicod, C.; Deschamps, M.; Ferrand, C. Overcoming Target Epitope Masking Resistance That Can Occur on Low-Antigen-Expresser AML Blasts after IL-1RAP Chimeric Antigen Receptor T Cell Therapy Using the Inducible Caspase 9 Suicide Gene Safety Switch. Cancer Gene Ther. 2021, 28, 1365–1375. [Google Scholar] [CrossRef]
- Minagawa, K.; Al-Obaidi, M.; Di Stasi, A. Generation of Suicide Gene-Modified Chimeric Antigen Receptor-Redirected T-Cells for Cancer Immunotherapy. In Suicide Gene Therapy; Düzgüneş, N., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2019; Volume 1895, pp. 57–73. [Google Scholar] [CrossRef]
- Amatya, C.; Pegues, M.A.; Lam, N.; Vanasse, D.; Geldres, C.; Choi, S.; Hewitt, S.M.; Feldman, S.A.; Kochenderfer, J.N. Development of CAR T Cells Expressing a Suicide Gene Plus a Chimeric Antigen Receptor Targeting Signaling Lymphocytic-Activation Molecule F7. Mol. Ther. 2021, 29, 702–717. [Google Scholar] [CrossRef]
- Diaconu, I.; Ballard, B.; Zhang, M.; Chen, Y.; West, J.; Dotti, G.; Savoldo, B. Inducible Caspase-9 Selectively Modulates the Toxicities of CD19-Specific Chimeric Antigen Receptor-Modified T Cells. Mol. Ther. 2017, 25, 580–592. [Google Scholar] [CrossRef] [Green Version]
- Warda, W.; Larosa, F.; Neto Da Rocha, M.; Trad, R.; Deconinck, E.; Fajloun, Z.; Faure, C.; Caillot, D.; Moldovan, M.; Valmary-Degano, S.; et al. CML Hematopoietic Stem Cells Expressing IL1RAP Can Be Targeted by Chimeric Antigen Receptor–Engineered T Cells. Cancer Res. 2019, 79, 663–675. [Google Scholar] [CrossRef] [Green Version]
- Duong, M.T.; Collinson-Pautz, M.R.; Morschl, E.; Lu, A.; Szymanski, S.P.; Zhang, M.; Brandt, M.E.; Chang, W.-C.; Sharp, K.L.; Toler, S.M.; et al. Two-Dimensional Regulation of CAR-T Cell Therapy with Orthogonal Switches. Mol. Ther. Oncolytics 2019, 12, 124–137. [Google Scholar] [CrossRef] [Green Version]
- Webster, A.C.; Lee, V.W.S.; Chapman, J.R.; Craig, J.C. Target of Rapamycin Inhibitors (Sirolimus and Everolimus) for Primary Immunosuppression of Kidney Transplant Recipients: A Systematic Review and Meta-Analysis of Randomized Trials. Transplantation 2006, 81, 1234–1248. [Google Scholar] [CrossRef]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Tong, Y.; Dotti, G.; Shaim, H.; Savoldo, B.; Mukherjee, M.; Orange, J.; Wan, X.; Lu, X.; Reynolds, A.; et al. Cord Blood NK Cells Engineered to Express IL-15 and a CD19-Targeted CAR Show Long-Term Persistence and Potent Antitumor Activity. Leukemia 2018, 32, 520–531. [Google Scholar] [CrossRef]
- Dey, D.; Evans, G.R. Suicide Gene Therapy by Herpes Simplex Virus-1 Thymidine Kinase (HSV-TK). In Targets in Gene Therapy; You, Y., Ed.; InTech: London, UK, 2011. [Google Scholar] [CrossRef] [Green Version]
- Greco, R.; Oliveira, G.; Stanghellini, M.T.L.; Vago, L.; Bondanza, A.; Peccatori, J.; Cieri, N.; Marktel, S.; Mastaglio, S.; Bordignon, C.; et al. Improving the Safety of Cell Therapy with the TK-Suicide Gene. Front. Pharmacol. 2015, 6, 95. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Shen, F.; Xu, X.; Zhang, H.; Yang, X.; Liu, W. Gene Therapy with HSV1-Sr39TK/GCV Exhibits a Stronger Therapeutic Efficacy Than HSV1-TK/GCV in Rat C6 Glioma Cells. Sci. World J. 2013, 2013, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, H.; Gilmour, K.; Chan, L.; Farzaneh, F.; McNicol, A.M.; Xu, J.-H.; Adams, S.; Fehse, B.; Veys, P.; Thrasher, A.; et al. Production and First-in-Man Use of T Cells Engineered to Express a HSVTK-CD34 Sort-Suicide Gene. PLoS ONE 2013, 8, e77106. [Google Scholar] [CrossRef]
