
Genomic Aberrations Associated with the Pathophysiological Mechanisms of Neurodevelopmental Disorders


Abstract
:1. Introduction
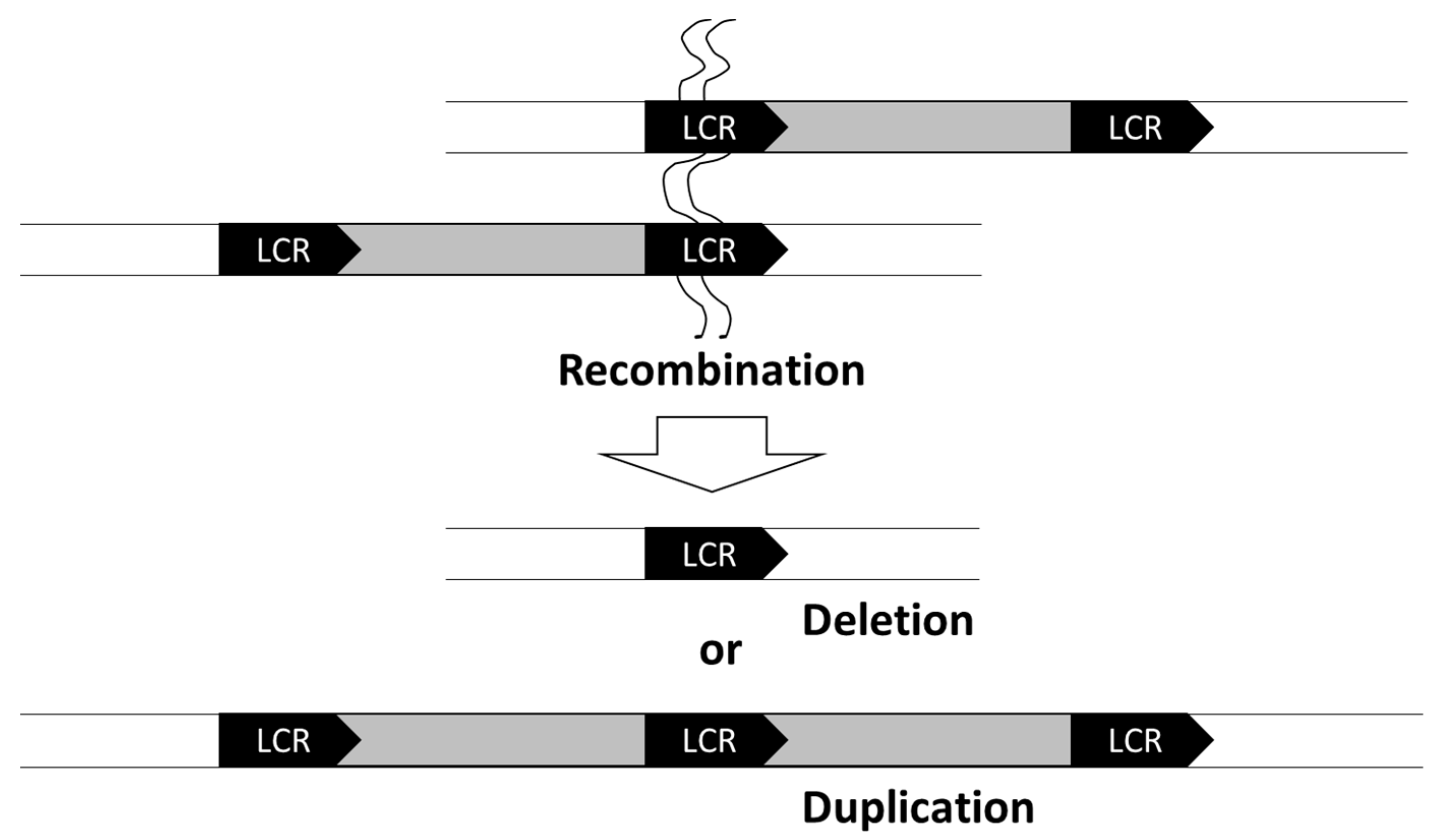
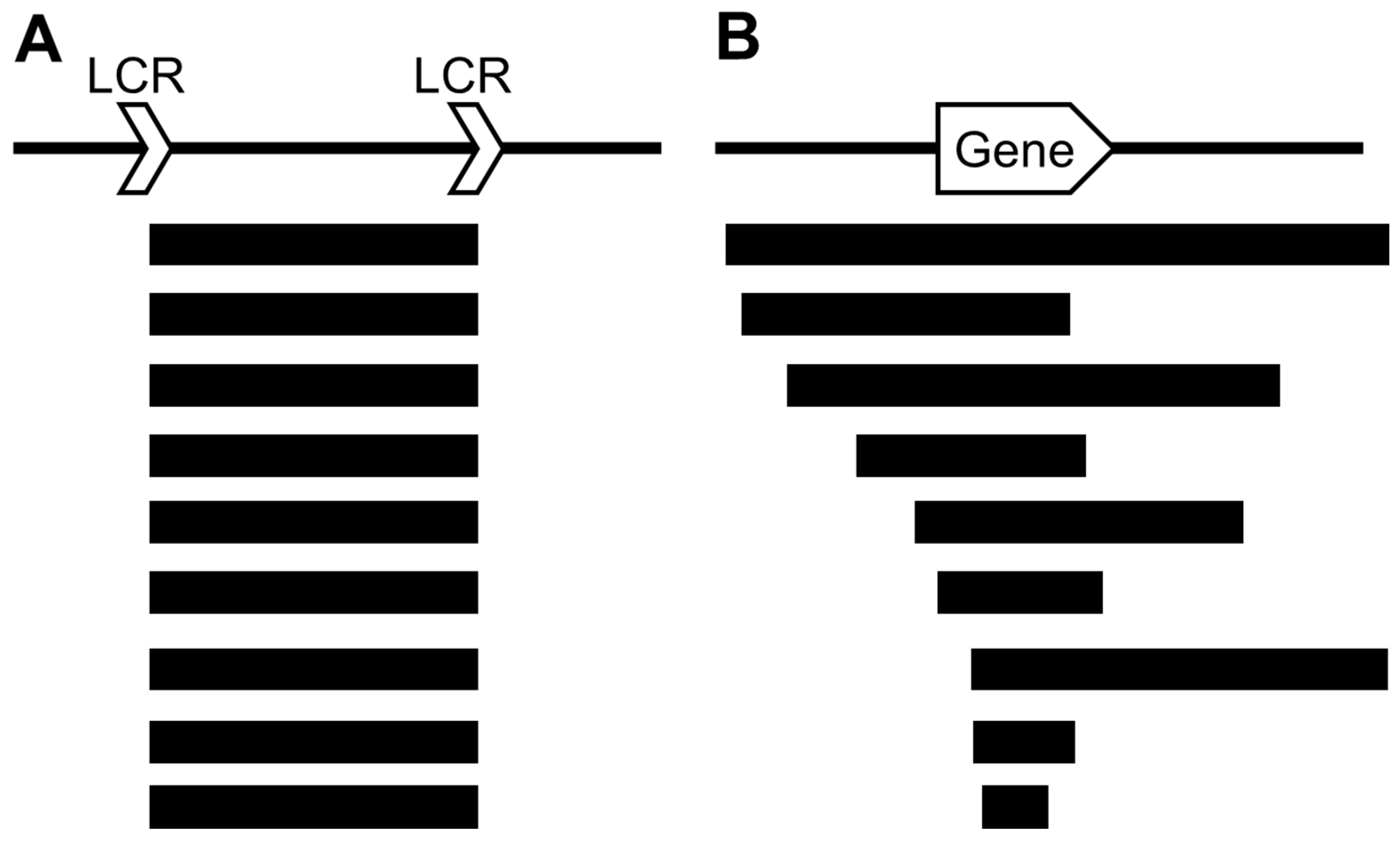
2. Chromosomal Deletions
3. Microduplications
4. Different Symptoms Are Associated with Deletion and Duplication of Certain Genes
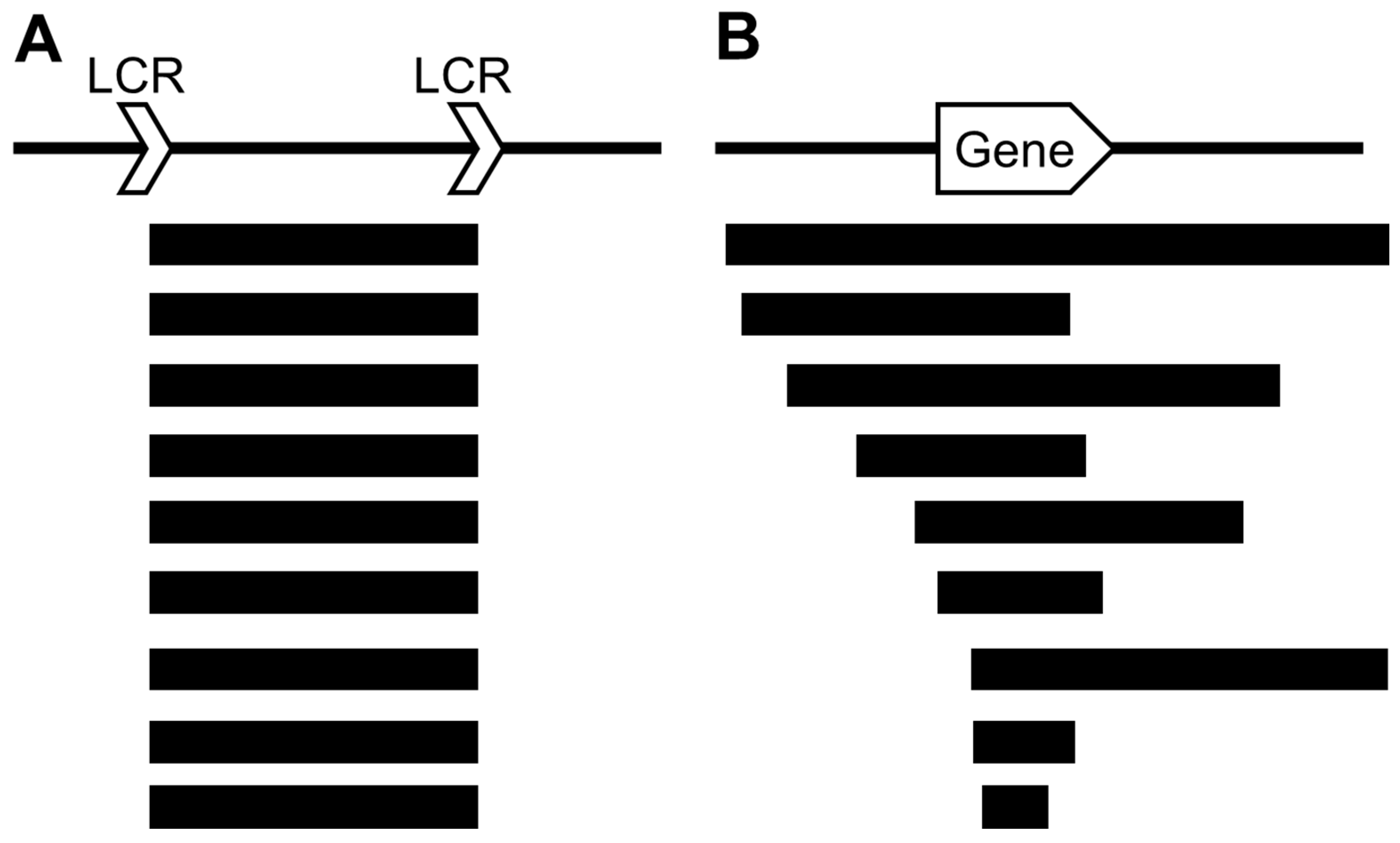
5. Significance of Microarray in Detecting Chromosomal Aberrations
6. Genes Identified Based on Their Genomic Copy Number Changes
7. Genes Whose Phenotypes Are Not Affected by Genomic Copy Number Changes
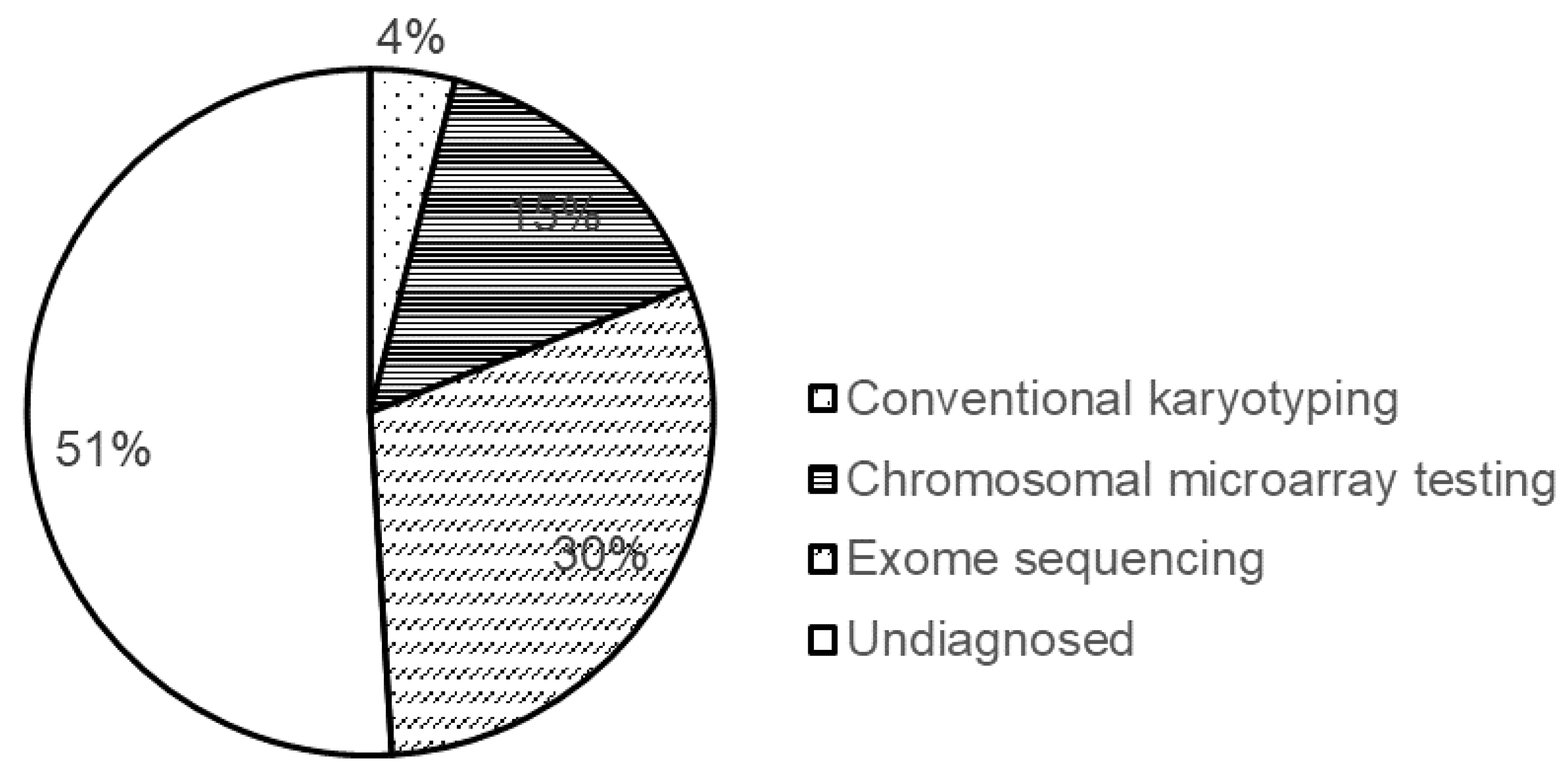
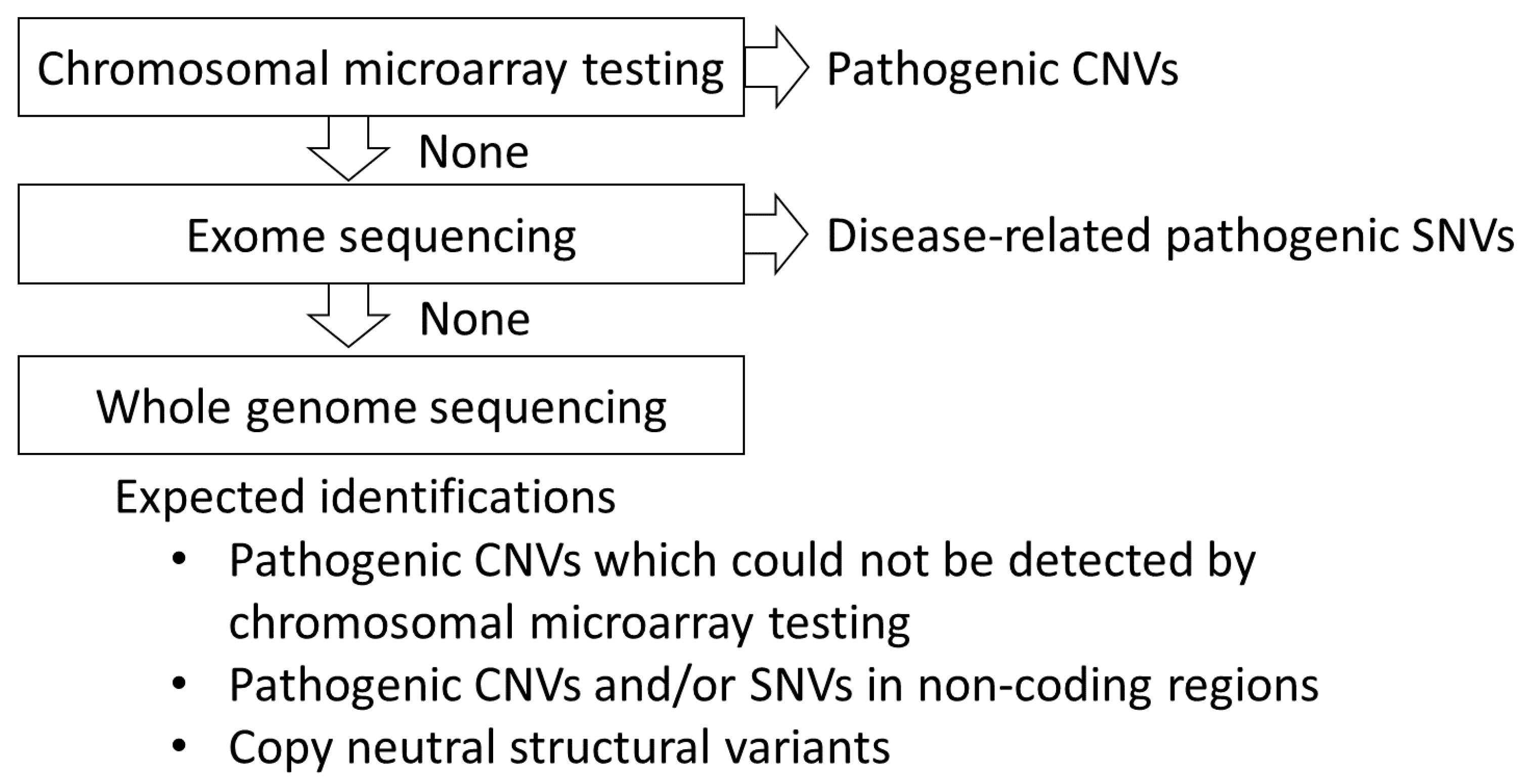
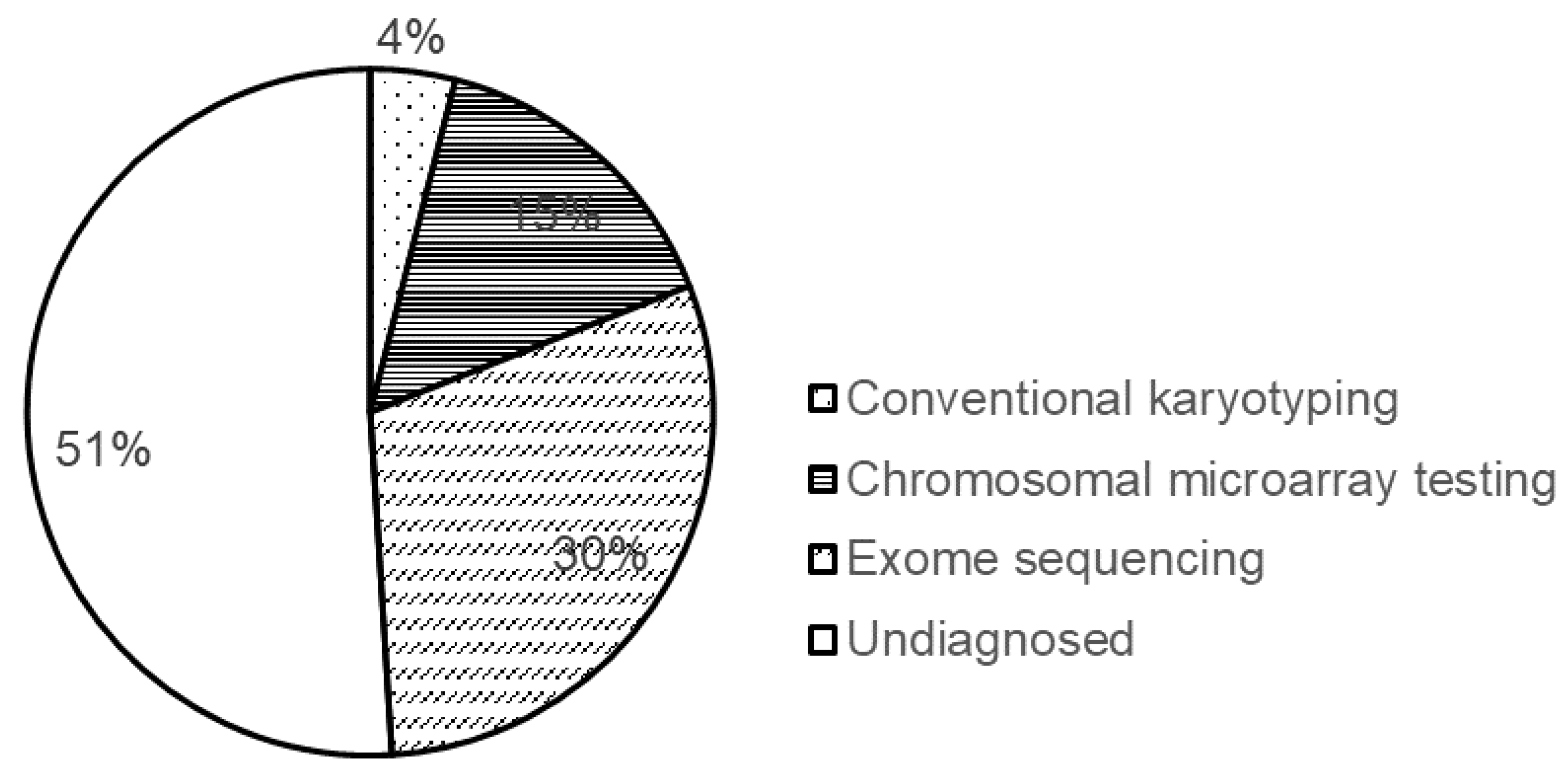
8. Diagnostic Yield of the Methods Used for Genetic Testing
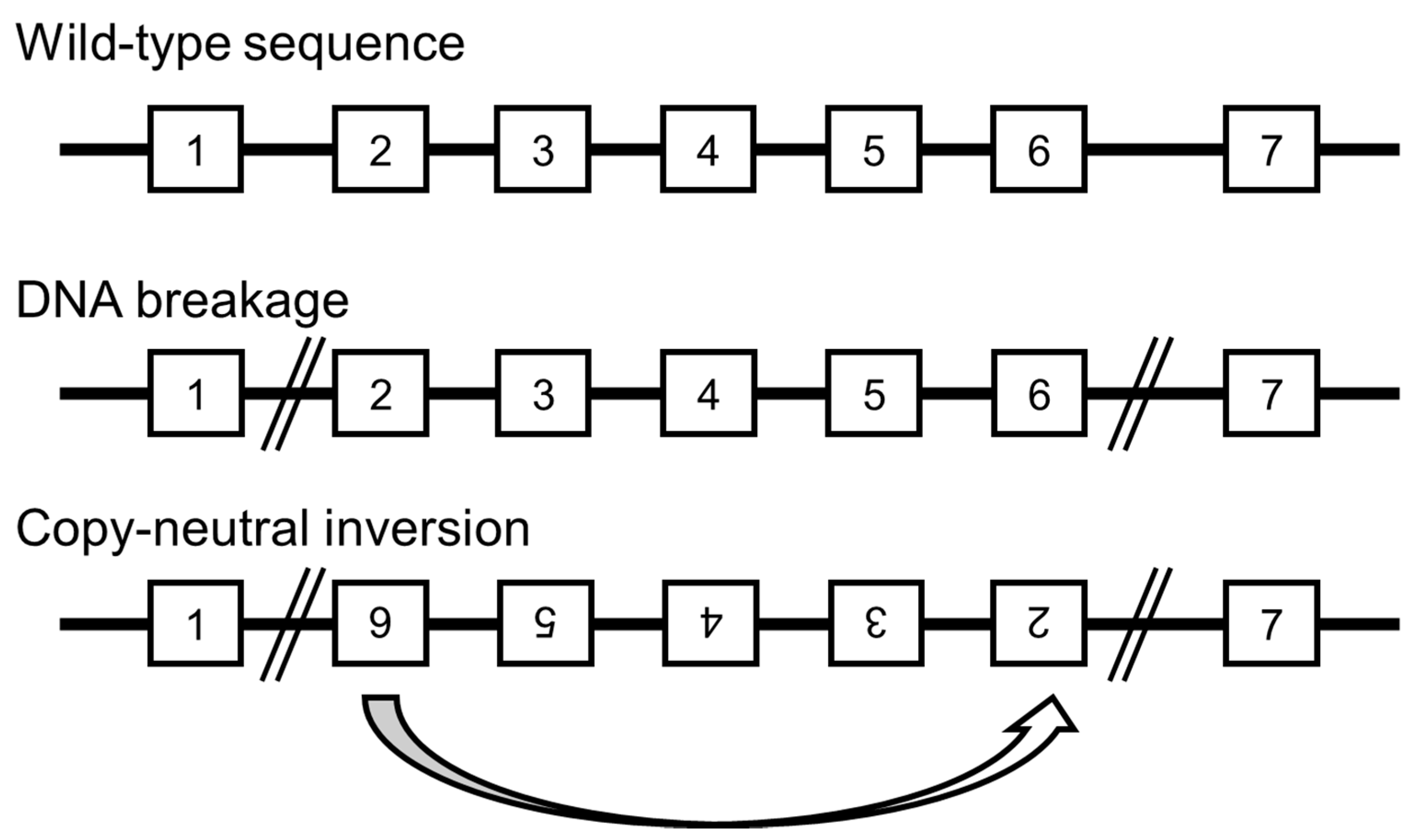
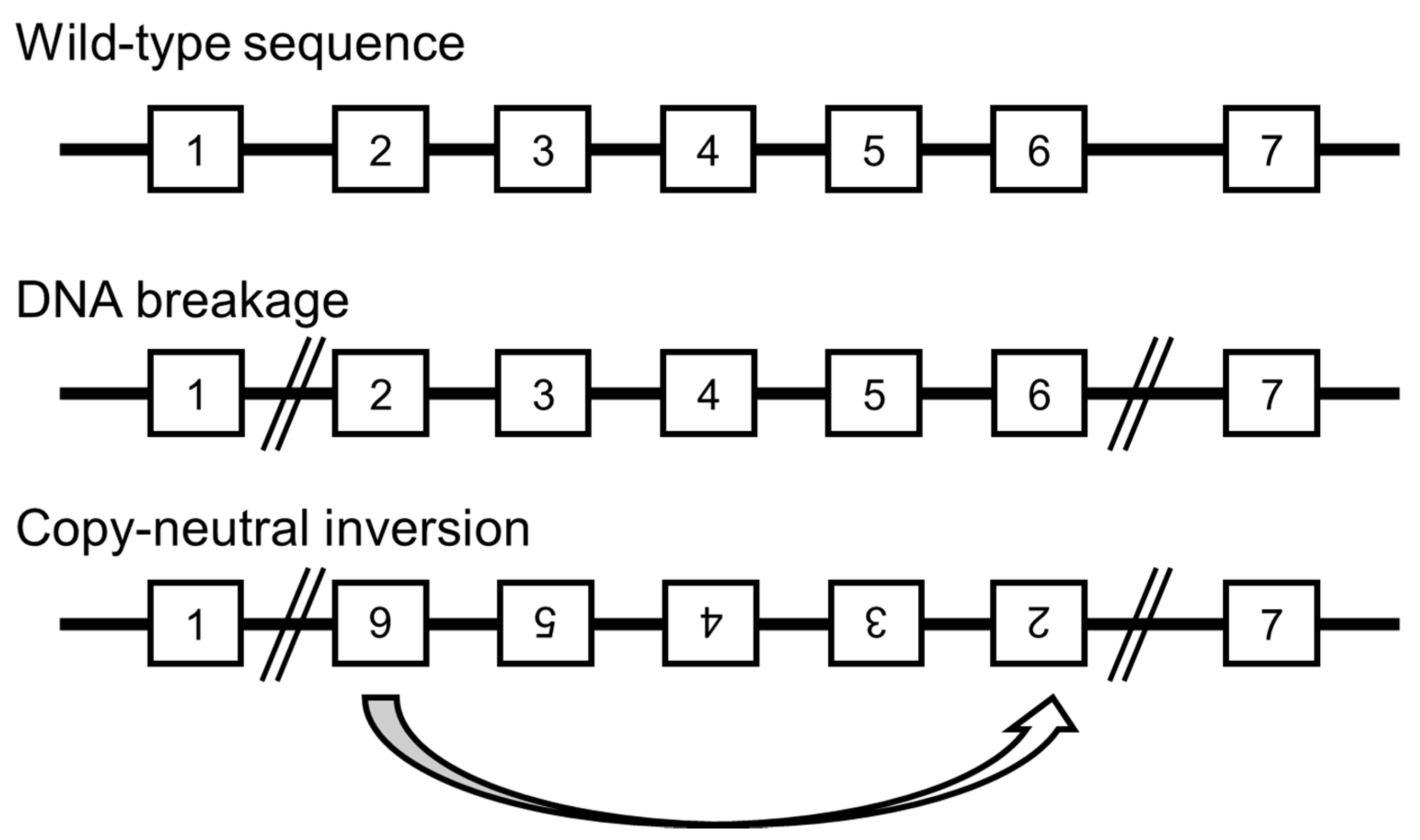
9. Undetected Genomic Backgrounds
10. Novel Developments Expected in Whole Genome Analysis for the Detection of Chromosomal Aberrations
11. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Savatt, J.M.; Myers, S.M. Genetic Testing in Neurodevelopmental Disorders. Front. Pediatr. 2021, 9, 526779. [Google Scholar] [CrossRef]
- Blesson, A.; Cohen, J.S. Genetic Counseling in Neurodevelopmental Disorders. Cold Spring Harb. Perspect. Med. 2020, 10, a036533. [Google Scholar] [CrossRef]
- Henstridge, C.M.; Pickett, E.; Spires-Jones, T.L. Synaptic pathology: A shared mechanism in neurological disease. Ageing Res. Rev. 2016, 28, 72–84. [Google Scholar] [CrossRef] [Green Version]
- Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature 2017, 542, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Moyses-Oliveira, M.; Yadav, R.; Erdin, S.; Talkowski, M.E. New gene discoveries highlight functional convergence in autism and related neurodevelopmental disorders. Curr. Opin. Genet. Dev. 2020, 65, 195–206. [Google Scholar] [CrossRef]
- Yamamoto, T.; Imaizumi, T.; Yamamoto-Shimojima, K.; Lu, Y.; Yanagishita, T.; Shimada, S.; Chong, P.F.; Kira, R.; Ueda, R.; Ishiyama, A.; et al. Genomic backgrounds of Japanese patients with undiagnosed neurodevelopmental disorders. Brain Dev. 2019, 41, 776–782. [Google Scholar] [CrossRef] [PubMed]
- Emanuel, B.S. Molecular mechanisms and diagnosis of chromosome 22q11.2 rearrangements. Dev. Disabil. Res. Rev. 2008, 14, 11–18. [Google Scholar] [CrossRef] [Green Version]
