
Pathophysiological Roles of Abnormal Axon Initial Segments in Neurodevelopmental Disorders


Abstract
:1. Introduction
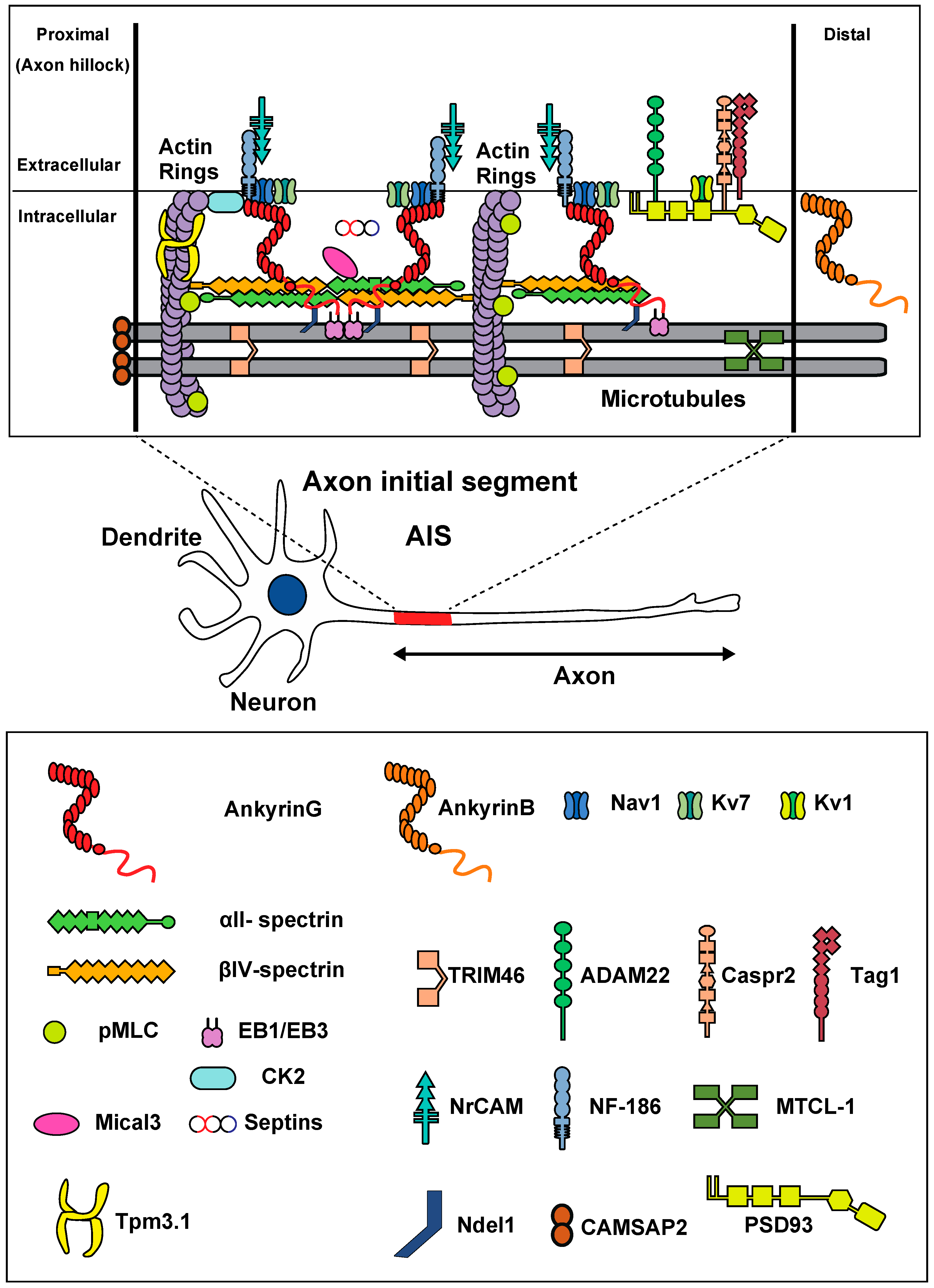
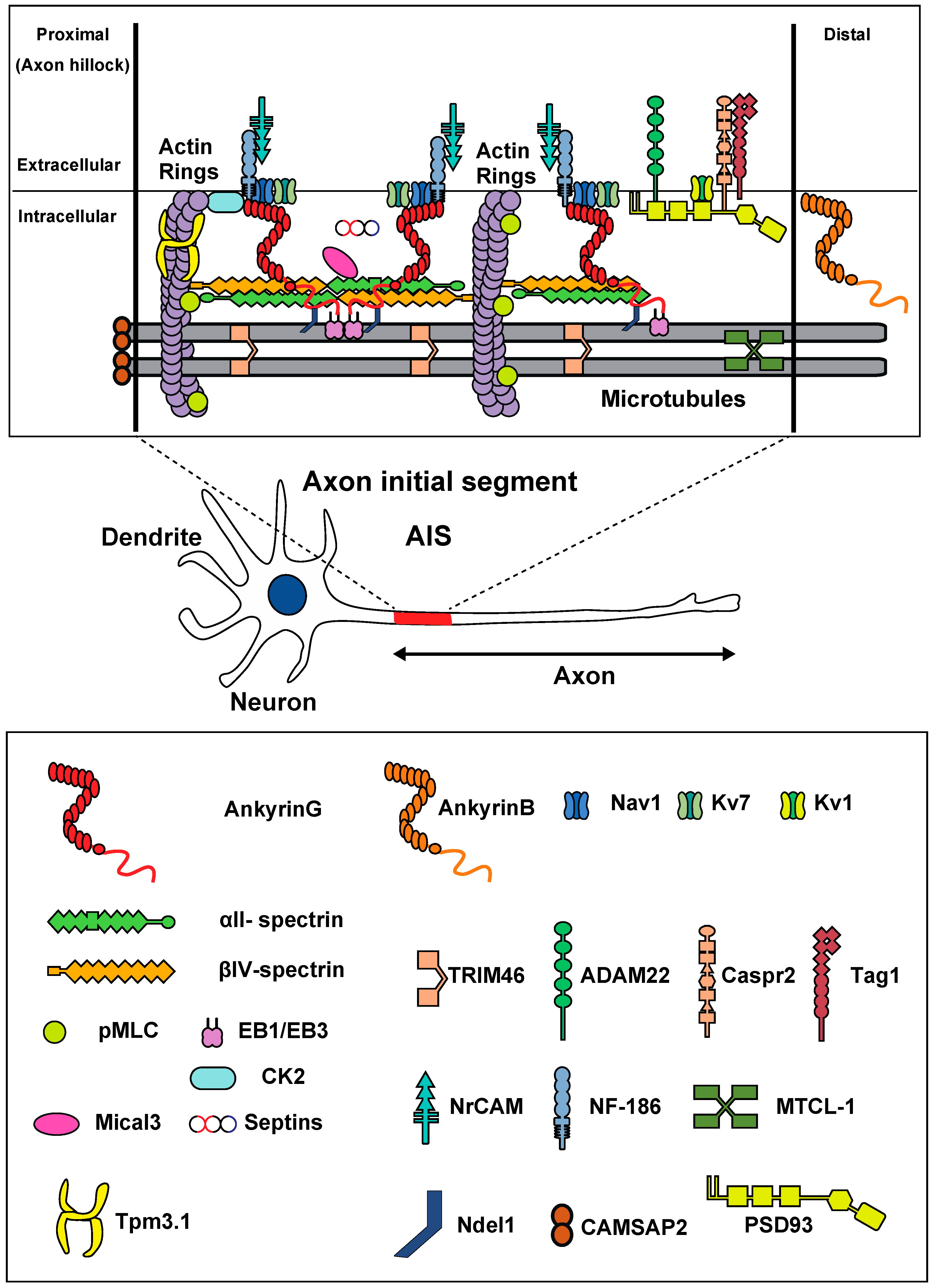
2. Molecular Characteristics of the AIS
3. Structural Characteristics of the AIS
4. Ion Channel Properties of the AIS
5. Non-Cell-Autonomous AIS Regulation through Axo-Axonic Synapse and Axo-Glial Interactions
6. Activity-Responding Plasticity of AIS in Development and Disease Models
7. Association and Mutation Studies on AIS-Related Genes
8. Abnormal AIS Characteristics in Neurodevelopmental Disorders
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huang, C.Y.; Rasband, M.N. Axon Initial Segments: Structure, Function, and Disease. Ann. N. Y. Acad. Sci. 2018, 1420, 46–61. [Google Scholar] [CrossRef] [PubMed]
- Leterrier, C. The Axon Initial Segment: An Updated Viewpoint. J. Neurosci. 2018, 38, 2135–2145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, V.; Lorenzo, D.N. Spectrin- and Ankyrin-Based Membrane Domains and the Evolution of Vertebrates. Curr. Top. Membr. 2013, 72, 1–37. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.D.; Jenkins, P.M. Axonal Membranes and Their Domains: Assembly and Function of the Axon Initial Segment and Node of Ranvier. Front. Cell. Neurosci. 2017, 11, 136. [Google Scholar] [CrossRef] [Green Version]
- Palay, S.L.; Sotelo, C.; Peters, A.; Orkand, P.M. The Axon Hillock and the Initial Segment. J. Cell Biol. 1968, 38, 193–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, A.; Proskauer, C.C.; Kaiserman-Abramof, I.R. The Small Pyramidal Neuron of the Rat Cerebral Cortex. The Axon Hillock and Initial Segment. J. Cell Biol. 1968, 39, 604–619. [Google Scholar] [CrossRef] [Green Version]
- Lorincz, A.; Nusser, Z. Cell-Type-Dependent Molecular Composition of the Axon Initial Segment. J. Neurosci. 2008, 28, 14329–14340. [Google Scholar] [CrossRef]
- Kuba, H.; Ishii, T.M.; Ohmori, H. Axonal Site of Spike Initiation Enhances Auditory Coincidence Detection. Nature 2006, 444, 1069–1072. [Google Scholar] [CrossRef]
- Bender, K.J.; Trussell, L.O. The Physiology of the Axon Initial Segment. Annu. Rev. Neurosci. 2012, 35, 249–265. [Google Scholar] [CrossRef]
- Kole, M.H.; Stuart, G.J. Signal Processing in the Axon Initial Segment. Neuron 2012, 73, 235–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, R.; Kuba, H. Structural and Functional Plasticity at the Axon Initial Segment. Front. Cell. Neurosci. 2016, 10, 250. [Google Scholar] [CrossRef] [Green Version]
- Kordeli, E.; Lambert, S.; Bennett, V.; Ankyrin, G. AnkyrinG. A New Ankyrin Gene with Neural-Specific Isoforms Localized at the Axonal Initial Segment and Node of Ranvier. J. Biol. Chem. 1995, 270, 2352–2359. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, P.M.; Kim, N.; Jones, S.L.; Tseng, W.C.; Svitkina, T.M.; Yin, H.H.; Bennett, V. Giant Ankyrin-G: A Critical Innovation in Vertebrate Evolution of Fast and Integrated Neuronal Signaling. Proc. Natl. Acad. Sci. USA 2015, 112, 957–964. [Google Scholar] [CrossRef] [Green Version]
- Boiko, T.; Vakulenko, M.; Ewers, H.; Yap, C.C.; Norden, C.; Winckler, B. Ankyrin-Dependent and -Independent Mechanisms Orchestrate Axonal Compartmentalization of L1 Family Members Neurofascin and L1/Neuron-Glia Cell Adhesion Molecule. J. Neurosci. 2007, 27, 590–603. [Google Scholar] [CrossRef] [Green Version]
- Galiano, M.R.; Jha, S.; Ho, T.S.; Zhang, C.; Ogawa, Y.; Chang, K.J.; Stankewich, M.C.; Mohler, P.J.; Rasband, M.N. A Distal Axonal Cytoskeleton Forms an Intra-Axonal Boundary That Controls Axon Initial Segment Assembly. Cell 2012, 149, 1125–1139. [Google Scholar] [CrossRef] [Green Version]
- Zhou, D.X.; Lambert, S.; Malen, P.L.; Carpenter, S.; Boland, L.M.; Bennett, V. Ankyring Is Required for Clustering of Voltage-Gated Na Channels at Axon Initial Segments and for Normal Action Potential Firing. Mol. Biol. Cell 1998, 143, 1295–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkins, S.M.; Bennett, V. Ankyrin-G Coordinates Assembly of the Spectrin-Based Membrane Skeleton, Voltage-Gated Sodium Channels, and L1 CAMs at Purkinje Neuron Initial Segments. J. Cell Biol. 2001, 155, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Hedstrom, K.L.; Xu, X.; Ogawa, Y.; Frischknecht, R.; Seidenbecher, C.I.; Shrager, P.; Rasband, M.N. Neurofascin Assembles a Specialized Extracellular Matrix at the Axon Initial Segment. J. Cell Biol. 2007, 178, 875–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legendre, K.; Safieddine, S.; Küssel-Andermann, P.; Petit, C.; El-Amraoui, A. alphaII-betaV Spectrin Bridges the Plasma Membrane and Cortical Lattice in the Lateral Wall of the Auditory Outer Hair Cells. J. Cell Sci. 2008, 121, 3347–3356. [Google Scholar] [CrossRef] [Green Version]