- Casucci, M.; Falcone, L.; Camisa, B.; Norelli, M.; Porcellini, S.; Stornaiuolo, A.; Ciceri, F.; Traversari, C.; Bordignon, C.; Bonini, C.; et al. Extracellular NGFR Spacers Allow Efficient Tracking and Enrichment of Fully Functional CAR-T Cells Co-Expressing a Suicide Gene. Front. Immunol. 2018, 9, 507. [Google Scholar] [CrossRef] [Green Version]
- Klopp, A.; Schreiber, S.; Kosinska, A.D.; Pulé, M.; Protzer, U.; Wisskirchen, K. Depletion of T Cells via Inducible Caspase 9 Increases Safety of Adoptive T-Cell Therapy Against Chronic Hepatitis B. Front. Immunol. 2021, 12, 734246. [Google Scholar] [CrossRef]
- Murty, S.; Labanieh, L.; Murty, T.; Gowrishankar, G.; Haywood, T.; Alam, I.S.; Beinat, C.; Robinson, E.; Aalipour, A.; Klysz, D.D.; et al. PET Reporter Gene Imaging and Ganciclovir-Mediated Ablation of Chimeric Antigen Receptor T Cells in Solid Tumors. Cancer Res. 2020, 80, 4731–4740. [Google Scholar] [CrossRef]
- Najjar, A.M.; Manuri, P.R.; Olivares, S.; Flores, L.; Mi, T.; Huls, H.; Shpall, E.J.; Champlin, R.E.; Turkman, N.; Paolillo, V.; et al. Imaging of Sleeping Beauty-Modified CD19-Specific T Cells Expressing HSV1-Thymidine Kinase by Positron Emission Tomography. Mol. Imaging Biol. 2016, 18, 838–848. [Google Scholar] [CrossRef] [Green Version]
- Sommer, C.; Cheng, H.-Y.; Nguyen, D.; Dettling, D.; Yeung, Y.A.; Sutton, J.; Hamze, M.; Valton, J.; Smith, J.; Djuretic, I.; et al. Allogeneic FLT3 CAR T Cells with an Off-Switch Exhibit Potent Activity against AML and Can Be Depleted to Expedite Bone Marrow Recovery. Mol. Ther. 2020, 28, 2237–2251. [Google Scholar] [CrossRef]
- Wang, Q.; He, F.; He, W.; Huang, Y.; Zeng, J.; Zi, F.; Zheng, J.; Fei, Y.; Xu, J.; Song, Y.; et al. A Transgene-Encoded Truncated Human Epidermal Growth Factor Receptor for Depletion of Anti- B-Cell Maturation Antigen CAR-T Cells. Cell. Immunol. 2021, 363, 104342. [Google Scholar] [CrossRef] [PubMed]
- Kao, R.L.; Truscott, L.C.; Chiou, T.-T.; Tsai, W.; Wu, A.M.; De Oliveira, S.N. A Cetuximab-Mediated Suicide System in Chimeric Antigen Receptor–Modified Hematopoietic Stem Cells for Cancer Therapy. Hum. Gene Ther. 2019, 30, 413–428. [Google Scholar] [CrossRef] [PubMed]
- Tran, V.L.; Novell, A.; Tournier, N.; Gerstenmayer, M.; Schweitzer-Chaput, A.; Mateos, C.; Jego, B.; Bouleau, A.; Nozach, H.; Winkeler, A.; et al. Impact of Blood-Brain Barrier Permeabilization Induced by Ultrasound Associated to Microbubbles on the Brain Delivery and Kinetics of Cetuximab: An ImmunoPET Study Using 89Zr-Cetuximab. J. Control. Release 2020, 328, 304–312. [Google Scholar] [CrossRef]