- Gibbs, R.A. The Human Genome Project changed everything. Nat. Rev. Genet. 2020, 21, 575–576. [Google Scholar] [CrossRef]
- Rive Le Gouard, N.; Jacquinet, A.; Ruaud, L.; Deleersnyder, H.; Ageorges, F.; Gallard, J.; Lacombe, D.; Odent, S.; Mikaty, M.; Manouvrier-Hanu, S.; et al. Smith-Magenis syndrome: Clinical and behavioral characteristics in a large retrospective cohort. Clin. Genet. 2021, 99, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Ciaccio, C.; Pantaleoni, C.; Milani, D.; Alfei, E.; Sciacca, F.L.; Canafoglia, L.; Erbetta, A.; D’Arrigo, S. Neurological phenotype of Potocki-Lupski syndrome. Am. J. Med. Genet. A 2020, 182, 2317–2324. [Google Scholar] [CrossRef]
- Garay, P.M.; Chen, A.; Tsukahara, T.; Rodríguez Díaz, J.C.; Kohen, R.; Althaus, J.C.; Wallner, M.A.; Giger, R.J.; Jones, K.S.; Sutton, M.A.; et al. RAI1 Regulates Activity-Dependent Nascent Transcription and Synaptic Scaling. Cell Rep. 2020, 32, 108002. [Google Scholar] [CrossRef] [PubMed]
- Pantera, H.; Shy, M.E.; Svaren, J. Regulating PMP22 expression as a dosage sensitive neuropathy gene. Brain Res. 2020, 1726, 146491. [Google Scholar] [CrossRef]
- Marinko, J.T.; Carter, B.D.; Sanders, C.R. Direct relationship between increased expression and mistrafficking of the Charcot-Marie-Tooth-associated protein PMP22. J. Biol. Chem. 2020, 295, 11963–11970. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K. PLP1-related inherited dysmyelinating disorders: Pelizaeus-Merzbacher disease and spastic paraplegia type 2. Neurogenetics 2005, 6, 1–16. [Google Scholar] [CrossRef]
- Inoue, K. Pelizaeus-Merzbacher Disease: Molecular and Cellular Pathologies and Associated Phenotypes. Adv. Exp. Med. Biol. 2019, 1190, 201–216. [Google Scholar] [CrossRef]
- Yamamoto, T.; Shimojima, K. Pelizaeus-Merzbacher disease as a chromosomal disorder. Congenit Anom. 2013, 53, 3–8. [Google Scholar] [CrossRef]
- Yamamoto, T.; Shimojima, K.; Shimada, S.; Yokochi, K.; Yoshitomi, S.; Yanagihara, K.; Imai, K.; Okamoto, N. Clinical impacts of genomic copy number gains at Xq28. Hum. Genome Var. 2014, 1, 14001. [Google Scholar] [CrossRef] [Green Version]