- Berghs, S.; Aggujaro, D.; Dirkx, R.; Maksimova, E.; Stabach, P.; Hermel, J.M.; Zhang, J.P.; Philbrick, W.; Slepnev, V.; Ort, T.; et al. Beta IV Spectrin, a New Spectrin Localized at Axon Initial Segments and Nodes of Ranvier in the Central and Peripheral Nervous System. J. Cell Biol. 2000, 151, 985–1002. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.Y.; Zhang, C.; Zollinger, D.R.; Leterrier, C.; Rasband, M.N. An alphaII Spectrin-Based Cytoskeleton Protects Large-Diameter Myelinated Axons from Degeneration. J. Neurosci. 2017, 37, 11323–11334. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.Y.; Zhang, C.; Ho, T.S.; Oses-Prieto, J.; Burlingame, A.L.; Lalonde, J.; Noebels, J.L.; Leterrier, C.; Rasband, M.N. alphaII Spectrin Forms a Periodic Cytoskeleton at the Axon Initial Segment and Is Required for Nervous System Function. J. Neurosci. 2017, 37, 11311–11322. [Google Scholar] [CrossRef] [Green Version]
- Stankewich, M.C.; Cianci, C.D.; Stabach, P.R.; Ji, L.; Nath, A.; Morrow, J.S. Cell Organization, Growth, and Neural and Cardiac Development Require alphaII-spectrin. J. Cell Sci. 2011, 124, 3956–3966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Ji, T.; Nelson, A.D.; Glanowska, K.; Murphy, G.G.; Jenkins, P.M.; Parent, J.M. Critical Roles of alphaII Spectrin in Brain Development and Epileptic Encephalopathy. J. Clin. Investig. 2018, 128, 760–773. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.H.; Seo, R.; Ho, T.S.; Stankewich, M.; Mohler, P.J.; Hund, T.J.; Noebels, J.L.; Rasband, M.N.; Rasband, M.N. Beta Spectrin-Dependent and Domain Specific Mechanisms for Na+ Channel Clustering. eLife 2020, 9, e56629. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, T.; Rasband, M.N. Axon Initial Segments: Diverse and Dynamic Neuronal Compartments. Curr. Opin. Neurobiol. 2014, 27, 96–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuijpers, M.; van de Willige, D.; Freal, A.; Chazeau, A.; Franker, M.A.; Hofenk, J.; Rodrigues, R.J.; Kapitein, L.C.; Akhmanova, A.; Jaarsma, D.; et al. Dynein Regulator NDEL1 Controls Polarized Cargo Transport at the Axon Initial Segment. Neuron 2016, 89, 461–471. [Google Scholar] [CrossRef] [Green Version]
- Davis, J.Q.; Lambert, S.; Bennett, V. Molecular Composition of the Node of Ranvier: Identification of Ankyrin-Binding Cell Adhesion Molecules Neurofascin (Mucin+/third FNIII Domain-) and NrCAM at Nodal Axon Segments. J. Cell Biol. 1996, 135, 1355–1367. [Google Scholar] [CrossRef] [Green Version]
- Zonta, B.; Desmazieres, A.; Rinaldi, A.; Tait, S.; Sherman, D.L.; Nolan, M.F.; Brophy, P.J. A Critical Role for Neurofascin in Regulating Action Potential Initiation Through Maintenance of the Axon Initial Segment. Neuron 2011, 69, 945–956. [Google Scholar] [CrossRef] [Green Version]
- Alpizar, S.A.; Baker, A.L.; Gulledge, A.T.; Hoppa, M.B. Loss of Neurofascin-186 Disrupts Alignment of AnkyrinG Relative to Its Binding Partners in the Axon Initial Segment. Front. Cell. Neurosci. 2019, 13, 1. [Google Scholar] [CrossRef] [Green Version]
- Leterrier, C.; Clerc, N.; Rueda-Boroni, F.; Montersino, A.; Dargent, B.; Castets, F.; Ankyrin, G. Ankyrin G Membrane Partners Drive the Establishment and Maintenance of the Axon Initial Segment. Front. Cell. Neurosci. 2017, 11, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leterrier, C.; Vacher, H.; Fache, M.P.; d’Ortoli, S.A.; Castets, F.; Autillo-Touati, A.; Dargent, B. End-Binding Proteins EB3 and EB1 Link Microtubules to Ankyrin G in the Axon Initial Segment. Proc. Natl. Acad. Sci. USA 2011, 108, 8826–8831. [Google Scholar] [CrossRef] [Green Version]
- Van Beuningen, S.F.B.; Will, L.; Harterink, M.; Chazeau, A.; van Battum, E.Y.; Frias, C.P.; Franker, M.A.M.; Katrukha, E.A.; Stucchi, R.; Vocking, K.; et al. TRIM46 Controls Neuronal Polarity and Axon Specification by Driving the Formation of Parallel Microtubule Arrays. Neuron 2015, 88, 1208–1226. [Google Scholar] [CrossRef] [Green Version]
- Satake, T.; Yamashita, K.; Hayashi, K.; Miyatake, S.; Tamura-Nakano, M.; Doi, H.; Furuta, Y.; Shioi, G.; Miura, E.; Takeo, Y.H.; et al. MTCL1 Plays an Essential Role in Maintaining Purkinje Neuron Axon Initial Segment. EMBO J. 2017, 36, 1227–1242. [Google Scholar] [CrossRef]