- Bonnan, M.; Ferrari, S.; Bertandeau, E.; Demasles, S.; Krim, E.; Miquel, M.; Barroso, B. Intrathecal Rituximab Therapy in Multiple Sclerosis: Review of Evidence Supporting the Need for Future Trials. CDT 2014, 15, 1205–1214. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Sun, J.; Wu, Z.; Yu, J.; Cui, Q.; Pu, C.; Liang, B.; Luo, Y.; Shi, J.; Jin, A.; et al. Predominant Cerebral Cytokine Release Syndrome in CD19-Directed Chimeric Antigen Receptor-Modified T Cell Therapy. J. Hematol. Oncol. 2016, 9, 70. [Google Scholar] [CrossRef] [Green Version]
- Gust, J.; Hay, K.A.; Hanafi, L.-A.; Li, D.; Myerson, D.; Gonzalez-Cuyar, L.F.; Yeung, C.; Liles, W.C.; Wurfel, M.; Lopez, J.A.; et al. Endothelial Activation and Blood–Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells. Cancer Discov. 2017, 7, 1404–1419. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Gao, Q.; Liu, H.; Lin, S.; Chen, H.; Ding, R.; Gu, Y.; Chao, C.; Dong, X. The Targeting Effect of Cetuximab Combined with PD-L1 Blockade against EGFR-Expressing Tumors in a Tailored CD16-CAR T-Cell Reporter System. Cancer Investig. 2021, 39, 285–296. [Google Scholar] [CrossRef]
- Caratelli, S.; Arriga, R.; Sconocchia, T.; Ottaviani, A.; Lanzilli, G.; Pastore, D.; Cenciarelli, C.; Venditti, A.; Del Principe, M.I.; Lauro, D.; et al. In Vitro Elimination of Epidermal Growth Factor Receptor-overexpressing Cancer Cells by CD32A-chimeric Receptor T Cells in Combination with Cetuximab or Panitumumab. Int. J. Cancer 2020, 146, 236–247. [Google Scholar] [CrossRef]
- Shah, N.N.; Johnson, B.D.; Schneider, D.; Zhu, F.; Szabo, A.; Keever-Taylor, C.A.; Krueger, W.; Worden, A.A.; Kadan, M.J.; Yim, S.; et al. Bispecific Anti-CD20, Anti-CD19 CAR T Cells for Relapsed B Cell Malignancies: A Phase 1 Dose Escalation and Expansion Trial. Nat. Med. 2020, 26, 1569–1575. [Google Scholar] [CrossRef]
- Wermke, M.; Kraus, S.; Ehninger, A.; Bargou, R.C.; Goebeler, M.-E.; Middeke, J.M.; Kreissig, C.; von Bonin, M.; Koedam, J.; Pehl, M.; et al. Proof of Concept for a Rapidly Switchable Universal CAR-T Platform with UniCAR-T-CD123 in Relapsed/Refractory AML. Blood 2021, 137, 3145–3148. [Google Scholar] [CrossRef]
- Jureczek, J.; Feldmann, A.; Bergmann, R.; Arndt, C.; Berndt, N.; Koristka, S.; Loureiro, L.R.; Mitwasi, N.; Hoffmann, A.; Kegler, A.; et al. Highly Efficient Targeting of EGFR-Expressing Tumor Cells with UniCAR T Cells via Target Modules Based on Cetuximab®. OncoTargets Ther. 2020, 13, 5515–5527. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.G.; Chu, H.; Lu, Y.; Leamon, C.P.; Srinivasarao, M.; Putt, K.S.; Low, P.S. Regulation of CAR T Cell-Mediated Cytokine Release Syndrome-like Toxicity Using Low Molecular Weight Adapters. Nat. Commun. 2019, 10, 2681. [Google Scholar] [CrossRef]
- Cho, J.H.; Collins, J.J.; Wong, W.W. Universal Chimeric Antigen Receptors for Multiplexed and Logical Control of T Cell Responses. Cell 2018, 173, 1426–1438.e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohmueller, J.J.; Ham, J.D.; Kvorjak, M.; Finn, O.J. MSA2 Affinity-Enhanced Biotin-Binding CAR T Cells for Universal Tumor Targeting. OncoImmunology 2018, 7, e1368604. [Google Scholar] [CrossRef]