- Vermudez, S.A.D.; Gogliotti, R.G.; Arthur, B.; Buch, A.; Morales, C.; Moxley, Y.; Rajpal, H.; Conn, P.J.; Niswender, C.M. Profiling beneficial and potential adverse effects of MeCP2 overexpression in a hypomorphic Rett syndrome mouse model. Genes Brain Behav. 2021, e12752. [Google Scholar] [CrossRef]
- Battaglia, A.; Doccini, V.; Bernardini, L.; Novelli, A.; Loddo, S.; Capalbo, A.; Filippi, T.; Carey, J.C. Confirmation of chromosomal microarray as a first-tier clinical diagnostic test for individuals with developmental delay, intellectual disability, autism spectrum disorders and dysmorphic features. Eur. J. Paediatr. Neurol. 2013, 17, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.T.; Adam, M.P.; Aradhya, S.; Biesecker, L.G.; Brothman, A.R.; Carter, N.P.; Church, D.M.; Crolla, J.A.; Eichler, E.E.; Epstein, C.J.; et al. Consensus statement: Chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet. 2010, 86, 749–764. [Google Scholar] [CrossRef]
- Kumar, R.A.; KaraMohamed, S.; Sudi, J.; Conrad, D.F.; Brune, C.; Badner, J.A.; Gilliam, T.C.; Nowak, N.J.; Cook, E.H., Jr.; Dobyns, W.B.; et al. Recurrent 16p11.2 microdeletions in autism. Hum. Mol. Genet. 2008, 17, 628–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimojima, K.; Sugawara, M.; Shichiji, M.; Mukaida, S.; Takayama, R.; Imai, K.; Yamamoto, T. Loss-of-function mutation of collybistin is responsible for X-linked mental retardation associated with epilepsy. J. Hum. Genet. 2011, 56, 561–565. [Google Scholar] [CrossRef]
- Alber, M.; Kalscheuer, V.M.; Marco, E.; Sherr, E.; Lesca, G.; Till, M.; Gradek, G.; Wiesener, A.; Korenke, C.; Mercier, S.; et al. ARHGEF9 disease: Phenotype clarification and genotype-phenotype correlation. Neurol. Genet. 2017, 3, e148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimojima, K.; Isidor, B.; Le Caignec, C.; Kondo, A.; Sakata, S.; Ohno, K.; Yamamoto, T. A new microdeletion syndrome of 5q31.3 characterized by severe developmental delays, distinctive facial features, and delayed myelination. Am. J. Med. Genet. A 2011, 155, 732–736. [Google Scholar] [CrossRef]
- Hosoki, K.; Ohta, T.; Natsume, J.; Imai, S.; Okumura, A.; Matsui, T.; Harada, N.; Bacino, C.A.; Scaglia, F.; Jones, J.Y.; et al. Clinical phenotype and candidate genes for the 5q31.3 microdeletion syndrome. Am. J. Med. Genet. A 2012, 158a, 1891–1896. [Google Scholar] [CrossRef]