- Ogawa, Y.; Oses-Prieto, J.; Kim, M.Y.; Horresh, I.; Peles, E.; Burlingame, A.L.; Trimmer, J.S.; Meijer, D.; Rasband, M.N.A. ADAM22, a Kv1 Channel-Interacting Protein, Recruits Membrane-Associated Guanylate Kinases to Juxtaparanodes of Myelinated Axons. J. Neurosci. 2010, 30, 1038–1048. [Google Scholar] [CrossRef]
- Bréchet, A.; Fache, M.P.; Brachet, A.; Ferracci, G.; Baude, A.; Irondelle, M.; Pereira, S.; Leterrier, C.; Dargent, B. Protein Kinase CK2 Contributes to the Organization of Sodium Channels in Axonal Membranes by Regulating Their Interactions with Ankyrin G. J. Cell Biol. 2008, 183, 1101–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, S.L.; Leo-Macias, A.; Yuen, S.; Khatri, L.; Pfennig, S.; Zhang, Y.; Agullo-Pascual, E.; Caillol, G.; Zhu, M.S.; Rothenberg, E.; et al. Localized Myosin II Activity Regulates Assembly and Plasticity of the Axon Initial Segment. Neuron 2018, 97, 555–570.e6. [Google Scholar] [CrossRef] [Green Version]
- Abouelezz, A.; Stefen, H.; Segerstråle, M.; Micinski, D.; Minkeviciene, R.; Lahti, L.; Hardeman, E.C.; Gunning, P.W.; Hoogenraad, C.C.; Taira, T.; et al. Tropomyosin Tpm3.1 Is Required to Maintain the Structure and Function of the Axon Initial Segment. iScience 2020, 23, 101053. [Google Scholar] [CrossRef] [PubMed]
- Hamdan, H.; Lim, B.C.; Torii, T.; Joshi, A.; Konning, M.; Smith, C.; Palmer, D.J.; Ng, P.; Leterrier, C.; Oses-Prieto, J.A.; et al. Mapping Axon Initial Segment Structure and Function by Multiplexed Proximity Biotinylation. Nat. Commun. 2020, 11, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, K.; Zhong, G.; Zhuang, X. Actin, Spectrin, and Associated Proteins Form a Periodic Cytoskeletal Structure in Axons. Science 2013, 339, 452–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, G.; He, J.; Zhou, R.; Lorenzo, D.; Babcock, H.P.; Bennett, V.; Zhuang, X. Developmental Mechanism of the Periodic Membrane Skeleton in Axons. eLife 2014, 3, e04581. [Google Scholar] [CrossRef]
- Leterrier, C.; Potier, J.; Caillol, G.; Debarnot, C.; Rueda Boroni, F.; Dargent, B. Nanoscale Architecture of the Axon Initial Segment Reveals an Organized and Robust Scaffold. Cell Rep. 2015, 13, 2781–2793. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.L.; Korobova, F.; Svitkina, T. Axon Initial Segment Cytoskeleton Comprises a Multiprotein Submembranous Coat Containing Sparse Actin Filaments. J. Cell Biol. 2014, 205, 67–81. [Google Scholar] [CrossRef] [Green Version]
- Vassilopoulos, S.; Gibaud, S.; Jimenez, A.; Caillol, G.; Leterrier, C. Ultrastructure of the Axonal Periodic Scaffold Reveals a Braid-Like Organization of Actin Rings. Nat. Commun. 2019, 10, 5803. [Google Scholar] [CrossRef] [Green Version]
- Leterrier, C. Putting the Axonal Periodic Scaffold in Order. Curr. Opin. Neurobiol. 2021, 69, 33–40. [Google Scholar] [CrossRef]
- Boiko, T.; Van Wart, A.; Caldwell, J.H.; Levinson, S.R.; Trimmer, J.S.; Matthews, G. Functional Specialization of the Axon Initial Segment by Isoform-Specific Sodium Channel Targeting. J. Neurosci. 2003, 23, 2306–2313. [Google Scholar] [CrossRef] [Green Version]
- Duflocq, A.; Le Bras, B.; Bullier, E.; Couraud, F.; Davenne, M. Nav1.1 Is Predominantly Expressed in Nodes of Ranvier and Axon Initial Segments. Mol. Cell. Neurosci. 2008, 39, 180–192. [Google Scholar] [CrossRef]
- Kole, M.H.; Ilschner, S.U.; Kampa, B.M.; Williams, S.R.; Ruben, P.C.; Stuart, G.J. Action Potential Generation Requires a High Sodium Channel Density in the Axon Initial Segment. Nat. Neurosci. 2008, 11, 178–186. [Google Scholar] [CrossRef]
- Schmidt-Hieber, C.; Bischofberger, J. Fast Sodium Channel Gating Supports Localized and Efficient Axonal Action Potential Initiation. J. Neurosci. 2010, 30, 10233–10242. [Google Scholar] [CrossRef] [Green Version]
- Nathanson, A.J.; Davies, P.A.; Moss, S.J. Inhibitory Synapse Formation at the Axon Initial Segment. Front. Mol. Neurosci. 2019, 12, 266. [Google Scholar] [CrossRef] [Green Version]
- DeFelipe, J.; Hendry, S.H.; Jones, E.G.; Schmechel, D. Variability in the Terminations of GABAergic Chandelier Cell Axons on Initial Segments of Pyramidal Cell Axons in the Monkey Sensory-Motor Cortex. J. Comp. Neurol. 1985, 231, 364–384. [Google Scholar] [CrossRef] [PubMed]
- Somogyi, P. A Specific ‘Axo-Axonal’ Interneuron in the Visual Cortex of the Rat. Brain Res. 1977, 136, 345–350. [Google Scholar] [CrossRef]
- Hines, R.M.; Maric, H.M.; Hines, D.J.; Modgil, A.; Panzanelli, P.; Nakamura, Y.; Nathanson, A.J.; Cross, A.; Deeb, T.; Brandon, N.J.; et al. Developmental Seizures and Mortality Result from Reducing GABAA Receptor alpha2-Subunit Interaction with Collybistin. Nat. Commun. 2018, 9, 3130. [Google Scholar] [CrossRef]