- Lee, Y.G.; Marks, I.; Srinivasarao, M.; Kanduluru, A.K.; Mahalingam, S.M.; Liu, X.; Chu, H.; Low, P.S. Use of a Single CAR T Cell and Several Bispecific Adapters Facilitates Eradication of Multiple Antigenically Different Solid Tumors. Cancer Res. 2019, 79, 387–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raj, D.; Yang, M.-H.; Rodgers, D.; Hampton, E.N.; Begum, J.; Mustafa, A.; Lorizio, D.; Garces, I.; Propper, D.; Kench, J.G.; et al. Switchable CAR-T Cells Mediate Remission in Metastatic Pancreatic Ductal Adenocarcinoma. Gut 2019, 68, 1052–1064. [Google Scholar] [CrossRef] [Green Version]
- Viaud, S.; Ma, J.S.Y.; Hardy, I.R.; Hampton, E.N.; Benish, B.; Sherwood, L.; Nunez, V.; Ackerman, C.J.; Khialeeva, E.; Weglarz, M.; et al. Switchable Control over in Vivo CAR T Expansion, B Cell Depletion, and Induction of Memory. Proc. Natl. Acad. Sci. USA 2018, 115, E10898–E10906. [Google Scholar] [CrossRef] [Green Version]
- Mitwasi, N.; Feldmann, A.; Arndt, C.; Koristka, S.; Berndt, N.; Jureczek, J.; Loureiro, L.R.; Bergmann, R.; Máthé, D.; Hegedüs, N.; et al. “UniCAR”-Modified off-the-Shelf NK-92 Cells for Targeting of GD2-Expressing Tumour Cells. Sci. Rep. 2020, 10, 2141. [Google Scholar] [CrossRef] [Green Version]
- Loff, S.; Dietrich, J.; Meyer, J.-E.; Riewaldt, J.; Spehr, J.; von Bonin, M.; Gründer, C.; Swayampakula, M.; Franke, K.; Feldmann, A.; et al. Rapidly Switchable Universal CAR-T Cells for Treatment of CD123-Positive Leukemia. Mol. Ther. Oncolytics 2020, 17, 408–420. [Google Scholar] [CrossRef]
- Cartellieri, M.; Feldmann, A.; Koristka, S.; Arndt, C.; Loff, S.; Ehninger, A.; von Bonin, M.; Bejestani, E.P.; Ehninger, G.; Bachmann, M.P. Switching CAR T Cells on and off: A Novel Modular Platform for Retargeting of T Cells to AML Blasts. Blood Cancer J. 2016, 6, e458. [Google Scholar] [CrossRef] [Green Version]
- Qi, J.; Tsuji, K.; Hymel, D.; Burke, T.R.; Hudecek, M.; Rader, C.; Peng, H. Chemically Programmable and Switchable CAR-T Therapy. Angew. Chem. Int. Ed. 2020, 59, 12178–12185. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, Y.; Huang, S.; Sun, J.; Wang, M.; Ma, W.; You, Y.; Wu, L.; Hu, J.; Song, W.; et al. Photoswitchable CAR-T Cell Function In Vitro and In Vivo via a Cleavable Mediator. Cell Chem. Biol. 2021, 28, 60–69.e7. [Google Scholar] [CrossRef]
- Loureiro, L.R.; Feldmann, A.; Bergmann, R.; Koristka, S.; Berndt, N.; Máthé, D.; Hegedüs, N.; Szigeti, K.; Videira, P.A.; Bachmann, M.; et al. Extended Half-Life Target Module for Sustainable UniCAR T-Cell Treatment of STn-Expressing Cancers. J. Exp. Clin. Cancer Res. 2020, 39, 77. [Google Scholar] [CrossRef]
- Meyer, J.-E.; Loff, S.; Dietrich, J.; Spehr, J.; Jurado Jiménez, G.; von Bonin, M.; Ehninger, G.; Cartellieri, M.; Ehninger, A. Evaluation of Switch-Mediated Costimulation in Trans on Universal CAR-T Cells (UniCAR) Targeting CD123-Positive AML. OncoImmunology 2021, 10, 1945804. [Google Scholar] [CrossRef] [PubMed]
- Zajc, C.U.; Dobersberger, M.; Schaffner, I.; Mlynek, G.; Pühringer, D.; Salzer, B.; Djinović-Carugo, K.; Steinberger, P.; De Sousa Linhares, A.; Yang, N.J.; et al. A Conformation-Specific ON-Switch for Controlling CAR T Cells with an Orally Available Drug. Proc. Natl. Acad. Sci. USA 2020, 117, 14926–14935. [Google Scholar] [CrossRef] [PubMed]