- Brown, N.; Burgess, T.; Forbes, R.; McGillivray, G.; Kornberg, A.; Mandelstam, S.; Stark, Z. 5q31.3 Microdeletion syndrome: Clinical and molecular characterization of two further cases. Am. J. Med. Genet. A 2013, 161a, 2604–2608. [Google Scholar] [CrossRef] [PubMed]
- Lalani, S.R.; Zhang, J.; Schaaf, C.P.; Brown, C.W.; Magoulas, P.; Tsai, A.C.; El-Gharbawy, A.; Wierenga, K.J.; Bartholomew, D.; Fong, C.T.; et al. Mutations in PURA cause profound neonatal hypotonia, seizures, and encephalopathy in 5q31.3 microdeletion syndrome. Am. J. Hum. Genet. 2014, 95, 579–583. [Google Scholar] [CrossRef] [Green Version]
- Shimojima, K.; Ondo, Y.; Okamoto, N.; Yamamoto, T. A 15q14 microdeletion involving MEIS2 identified in a patient with autism spectrum disorder. Hum. Genome Var. 2017, 4, 17029. [Google Scholar] [CrossRef]
- Fujita, A.; Isidor, B.; Piloquet, H.; Corre, P.; Okamoto, N.; Nakashima, M.; Tsurusaki, Y.; Saitsu, H.; Miyake, N.; Matsumoto, N. De novo MEIS2 mutation causes syndromic developmental delay with persistent gastro-esophageal reflux. J. Hum. Genet. 2016, 61, 835–838. [Google Scholar] [CrossRef]
- Yamamoto-Shimojima, K.; Imaizumi, T.; Akagawa, H.; Kanno, H.; Yamamoto, T. Primrose syndrome associated with unclassified immunodeficiency and a novel ZBTB20 mutation. Am. J. Med. Genet. A 2020, 182, 521–526. [Google Scholar] [CrossRef]
- Shimojima, K.; Saito, K.; Yamamoto, T. A de novo 1.9-Mb interstitial deletion of 3q13.2q13.31 in a girl with dysmorphic features, muscle hypotonia, and developmental delay. Am. J. Med. Genet. A 2009, 149a, 1818–1822. [Google Scholar] [CrossRef]
- Leoyklang, P.; Suphapeetiporn, K.; Siriwan, P.; Desudchit, T.; Chaowanapanja, P.; Gahl, W.A.; Shotelersuk, V. Heterozygous nonsense mutation SATB2 associated with cleft palate, osteoporosis, and cognitive defects. Hum. Mutat. 2007, 28, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, J.A.; Ballif, B.C.; Lucas, A.; Spence, E.J.; Powell, C.; Aylsworth, A.S.; Torchia, B.A.; Shaffer, L.G. Small deletions of SATB2 cause some of the clinical features of the 2q33.1 microdeletion syndrome. PLoS ONE 2009, 4, e6568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berko, E.R.; Cho, M.T.; Eng, C.; Shao, Y.; Sweetser, D.A.; Waxler, J.; Robin, N.H.; Brewer, F.; Donkervoort, S.; Mohassel, P.; et al. De novo missense variants in HECW2 are associated with neurodevelopmental delay and hypotonia. J. Med. Genet. 