- Inda, M.C.; Defelipe, J.; Muñoz, A. The Distribution of Chandelier Cell Axon Terminals That Express the GABA Plasma Membrane Transporter GAT-1 in the Human Neocortex. Cereb. Cortex 2007, 17, 2060–2071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tai, Y.; Gallo, N.B.; Wang, M.; Yu, J.R.; Van Aelst, L. Axo-Axonic Innervation of Neocortical Pyramidal Neurons by GABAergic Chandelier Cells Requires AnkyrinG-Associated L1CAM. Neuron 2019, 102, 358–372.e9. [Google Scholar] [CrossRef] [Green Version]
- Pan-Vazquez, A.; Wefelmeyer, W.; Gonzalez Sabater, V.; Neves, G.; Burrone, J. Activity-Dependent Plasticity of Axo-Axonic Synapses at the Axon Initial Segment. Neuron 2020, 106, 265–276.e6. [Google Scholar] [CrossRef] [PubMed]
- Baalman, K.; Marin, M.A.; Ho, T.S.; Godoy, M.; Cherian, L.; Robertson, C.; Rasband, M.N. Axon Initial Segment-Associated Microglia. J. Neurosci. 2015, 35, 2283–2292. [Google Scholar] [CrossRef] [Green Version]
- Tamada, H.; Kiryu-Seo, S.; Sawada, S.; Kiyama, H. Axonal Injury Alters the Extracellular Glial Environment of the Axon Initial Segment (AIS) and Allows Substantial Mitochondrial Influx into AIS. J. Comp. Neurol. 2021, 1–12. [Google Scholar] [CrossRef]
- Molofsky, A.V.; Kelley, K.W.; Tsai, H.H.; Redmond, S.A.; Chang, S.M.; Madireddy, L.; Chan, J.R.; Baranzini, S.E.; Ullian, E.M.; Rowitch, D.H. Astrocyte-Encoded Positional Cues Maintain Sensorimotor Circuit Integrity. Nature 2014, 509, 189–194. [Google Scholar] [CrossRef] [Green Version]
- Hamada, M.S.; Kole, M.H. Myelin Loss and Axonal Ion Channel Adaptations Associated with Gray Matter Neuronal Hyperexcitability. J. Neurosci. 2015, 35, 7272–7286. [Google Scholar] [CrossRef] [Green Version]
- Moore, S.; Meschkat, M.; Ruhwedel, T.; Trevisiol, A.; Tzvetanova, I.D.; Battefeld, A.; Kusch, K.; Kole, M.H.P.; Strenzke, N.; Möbius, W.; et al. A Role of Oligodendrocytes in Information Processing. Nat. Commun. 2020, 11, 5497. [Google Scholar] [CrossRef] [PubMed]
- Jamann, N.; Jordan, M.; Engelhardt, M. Activity-Dependent Axonal Plasticity in Sensory Systems. Neuroscience 2018, 368, 268–282. [Google Scholar] [CrossRef]
- Evans, M.D.; Dumitrescu, A.S.; Kruijssen, D.L.H.; Taylor, S.E.; Grubb, M.S. Rapid Modulation of Axon Initial Segment Length Influences Repetitive Spike Firing. Cell Rep. 2015, 13, 1233–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grubb, M.S.; Burrone, J. Activity-Dependent Relocation of the Axon Initial Segment Fine-Tunes Neuronal Excitability. Nature 2010, 465, 1070–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuba, H.; Oichi, Y.; Ohmori, H. Presynaptic Activity Regulates Na+ Channel Distribution at the Axon Initial Segment. Nature 2010, 465, 1075–1078. [Google Scholar] [CrossRef]
- Akter, N.; Fukaya, R.; Adachi, R.; Kawabe, H.; Kuba, H. Structural and Functional Refinement of the Axon Initial Segment iNavian Cochlear Nucleus During Development. J. Neurosci. 2020, 40, 6709–6721. [Google Scholar] [CrossRef]
- Yermakov, L.M.; Drouet, D.E.; Griggs, R.B.; Elased, K.M.; Susuki, K. Type 2 Diabetes Leads to Axon Initial Segment Shortening in Db/Db Mice. Front. Cell. Neurosci. 2018, 12, 146. [Google Scholar] [CrossRef] [PubMed]
- Harty, R.C.; Kim, T.H.; Thomas, E.A.; Cardamone, L.; Jones, N.C.; Petrou, S.; Wimmer, V.C. Axon Initial Segment Structural Plasticity in Animal Models of Genetic and Acquired Epilepsy. Epilepsy Res. 2013, 105, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Hinman, J.D.; Rasband, M.N.; Carmichael, S.T. Remodeling of the Axon Initial Segment After Focal Cortical and White Matter Stroke. Stroke 2013, 44, 182–189. [Google Scholar] [CrossRef]
- Benusa, S.D.; George, N.M.; Sword, B.A.; DeVries, G.H.; Dupree, J.L. Acute Neuroinflammation Induces AIS Structural Plasticity in a NOX2-Dependent Manner. J. Neuroinflamm. 2017, 14, 116. [Google Scholar] [CrossRef] [Green Version]
- Vascak, M.; Sun, J.; Baer, M.; Jacobs, K.M.; Povlishock, J.T. Mild Traumatic Brain Injury Evokes Pyramidal Neuron Axon Initial Segment Plasticity and Diffuse Presynaptic Inhibitory Terminal Loss. Front. Cell. Neurosci. 2017, 11, 157. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Wu, Y.; Gu, M.; Liu, Z.; Ma, Y.; Li, J.; Zhang, Y. Selective Filtering Defect at the Axon Initial Segment in Alzheimer’s Disease Mouse Models. Proc. Natl. Acad. Sci. USA 2014, 111, 14271–14276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jørgensen, H.S.; Jensen, D.B.; Dimintiyanova, K.P.; Bonnevie, V.S.; Hedegaard, A.; Lehnhoff, J.; Moldovan, M.; Grondahl, L.; Meehan, C.F. Increased Axon Initial Segment Length Results in Increased Na+ Currents in Spinal Motoneurones at Symptom Onset in the G127X SOD1 Mouse Model of Amyotrophic Lateral Sclerosis. Neuroscience 2021, 468, 247–264. [Google Scholar] [CrossRef]