- Gardner, R.; Wu, D.; Cherian, S.; Fang, M.; Hanafi, L.-A.; Finney, O.; Smithers, H.; Jensen, M.C.; Riddell, S.R.; Maloney, D.G.; et al. Acquisition of a CD19-Negative Myeloid Phenotype Allows Immune Escape of MLL-Rearranged B-ALL from CD19 CAR-T-Cell Therapy. Blood 2016, 127, 2406–2410. [Google Scholar] [CrossRef] [Green Version]
- Rodgers, D.T.; Mazagova, M.; Hampton, E.N.; Cao, Y.; Ramadoss, N.S.; Hardy, I.R.; Schulman, A.; Du, J.; Wang, F.; Singer, O.; et al. Switch-Mediated Activation and Retargeting of CAR-T Cells for B-Cell Malignancies. Proc. Natl. Acad. Sci. USA 2016, 113, E459–E468. [Google Scholar] [CrossRef] [Green Version]
- Khanali, J.; Azangou-Khyavy, M.; Boroomand-Saboor, M.; Ghasemi, M.; Niknejad, H. JAK/STAT-Dependent Chimeric Antigen Receptor (CAR) Expression: A Design Benefiting from a Dual AND/OR Gate Aiming to Increase Specificity, Reduce Tumor Escape and Affect Tumor Microenvironment. Front. Immunol. 2021, 12, 638639. [Google Scholar] [CrossRef]
- Cho, J.H.; Okuma, A.; Sofjan, K.; Lee, S.; Collins, J.J.; Wong, W.W. Engineering Advanced Logic and Distributed Computing in Human CAR Immune Cells. Nat. Commun. 2021, 12, 792. [Google Scholar] [CrossRef]
- Golubovskaya, V.M. GITR Domain inside CAR Co-Stimulates Activity of CAR-T Cells against Cancer. Front. Biosci. 2018, 23, 2245–2254. [Google Scholar] [CrossRef]
- Sun, M.; Xu, P.; Wang, E.; Zhou, M.; Xu, T.; Wang, J.; Wang, Q.; Wang, B.; Lu, K.; Wang, C.; et al. Novel Two-Chain Structure Utilizing KIRS2/DAP12 Domain Improves the Safety and Efficacy of CAR-T Cells in Adults with r/r B-ALL. Mol. Ther. Oncolytics 2021, 23, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Lanitis, E.; Rota, G.; Kosti, P.; Ronet, C.; Spill, A.; Seijo, B.; Romero, P.; Dangaj, D.; Coukos, G.; Irving, M. Optimized Gene Engineering of Murine CAR-T Cells Reveals the Beneficial Effects of IL-15 Coexpression. J. Exp. Med. 2021, 218, e20192203. [Google Scholar] [CrossRef] [PubMed]
- Voß, S.; Klewer, L.; Wu, Y.-W. Chemically Induced Dimerization: Reversible and Spatiotemporal Control of Protein Function in Cells. Curr. Opin. Chem. Biol. 2015, 28, 194–201. [Google Scholar] [CrossRef]
- Mata, M.; Gerken, C.; Nguyen, P.; Krenciute, G.; Spencer, D.M.; Gottschalk, S. Inducible Activation of MyD88 and CD40 in CAR T Cells Results in Controllable and Potent Antitumor Activity in Preclinical Solid Tumor Models. Cancer Discov. 2017, 7, 1306–1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaczanowska, S.; Joseph, A.M.; Davila, E. TLR Agonists: Our Best Frenemy in Cancer Immunotherapy. J. Leukoc. Biol. 2013, 93, 847–863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isogawa, M.; Chung, J.; Murata, Y.; Kakimi, K.; Chisari, F.V. CD40 Activation Rescues Antiviral CD8+ T Cells from PD-1-Mediated Exhaustion. PLoS Pathog. 2013, 9, e1003490. [Google Scholar] [CrossRef] [Green Version]