2017, 54, 84–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halvardson, J.; Zhao, J.J.; Zaghlool, A.; Wentzel, C.; Georgii-Hemming, P.; Månsson, E.; Ederth Sävmarker, H.; Brandberg, G.; Soussi Zander, C.; Thuresson, A.C.; et al. Mutations in HECW2 are associated with intellectual disability and epilepsy. J. Med. Genet. 2016, 53, 697–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanagishita, T.; Hirade, T.; Shimojima Yamamoto, K.; Funatsuka, M.; Miyamoto, Y.; Maeda, M.; Yanagi, K.; Kaname, T.; Nagata, S.; Nagata, M.; et al. HECW2-related disorder in four Japanese patients. Am. J. Med. Genet. A 2021. [Google Scholar] [CrossRef]
- Hochstenbach, R.; van Binsbergen, E.; Engelen, J.; Nieuwint, A.; Polstra, A.; Poddighe, P.; Ruivenkamp, C.; Sikkema-Raddatz, B.; Smeets, D.; Poot, M. Array analysis and karyotyping: Workflow consequences based on a retrospective study of 36,325 patients with idiopathic developmental delay in the Netherlands. Eur. J. Med. Genet. 2009, 52, 161–169. [Google Scholar] [CrossRef]
- Martin, C.L.; Ledbetter, D.H. Chromosomal Microarray Testing for Children With Unexplained Neurodevelopmental Disorders. JAMA 2017, 317, 2545–2546. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.R.Q.; Nguyen, Q.; Yokota, T. Genotype-Phenotype Correlations in Duchenne and Becker Muscular Dystrophy Patients from the Canadian Neuromuscular Disease Registry. J. Pers. Med. 2020, 10, 241. [Google Scholar] [CrossRef]
- Yamamoto, T.; Shimojima, K.; Ondo, Y.; Shimakawa, S.; Okamoto, N. MED13L haploinsufficiency syndrome: A de novo frameshift and recurrent intragenic deletions due to parental mosaicism. Am. J. Med. Genet. A 2017, 173, 1264–1269. [Google Scholar] [CrossRef] [PubMed]
- Dehainault, C.; Michaux, D.; Pagès-Berhouet, S.; Caux-Moncoutier, V.; Doz, F.; Desjardins, L.; Couturier, J.; Parent, P.; Stoppa-Lyonnet, D.; Gauthier-Villars, M.; et al. A deep intronic mutation in the RB1 gene leads to intronic sequence exonisation. Eur. J. Hum. Genet. 2007, 15, 473–477. [Google Scholar] [CrossRef]