- Sohn, P.D.; Huang, C.T.; Yan, R.; Fan, L.; Tracy, T.E.; Camargo, C.M.; Montgomery, K.M.; Arhar, T.; Mok, S.A.; Freilich, R.; et al. Pathogenic Tau Impairs Axon Initial Segment Plasticity and Excitability Homeostasis. Neuron 2019, 104, 458–470.e5. [Google Scholar] [CrossRef]
- Gutzmann, A.; Ergül, N.; Grossmann, R.; Schultz, C.; Wahle, P.; Engelhardt, M. A Period of Structural Plasticity at the Axon Initial Segment in Developing Visual Cortex. Front. Neuroanat. 2014, 8, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlüter, A.; Del Turco, D.; Deller, T.; Gutzmann, A.; Schultz, C.; Engelhardt, M. Structural Plasticity of Synaptopodin in the Axon Initial Segment During Visual Cortex Development. Cereb. Cortex 2017, 27, 4662–4675. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Feng, C.; Santamaria, F.; Kim, J.H. Impact of Auditory Experience on the Structural Plasticity of the AIS in the Mouse Brainstem Throughout the Lifespan. Front. Cell. Neurosci. 2019, 13, 456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamann, N.; Dannehl, D.; Lehmann, N.; Wagener, R.; Thielemann, C.; Schultz, C.; Staiger, J.; Kole, M.H.P.; Engelhardt, M. Sensory Input Drives Rapid Homeostatic Scaling of the Axon Initial Segment in Mouse Barrel Cortex. Nat. Commun. 2021, 12, 23. [Google Scholar] [CrossRef]
- Galliano, E.; Hahn, C.; Browne, L.P.; Villamayor, P.R.; Tufo, C.; Crespo, A.; Grubb, M.S. Brief Sensory Deprivation Triggers Cell Type-Specific Structural and Functional Plasticity in Olfactory Bulb Neurons. J. Neurosci. 2021, 41, 2135–2151. [Google Scholar] [CrossRef]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM-5), 5th ed.; American Psychiatric Association: Arlington County, VA, USA, 2013. [Google Scholar]
- Thapar, A.; Cooper, M.; Rutter, M. Neurodevelopmental Disorders. Lancet Psychiatry 2017, 4, 339–346. [Google Scholar] [CrossRef] [Green Version]
- Spillane, J.; Kullmann, D.M.; Hanna, M.G. Genetic Neurological Channelopathies: Molecular Genetics and Clinical Phenotypes. J. Neurol. Neurosurg. Psychiatry 2016, 87, 37–48. [Google Scholar] [CrossRef]
- Wimmer, V.C.; Reid, C.A.; So, E.Y.; Berkovic, S.F.; Petrou, S. Axon Initial Segment Dysfunction in Epilepsy. J. Physiol. 2010, 588, 1829–1840. [Google Scholar] [CrossRef]
- Ferreira, M.A.; O’Donovan, M.C.; Meng, Y.A.; Jones, I.R.; Ruderfer, D.M.; Jones, L.; Fan, J.; Kirov, G.; Perlis, R.H.; Green, E.K.; et al. Wellcome Trust Case Control Consortium. Collaborative Genome-Wide Association Analysis Supports a Role for ANK3 and CACNA1C in Bipolar Disorder. Nat. Genet. 2008, 40, 1056–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leussis, M.P.; Madison, J.M.; Petryshen, T.L. Ankyrin 3: Genetic Association with Bipolar Disorder and Relevance to Disease Pathophysiology. Biol. Mood Anxiety Disord. 2012, 2, 18. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Xiao, X.; Li, T.; Li, M. Translational Genomics and Beyond in Bipolar Disorder. Mol. Psychiatry 2021, 26, 186–202. [Google Scholar] [CrossRef] [PubMed]
- Rueckert, E.H.; Barker, D.; Ruderfer, D.; Bergen, S.E.; O’Dushlaine, C.; Luce, C.J.; Sheridan, S.D.; Theriault, K.M.; Chambert, K.; Moran, J.; et al. cis-Acting Regulation of Brain-Specific ANK3 Gene Expression by a Genetic Variant Associated with Bipolar Disorder. Mol. Psychiatry 2013, 18, 922–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, A.Y.; Wang, X.; Xu, M.; Maheshwari, A.; Curry, D.; Lam, S.; Adesina, A.M.; Noebels, J.L.; Sun, Q.Q.; Cooper, E.C. Ankyrin-G Isoform Imbalance and Interneuronopathy Link Epilepsy and Bipolar Disorder. Mol. Psychiatry 2017, 22, 1464–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schizophrenia Psychiatric Genome-Wide Association Study (GWAS) Consortium. Genome-Wide Association Study Identifies Five New Schizophrenia Loci. Nat. Genet. 2011, 43, 969–976. [Google Scholar] [CrossRef]
- Roussos, P.; Katsel, P.; Davis, K.L.; Bitsios, P.; Giakoumaki, S.G.; Jogia, J.; Rozsnyai, K.; Collier, D.; Frangou, S.; Siever, L.J.; et al. Molecular and Genetic Evidence for Abnormalities in the Nodes of Ranvier in Schizophrenia. Arch. Gen. Psychiatry 2012, 69, 7–15. [Google Scholar] [CrossRef]