- Collinson-Pautz, M.R.; Chang, W.-C.; Lu, A.; Khalil, M.; Crisostomo, J.W.; Lin, P.-Y.; Mahendravada, A.; Shinners, N.P.; Brandt, M.E.; Zhang, M.; et al. Constitutively Active MyD88/CD40 Costimulation Enhances Expansion and Efficacy of Chimeric Antigen Receptor T Cells Targeting Hematological Malignancies. Leukemia 2019, 33, 2195–2207. [Google Scholar] [CrossRef] [Green Version]
- Prinzing, B.; Schreiner, P.; Bell, M.; Fan, Y.; Krenciute, G.; Gottschalk, S. MyD88/CD40 Signaling Retains CAR T Cells in a Less Differentiated State. JCI Insight 2020, 5, e136093. [Google Scholar] [CrossRef]
- Wang, X.; Jasinski, D.L.; Medina, J.L.; Spencer, D.M.; Foster, A.E.; Bayle, J.H. Inducible MyD88/CD40 Synergizes with IL-15 to Enhance Antitumor Efficacy of CAR-NK Cells. Blood Adv. 2020, 4, 1950–1964. [Google Scholar] [CrossRef]
- Vinanica, N.; Yong, A.; Wong, D.; Png, Y.T.; Seow, S.V.; Imamura, M.; Campana, D. Specific Stimulation of T Lymphocytes with Erythropoietin for Adoptive Immunotherapy. Blood 2020, 135, 668–679. [Google Scholar] [CrossRef]
- Zhang, Q.; Hresko, M.E.; Picton, L.K.; Su, L.; Hollander, M.J.; Nunez-Cruz, S.; Zhang, Z.; Assenmacher, C.-A.; Sockolosky, J.T.; Garcia, K.C.; et al. A Human Orthogonal IL-2 and IL-2Rβ System Enhances CAR T Cell Expansion and Antitumor Activity in a Murine Model of Leukemia. Sci. Transl. Med. 2021, 13, eabg6986. [Google Scholar] [CrossRef] [PubMed]
- Miller, I.C.; Zamat, A.; Sun, L.-K.; Phuengkham, H.; Harris, A.M.; Gamboa, L.; Yang, J.; Murad, J.P.; Priceman, S.J.; Kwong, G.A. Enhanced Intratumoural Activity of CAR T Cells Engineered to Produce Immunomodulators under Photothermal Control. Nat. Biomed. Eng. 2021, 5, 1348–1359. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, D.T.; Antony, J.; Martin, A.J.; Moser, R.J.; Hekele, A.; Wetzel, K.J.; Brown, A.E.; Triggiano, M.A.; Hux, J.A.; Pham, C.D.; et al. Integration of a CD19 CAR into the TCR Alpha Chain Locus Streamlines Production of Allogeneic Gene-Edited CAR T Cells. Mol. Ther. 2017, 25, 949–961. [Google Scholar] [CrossRef] [Green Version]
- Sommer, C.; Boldajipour, B.; Kuo, T.C.; Bentley, T.; Sutton, J.; Chen, A.; Geng, T.; Dong, H.; Galetto, R.; Valton, J.; et al. Preclinical Evaluation of Allogeneic CAR T Cells Targeting BCMA for the Treatment of Multiple Myeloma. Mol. Ther. 2019, 27, 1126–1138. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Smith, M.; James, S.E.; Davila, M.L.; Velardi, E.; Argyropoulos, K.V.; Gunset, G.; Perna, F.; Kreines, F.M.; Levy, E.R.; et al. Donor CD19 CAR T Cells Exert Potent Graft-versus-Lymphoma Activity with Diminished Graft-versus-Host Activity. Nat. Med. 2017, 23, 242–249. [Google Scholar] [CrossRef] [Green Version]
- Fisher, J.; Anderson, J. Engineering Approaches in Human Gamma Delta T Cells for Cancer Immunotherapy. Front. Immunol. 2018, 9, 1409. [Google Scholar] [CrossRef]
- Wang, D.; Quan, Y.; Yan, Q.; Morales, J.E.; Wetsel, R.A. Targeted Disruption of the β 2-Microglobulin Gene Minimizes the Immunogenicity of Human Embryonic Stem Cells. Stem Cells Transl. Med. 2015, 4, 1234–1245. [Google Scholar] [CrossRef]
- Xu, H.; Wang, B.; Ono, M.; Kagita, A.; Fujii, K.; Sasakawa, N.; Ueda, T.; Gee, P.; Nishikawa, M.; Nomura, M.; et al. Targeted Disruption of HLA Genes via CRISPR-Cas9 Generates IPSCs with Enhanced Immune Compatibility. Cell Stem Cell 2019, 24, 566–578.e7. [Google Scholar] [CrossRef] [Green Version]