- Encarnação, M.; Coutinho, M.F.; Cho, S.M.; Cardoso, M.T.; Ribeiro, I.; Chaves, P.; Santos, J.I.; Quelhas, D.; Lacerda, L.; Leão Teles, E.; et al. NPC1 silent variant induces skipping of exon 11 (p.V562V) and unfolded protein response was found in a specific Niemann-Pick type C patient. Mol. Genet. Genom. Med. 2020, 8, e1451. [Google Scholar] [CrossRef]
- Sun, Y.; Li, Y.; Chen, M.; Luo, Y.; Qian, Y.; Yang, Y.; Lu, H.; Lou, F.; Dong, M. A Novel Silent Mutation in the L1CAM Gene Causing Fetal Hydrocephalus Detected by Whole-Exome Sequencing. Front. Genet. 2019, 10, 817. [Google Scholar] [CrossRef] [PubMed]
- Imaizumi, T.; Yamamoto-Shimojima, K.; Yanagishita, T.; Ondo, Y.; Nishi, E.; Okamoto, N.; Yamamoto, T. Complex chromosomal rearrangements of human chromosome 21 in a patient manifesting clinical features partially overlapped with that of Down syndrome. Hum. Genet. 2020, 139, 1555–1563. [Google Scholar] [CrossRef]
- Imaizumi, T.; Yamamoto-Shimojima, K.; Yanagishita, T.; Ondo, Y.; Yamamoto, T. Analyses of breakpoint junctions of complex genomic rearrangements comprising multiple consecutive microdeletions by nanopore sequencing. J. Hum. Genet. 2020, 65, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Yanagishita, T.; Imaizumi, T.; Yamamoto-Shimojima, K.; Yano, T.; Okamoto, N.; Nagata, S.; Yamamoto, T. Breakpoint junction analysis for complex genomic rearrangements with the caldera volcano-like pattern. Hum. Mutat. 2020, 41, 2119–2127. [Google Scholar] [CrossRef] [PubMed]
- Morak, M.; Steinke-Lange, V.; Massdorf, T.; Benet-Pages, A.; Locher, M.; Laner, A.; Kayser, K.; Aretz, S.; Holinski-Feder, E. Prevalence of CNV-neutral structural genomic rearrangements in MLH1, MSH2, and PMS2 not detectable in routine NGS diagnostics. Fam. Cancer 2020, 19, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, R.; Gilbert, M.A.; McEldrew, D.A.; Nassur, J.A.; Loomes, K.M.; Piccoli, D.A.; Krantz, I.D.; Conlin, L.K.; Spinner, N.B. Genome sequencing increases diagnostic yield in clinically diagnosed Alagille syndrome patients with previously negative test results. Genet. Med. 2021, 23, 323–330. [Google Scholar] [CrossRef]
- Weisschuh, N.; Mazzola, P.; Heinrich, T.; Haack, T.; Wissinger, B.; Tonagel, F.; Kelbsch, C. First submicroscopic inversion of the OPA1 gene identified in dominant optic atrophy—A case report. BMC Med. Genet. 2020, 21, 236. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chromosomal Regions | Deletions | Duplications | ||