- Athanasiu, L.; Mattingsdal, M.; Kähler, A.K.; Brown, A.; Gustafsson, O.; Agartz, I.; Giegling, I.; Muglia, P.; Cichon, S.; Rietschel, M.; et al. Gene Variants Associated with Schizophrenia in a Norwegian Genome-Wide Study Are Replicated in a Large European Cohort. J. Psychiatr. Res. 2010, 44, 748–753. [Google Scholar] [CrossRef] [Green Version]
- Bi, C.; Wu, J.; Jiang, T.; Liu, Q.; Cai, W.; Yu, P.; Cai, T.; Zhao, M.; Jiang, Y.H.; Sun, Z.S. Mutations of ANK3 Identified by Exome Sequencing Are Associated with Autism Susceptibility. Hum. Mutat. 2012, 33, 1635–1638. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Zhang, X.; Golhar, R.; Otieno, F.G.; He, M.; Hou, C.; Kim, C.; Keating, B.; Lyon, G.J.; Wang, K.; et al. Whole-Genome Sequencing in an Autism Multiplex Family. Mol. Autism 2013, 4, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iqbal, Z.; Vandeweyer, G.; van der Voet, M.; Waryah, A.M.; Zahoor, M.Y.; Besseling, J.A.; Roca, L.T.; Vulto-van Silfhout, A.T.; Nijhof, B.; Kramer, J.M.; et al. Homozygous and Heterozygous Disruptions of ANK3: At the Crossroads of Neurodevelopmental and Psychiatric Disorders. Hum. Mol. Genet. 2013, 22, 1960–1970. [Google Scholar] [CrossRef] [Green Version]
- Karaca, E.; Harel, T.; Pehlivan, D.; Jhangiani, S.N.; Gambin, T.; Coban Akdemir, Z.; Gonzaga-Jauregui, C.; Erdin, S.; Bayram, Y.; Campbell, I.M.; et al. Genes That Affect Brain Structure and Function Identified by Rare Variant Analyses of Mendelian Neurologic Disease. Neuron 2015, 88, 499–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kloth, K.; Denecke, J.; Hempel, M.; Johannsen, J.; Strom, T.M.; Kubisch, C.; Lessel, D. First De Novo ANK3 Nonsense Mutation in a Boy with Intellectual Disability, Speech Impairment and Autistic Features. Eur. J. Med. Genet. 2017, 60, 494–498. [Google Scholar] [CrossRef]
- Hu, H.; Kahrizi, K.; Musante, L.; Fattahi, Z.; Herwig, R.; Hosseini, M.; Oppitz, C.; Abedini, S.S.; Suckow, V.; Larti, F.; et al. Genetics of Intellectual Disability in Consanguineous Families. Mol. Psychiatry 2019, 24, 1027–1039. [Google Scholar] [CrossRef]
- Zhu, X.; Petrovski, S.; Xie, P.; Ruzzo, E.K.; Lu, Y.F.; McSweeney, K.M.; Ben-Zeev, B.; Nissenkorn, A.; Anikster, Y.; Oz-Levi, D.; et al. Whole-Exome Sequencing in Undiagnosed Genetic Diseases: Interpreting 119 Trios. Genet. Med. 2015, 17, 774–781. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.; Walder-Christensen, K.K.; Lalani, S.; Yan, H.; García-Prieto, I.D.; Álvarez, S.; Fernández-Jaén, A.; Speltz, L.; Jiang, Y.H.; Bennett, V. Neurodevelopmental Mutation of Giant Ankyrin-G Disrupts a Core Mechanism for Axon Initial Segment Assembly. Proc. Natl. Acad. Sci. USA 2019, 116, 19717–19726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saitsu, H.; Tohyama, J.; Kumada, T.; Egawa, K.; Hamada, K.; Okada, I.; Mizuguchi, T.; Osaka, H.; Miyata, R.; Furukawa, T.; et al. Dominant-Negative Mutations in Alpha-II Spectrin Cause West Syndrome with Severe Cerebral Hypomyelination, Spastic Quadriplegia, and Developmental Delay. Am. J. Hum. Genet. 2010, 86, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Hamdan, F.F.; Saitsu, H.; Nishiyama, K.; Gauthier, J.; Dobrzeniecka, S.; Spiegelman, D.; Lacaille, J.C.; Décarie, J.C.; Matsumoto, N.; Rouleau, G.A.; et al. Identification of a Novel in-Frame De Novo Mutation in SPTAN1 in Intellectual Disability and Pontocerebellar Atrophy. Eur. J. Hum. Genet. 2012, 20, 796–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nonoda, Y.; Saito, Y.; Nagai, S.; Sasaki, M.; Iwasaki, T.; Matsumoto, N.; Ishii, M.; Saitsu, H. Progressive Diffuse Brain Atrophy in West Syndrome with Marked Hypomyelination Due to SPTAN1 Gene Mutation. Brain Dev. 2013, 35, 280–283. [Google Scholar] [CrossRef] [PubMed]
- Syrbe, S.; Harms, F.L.; Parrini, E.; Montomoli, M.; Mütze, U.; Helbig, K.L.; Polster, T.; Albrecht, B.; Bernbeck, U.; van Binsbergen, E.; et al. Delineating SPTAN1 Associated Phenotypes: From Isolated Epilepsy to Encephalopathy with Progressive Brain Atrophy. Brain 2017, 140, 2322–2336. [Google Scholar] [CrossRef]
- Ream, M.A.; Mikati, M.A. Clinical Utility of Genetic Testing in Pediatric Drug-Resistant Epilepsy: A Pilot Study. Epilepsy Behav. 2014, 37, 241–248. [Google Scholar] [CrossRef]
- Beijer, D.; Deconinck, T.; De Bleecker, J.L.; Dotti, M.T.; Malandrini, A.; Urtizberea, J.A.; Zulaica, M.; López de Munain, A.; Asselbergh, B.; De Jonghe, P.; et al. Nonsense Mutations in Alpha-II Spectrin in Three Families with Juvenile Onset Hereditary Motor Neuropathy. Brain 2019, 142, 2605–2616. [Google Scholar] [CrossRef] [Green Version]