- Koga, K.; Wang, B.; Kaneko, S. Current Status and Future Perspectives of HLA-Edited Induced Pluripotent Stem Cells. Inflamm. Regener. 2020, 40, 23. [Google Scholar] [CrossRef]
- Flahou, C.; Morishima, T.; Takizawa, H.; Sugimoto, N. Fit-For-All IPSC-Derived Cell Therapies and Their Evaluation in Humanized Mice with NK Cell Immunity. Front. Immunol. 2021, 12, 662360. [Google Scholar] [CrossRef]
- Athanasopoulos, T.; Munye, M.M.; Yáñez-Muñoz, R.J. Nonintegrating Gene Therapy Vectors. Hematol. Oncol. Clin. N. Am. 2017, 31, 753–770. [Google Scholar] [CrossRef] [PubMed]
- Baum, C.; Kustikova, O.; Modlich, U.; Li, Z.; Fehse, B. Mutagenesis and Oncogenesis by Chromosomal Insertion of Gene Transfer Vectors. Hum. Gene Ther. 2006, 17, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Ciuffi, A. The Benefits of Integration. Clin. Microbiol. Infect. 2016, 22, 324–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milone, M.C.; O’Doherty, U. Clinical Use of Lentiviral Vectors. Leukemia 2018, 32, 1529–1541. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Nakazawa, Y.; Huye, L.E.; Doherty, J.E.; Galvan, D.L.; Rooney, C.M.; Wilson, M.H. PiggyBac Transposon System Modification of Primary Human T Cells. JoVE 2012, 69, 4235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magnani, C.F.; Tettamanti, S.; Alberti, G.; Pisani, I.; Biondi, A.; Serafini, M.; Gaipa, G. Transposon-Based CAR T Cells in Acute Leukemias: Where Are We Going? Cells 2020, 9, 1337. [Google Scholar] [CrossRef]
- Tsukahara, T.; Iwase, N.; Kawakami, K.; Iwasaki, M.; Yamamoto, C.; Ohmine, K.; Uchibori, R.; Teruya, T.; Ido, H.; Saga, Y.; et al. The Tol2 Transposon System Mediates the Genetic Engineering of T-Cells with CD19-Specific Chimeric Antigen Receptors for B-Cell Malignancies. Gene Ther. 2015, 22, 209–215. [Google Scholar] [CrossRef]
- Bishop, D.C.; Caproni, L.; Gowrishankar, K.; Legiewicz, M.; Karbowniczek, K.; Tite, J.; Gottlieb, D.J.; Micklethwaite, K.P. CAR T Cell Generation by PiggyBac Transposition from Linear Doggybone DNA Vectors Requires Transposon DNA-Flanking Regions. Mol. Ther. Methods Clin. Dev. 2020, 17, 359–368. [Google Scholar] [CrossRef] [Green Version]
- Cooney, A.L.; Singh, B.K.; Sinn, P.L. Hybrid Nonviral/Viral Vector Systems for Improved PiggyBac DNA Transposon In Vivo Delivery. Mol. Ther. 2015, 23, 667–674. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Głowacki, P.; Rieske, P. Application and Design of Switches Used in CAR. Cells 2022, 11, 1910. https://doi.org/10.3390/cells11121910
Głowacki P, Rieske P. Application and Design of Switches Used in CAR. Cells. 2022; 11(12):1910. https://doi.org/10.3390/cells11121910
Chicago/Turabian StyleGłowacki, Paweł, and Piotr Rieske. 2022. "Application and Design of Switches Used in CAR" Cells 11, no. 12: 1910. https://doi.org/10.3390/cells11121910
APA StyleGłowacki, P., & Rieske, P. (2022). Application and Design of Switches Used in CAR. Cells, 11(12), 1910. https://doi.org/10.3390/cells11121910