|---|---|---|---|---|
| Microdeletion Syndromes | Main Clinical Features | Microduplication Syndromes | Main Clinical Features | |
| 22q11.2 | 22q11.2 deletion syndrome | Tetralogy of Fallot, language delay, distinctive facial features | 22q11.2 duplication syndrome | ADHD |
| 7q11.23 | Williams-Beuren syndrome | Supraventricular stenosis, intellectual disability, distinctive facial features | 7q11.23 duplication syndrome | Speech delay and autism spectrum behaviors |
| 15q11 | Prader-Willi syndrome | Developmental delay, hypotonia, obesity | 15q11 duplication | Intellectual disability, autism spectrum behaviors |
| Angelman syndrome | Developmental delay, epilepsy, distinctive facial features | |||
| 17p11 | Smith-Magenis syndrome | Congenital heart defects, developmental delay, distinctive facial features | Potocki-Lupski syndrome | Intellectual disability, autism spectrum behaviors |
| 5q35 | Sotos syndrome | Developmental delay, macrocephaly | 5q35 duplication | |
| Deletion | Duplication | |
|---|---|---|
| PMP22 | hereditary neuropathy with susceptibility to pressure palsies (HNPP) | Charcot-Marie-Tooth disease |
| PLP1 | spastic paraplegia | Pelizaeus-Merzbacher disease |
| MECP2 | Rett syndrome in female | MECP2 duplication syndrome in male |
| Regions | Responsible Gene(s) | Phenotypes | |
|---|---|---|---|
| Microdeletions/duplications derived from NAHR | |||
| 1q21.1 deletion/duplication | Developmental delay, distinctive features, congenital anomalies | ||
| 3q29 deletion | DLG1, PAK2 | Developmental delay, psychiatric symptoms | |
| 15q13.3 deletion | CHRNA7 | Intellectual disability, epilepsy | |
| 16p11.2 deletion/duplication | Developmental disorder | ||
| 17q12 deletion/duplication | HNF1B | Maturity onset diabetes of the young (MODY) | |
| 17q21.31 deletion/duplication | CRHR1, MAPT | Developmental delay, muscular hypotonia, distinctive features | |
| Microdeletions/duplications derived from random breakpoints | |||
| 1q32 deletion | IRF6 | Van der Woude syndrome | |
| 1q41q42 deletion | DISP1 | Developmental delay, epilepsy, distinctive features | |
| 2p15-p16.1 deletion | Autism spectrum disorder | ||
| 2q23.1 deletion | MBD5 | Severe developmental delay, epilepsy, microcephaly | |