- Dong, H.L.; Chen, L.; Wu, Z.Y. A Novel De Novo SPTAN1 Nonsense Variant Causes Hereditary Motor Neuropathy in a Chinese Family. Brain 2021, 144, e11. [Google Scholar] [CrossRef] [PubMed]
- Knierim, E.; Gill, E.; Seifert, F.; Morales-Gonzalez, S.; Unudurthi, S.D.; Hund, T.J.; Stenzel, W.; Schuelke, M. A Recessive Mutation in Beta-IV-Spectrin (SPTBN4) Associates with Congenital Myopathy, Neuropathy, and Central Deafness. Hum. Genet. 2017, 136, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Anazi, S.; Maddirevula, S.; Salpietro, V.; Asi, Y.T.; Alsahli, S.; Alhashem, A.; Shamseldin, H.E.; AlZahrani, F.; Patel, N.; Ibrahim, N.; et al. Expanding the Genetic Heterogeneity of Intellectual Disability. Hum. Genet. 2017, 136, 1419–1429. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.C.; Ortiz-González, X.R.; Yum, S.W.; Gill, S.M.; White, A.; Kelter, E.; Seaver, L.H.; Lee, S.; Wiley, G.; Gaffney, P.M.; et al. betaIV Spectrinopathies Cause Profound Intellectual Disability, Congenital Hypotonia, and Motor Axonal Neuropathy. Am. J. Hum. Genet. 2018, 102, 1158–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pehlivan, D.; Bayram, Y.; Gunes, N.; Coban Akdemir, Z.; Shukla, A.; Bierhals, T.; Tabakci, B.; Sahin, Y.; Gezdirici, A.; Fatih, J.M.; et al. The Genomics of Arthrogryposis, a Complex Trait: Candidate Genes and Further Evidence for Oligogenic Inheritance. Am. J. Hum. Genet. 2019, 105, 132–150. [Google Scholar] [CrossRef] [Green Version]
- Monies, D.; Abouelhoda, M.; Assoum, M.; Moghrabi, N.; Rafiullah, R.; Almontashiri, N.; Alowain, M.; Alzaidan, H.; Alsayed, M.; Subhani, S.; et al. Lessons Learned from Large-Scale, First-Tier Clinical Exome Sequencing in a Highly Consanguineous Population. Am. J. Hum. Genet. 2019, 104, 1182–1201. [Google Scholar] [CrossRef] [Green Version]
- Häusler, M.G.; Begemann, M.; Lidov, H.G.; Kurth, I.; Darras, B.T.; Elbracht, M. A Novel Homozygous Splice-Site Mutation in the SPTBN4 Gene Causes Axonal Neuropathy without Intellectual Disability. Eur. J. Med. Genet. 2020, 63, 103826. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Gonzalez, X.; Wierenga, K. SPTBN4 Disorder. Available online: https://www.ncbi.nlm.nih.gov/books/NBK559435/?report=classic (accessed on 12 August 2021).
- Kaphzan, H.; Buffington, S.A.; Jung, J.I.; Rasband, M.N.; Klann, E. Alterations in Intrinsic Membrane Properties and the Axon Initial Segment in a Mouse Model of Angelman Syndrome. J. Neurosci. 2011, 31, 17637–17648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Booker, S.A.; Simoes de Oliveira, L.; Anstey, N.J.; Kozic, Z.; Dando, O.R.; Jackson, A.D.; Baxter, P.S.; Isom, L.L.; Sherman, D.L.; Hardingham, G.E.; et al. Input-Output Relationship of CA1 Pyramidal Neurons Reveals Intact Homeostatic Mechanisms in a Mouse Model of Fragile X Syndrome. Cell Rep. 2020, 32, 107988. [Google Scholar] [CrossRef] [PubMed]
- Hong, T.; Falcone, C.; Dufour, B.; Amina, S.; Castro, R.P.; Regalado, J.; Pearson, W.; Noctor, S.C.; Martínez-Cerdeño, V. GABAARalpha2 Is Decreased in the Axon Initial Segment of Pyramidal Cells in Specific Areas of the Prefrontal Cortex in Autism. Neuroscience 2020, 437, 76–86. [Google Scholar] [CrossRef]
- Ariza, J.; Rogers, H.; Hashemi, E.; Noctor, S.C.; Martínez-Cerdeño, V. The Number of Chandelier and Basket Cells Are Differentially Decreased in Prefrontal Cortex in Autism. Cereb. Cortex 2018, 28, 411–420. [Google Scholar] [CrossRef]
- Tian, T.; Quintana-Urzainqui, I.; Kozić, Z.; Pratt, T.; Price, D.J. Pax6 Regulates the Morphological and Electrophysiological Development of Mouse Prethalamic Neurons. bioRxiv 2021. [Google Scholar] [CrossRef]
- Tang, X.; Jaenisch, R.; Sur, M. The Role of GABAergic Signalling in Neurodevelopmental Disorders. Nat. Rev. Neurosci. 2021, 22, 290–307. [Google Scholar] [CrossRef]
- Chen, R.; Gore, F.; Nguyen, Q.A.; Ramakrishnan, C.; Patel, S.; Kim, S.H.; Raffiee, M.; Kim, Y.S.; Hsueh, B.; Krook-Magnusson, E.; et al. Deep Brain Optogenetics without Intracranial Surgery. Nat. Biotechnol. 2021, 39, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.; Mendoza-Halliday, D.; Ting, J.T.; Kaiser, T.; Sun, X.; Bastos, A.M.; Wimmer, R.D.; Guo, B.; Chen, Q.; Zhou, Y.; et al. An Ultra-Sensitive Step-Function Opsin for Minimally Invasive Optogenetic Stimulation in Mice and Macaques. Neuron 2020, 107, 38–51.e8. [Google Scholar] [CrossRef]

{kind=link}
| Gene Name Accession # (Chromosome #) | Gene Mutation Locus | Mutated Protein | Symptoms | References |
|---|---|---|---|---|
| ANK-3 NM_020987.3 (10q21) | c.4705T > G c.11159C > T * c.12763A > C * c.11068G > A 46,XY,t(2;10)(q11.2;q21.2) c.10995delC c.9652C > T c.1990G > T c.11033del c.4960G > T, c.4465C > T | p.S1569A p.T3720M p.T4255P p.G3690R p.T3666LfsX2 p.L3218F p.G664 * p.P3678Lfs *45 p.D1654Y, p.P1489S | ASD ADHD, ASD ID, hypotonia, spasticity ID, brain atrophy, delayed myelination ID, ASD, macrocephaly ID Severe intractable seizures with DD | Bi et al., 2012 Shi et al., 2013 Iqbal et al., 2013 Karaca et al., 2015 Kloth et al., 2017 Hu et al., 2019 Zhu et al., 2020 |
SPTAN1 (SPECTRIN, ALPHAII) NM_0011304 (9q34.11) | c.6619-6621del c.6923-6928dup c.1697G > C c.6605-6607del c.6908-6916 dup c.6910_6918 dup c.5326C > T c.6184C > T c.6619_6621del c.6619_6621del c.6622_6624del c.6811G>A c.6850_6852del c.6908_6916dup c.6908_6916dup c.6908_6916dup c.6907_6915dup c.6907_6915dup c.6910_6918del c.6923_6928dup c.533G > A c.917C > T c.3716A > G c.4828C > T c.6908_6916del arr[hg19] 9q34.11 (131,349,701–131,351,531) x1 exon 20–21 deletion heterozygous c.415C4T heterozygous c.4615C4T heterozygous c.6385C4T heterozygous c.6781C4T | p.E2207del p.R2308_M2309dup p.R566P p.Q2202del p.D2303_L2305dup p.Q2304_G2306dup p.R1776W p.R2062W p.E2207del p.E2207del p.N2208del p.E2271K p.D2284del p.D2303_2305dup p.D2303_2305dup p.D2303_2305dup p.D2303_2305dup p.D2303_2305dup p.Q2304_G2306del p.R2308_M2309dup p.G178D p.A306V p.H1239R p.R1610W p.D2303_L2305del p.A927_L1002del p.R139 * p.Q1539 * p.Q2149 * p.R2261 * | WS, profound ID, spastic quadriplegia Mild ID, IS WS, severely impaired psychomotor development WS Infantile EE with IS and focal epilepsy, mild ID, ASD Infantile EE with tonic spasms and FDS, profound DD, severe hypotonia, microcephaly WS, profound DD, minimal interaction, hypotonia, hypokinesia; microcephaly WS, profound DD, hypotonia, microcephaly WS, profound DD, severe hypotonia, thermic dysregulation; microcephaly WS, profound DD, hypotonia, multifocal myoclonus, dyskinetic movement disorder, microcephaly Infantile EE with IS evolving to myoclonic seizures, severe DD, hypotonia, ataxic movement disorder WS, profound DD, hypotonia, ataxia, dyskinetic movement disorder, microcephaly WS (PEHO syndrome), profound DD, hypotonia, microcephaly WS, profound DD, hypotonia, microcephaly WS, profound DD, hypotonia, ataxia, dyskinetic movement disorder, microcephaly WS, profound DD, intermittent opisthotonus, hypotonia, microcephaly WS, mild ID, DD, delayed walking WS, profound DD, microcephaly Focal epilepsy Epilepsy with myoclonic and atonic seizures, moderate ID No epilepsy, mild DD, ID, ASD; mild dysmorphic signs Myoclonic epilepsy, mild–moderate DD, ID, ASD, hypotonia, mild spastic gait; hyperreflexia in lower limbs FDS, moderate ID, ADHD Focal seizures, mild DD, ID, mild diffuse hypotonia, slowly progressive and severe cerebellar ataxia Hereditary motor neuropathy | Saitsu et al., 2010 Hamdan et al., 2012 Nonoda et al., 2013 Ream and Mikati 2014 Syrbe et al., 2017 Beijer et al., 2019 Dong et al., 2021 |
| SPTBN4 (SPECTRIN, BETAIV) NM_020971.2 (19q13.2) | homozygous c.1597C > T homozygous c.3394del homozygous c.3820G > T homozygous c.2709G > A c.1511G > A; c.7303C > T homozygous c.7453del c.1813C > T; c.3829del homozygous c.1665+2T > C homozygous c.3949-1G > A | p.Q533 * p.H1132Tfs *39 p.E1274 * p.W903 * p.R504Q; p.R2435C p.A2485Lfs *31 p.Q605 *, p.Q1277Rfs *4 Intron Intron | Congenital myopathy, neuropathy, and central deafness Global DD, hypotonia, dysphasia, recurrent respiratory infections, blue sclerae, hyporeflexia Profound ID, congenital hypotonia, and motor axonal neuropathy Speech delay, ID, ataxia, seizures, cerebral atrophy Axonal neuropathy without ID | Knierim et al., 2017 Anazi et al., 2017 Wang et al., 2019 Monies et al., 2019 Hausler et al., 2020 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fujitani, M.; Otani, Y.; Miyajima, H. Pathophysiological Roles of Abnormal Axon Initial Segments in Neurodevelopmental Disorders. Cells 2021, 10, 2110. https://doi.org/10.3390/cells10082110
Fujitani M, Otani Y, Miyajima H. Pathophysiological Roles of Abnormal Axon Initial Segments in Neurodevelopmental Disorders. Cells. 2021; 10(8):2110. https://doi.org/10.3390/cells10082110
Chicago/Turabian StyleFujitani, Masashi, Yoshinori Otani, and Hisao Miyajima. 2021. "Pathophysiological Roles of Abnormal Axon Initial Segments in Neurodevelopmental Disorders" Cells 10, no. 8: 2110. https://doi.org/10.3390/cells10082110
APA StyleFujitani, M., Otani, Y., & Miyajima, H. (2021). Pathophysiological Roles of Abnormal Axon Initial Segments in Neurodevelopmental Disorders. Cells, 10(8), 2110. https://doi.org/10.3390/cells10082110