| 2q33 deletion/duplication | SATB2 | Intellectual disability | |
| 3p21.31 deletion | BSN | Developmental delay, white matter abnormality, hyperCKemia | |
| 3q13.31 deletion | ZBTB20 | Language delay | |
| 5q14 deletion | MEF2C | Severe developmental delay, epilepsy, brain abnormalities | |
| 5q31.3deletion | PURA, NRG2 | Severe developmental delay, epilepsy | |
| 8q24 deletion | EXT1, TRPS1 | Langer-Giedion syndrome | |
| 9q22.3 deletion | PTCH1 | Gorlin syndrome | |
| 10q22 deletion | KAT6B | Ohdo syndrome | |
| 10q23 deletion | PTEN | Juvenile polyposis | |
| 11p13 deletion | WT1, PAX6 | WAGR syndrome | |
| 11p11.2 deletion | EXT2, ALX4 | Potocki-Shaffer syndrome | |
| 12q24.21 deletion | MED13L | Intellectual disability | |
| 13q32 deletion | ZIC2 | Holoprosencephaly | |
| 15q22.2 deletion | NRG2, RORA | Developmental delay, epilepsy | |
| 16q24.3 deletion | ANKRD11, ZNF778 | Autism spectrum disorder | |
| 17p13.1 deletion | DLG4, GABARAP | Intellectual disability, epilepsy | |
| 18q12.3 deletion | SETBP1 | Language delay | |
| 18q21.2 deletion | TCF4 | Pitt-Hopkins syndrome | |
| 19p13.2 deletion | NFIX | Malan syndrome | |
| Xp22.3 deletion | KAL1 | Kallmann syndrome | |
| Xp21-22 deletion | CDKL5, ARX | Epileptic encephalopathy | |
| Xp11.4 deletion | CASK | Developmental delay, microcephaly | |
| Xp11.22 deletion | HUWE1 | Developmental delay | |
| Xq11.1 deletion | ARHGEF9 | Developmental delay, epilepsy | |
| Xq28 duplication | MECP2 | Developmental delay, epilepsy | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamamoto, T. Genomic Aberrations Associated with the Pathophysiological Mechanisms of Neurodevelopmental Disorders. Cells 2021, 10, 2317. https://doi.org/10.3390/cells10092317
Yamamoto T. Genomic Aberrations Associated with the Pathophysiological Mechanisms of Neurodevelopmental Disorders. Cells. 2021; 10(9):2317. https://doi.org/10.3390/cells10092317
Chicago/Turabian StyleYamamoto, Toshiyuki. 2021. "Genomic Aberrations Associated with the Pathophysiological Mechanisms of Neurodevelopmental Disorders" Cells 10, no. 9: 2317. https://doi.org/10.3390/cells10092317
APA StyleYamamoto, T. (2021). Genomic Aberrations Associated with the Pathophysiological Mechanisms of Neurodevelopmental Disorders. Cells, 10(9), 2317. https://doi.org/10.3390/cells10092317