
Phosphorylation in the Charged Linker Modulates Interactions and Secretion of Hsp90β

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmids
2.2. Mammalian Cell Culture
2.3. Cell Transfection
2.4. Cell Lysis of Transfected Cells for Co-Immunoprecipitation of HA Tagged Hsp90β
2.5. Analysis of Extracts of Transfected HEK293T-Hsp90βKO19 Cells (“Input” of Co-immunoprecipitations)
2.6. Co-Immunoprecipitation (Co-IP) of HA Tagged Hsp90β and Sample Preparation for Mass Spectrometry (Interactome) Analysis
2.7. Mass Spectrometry Data Analysis of HA-Hsp90βWT/HA-Hsp90βS226A/S255A/Hsp90βWT Co-Immunoprecipitation Experiments and Inputs
2.8. Total Hsp90β Occupancy Protocol
- Occupancy is then calculated in the light sample using value a and in the heavy sample using value b according to the following formulas:
- Occupancylight = ; Occupancyheavy =
2.9. Nuclear and Extracellular Hsp90β Occupancy Protocol
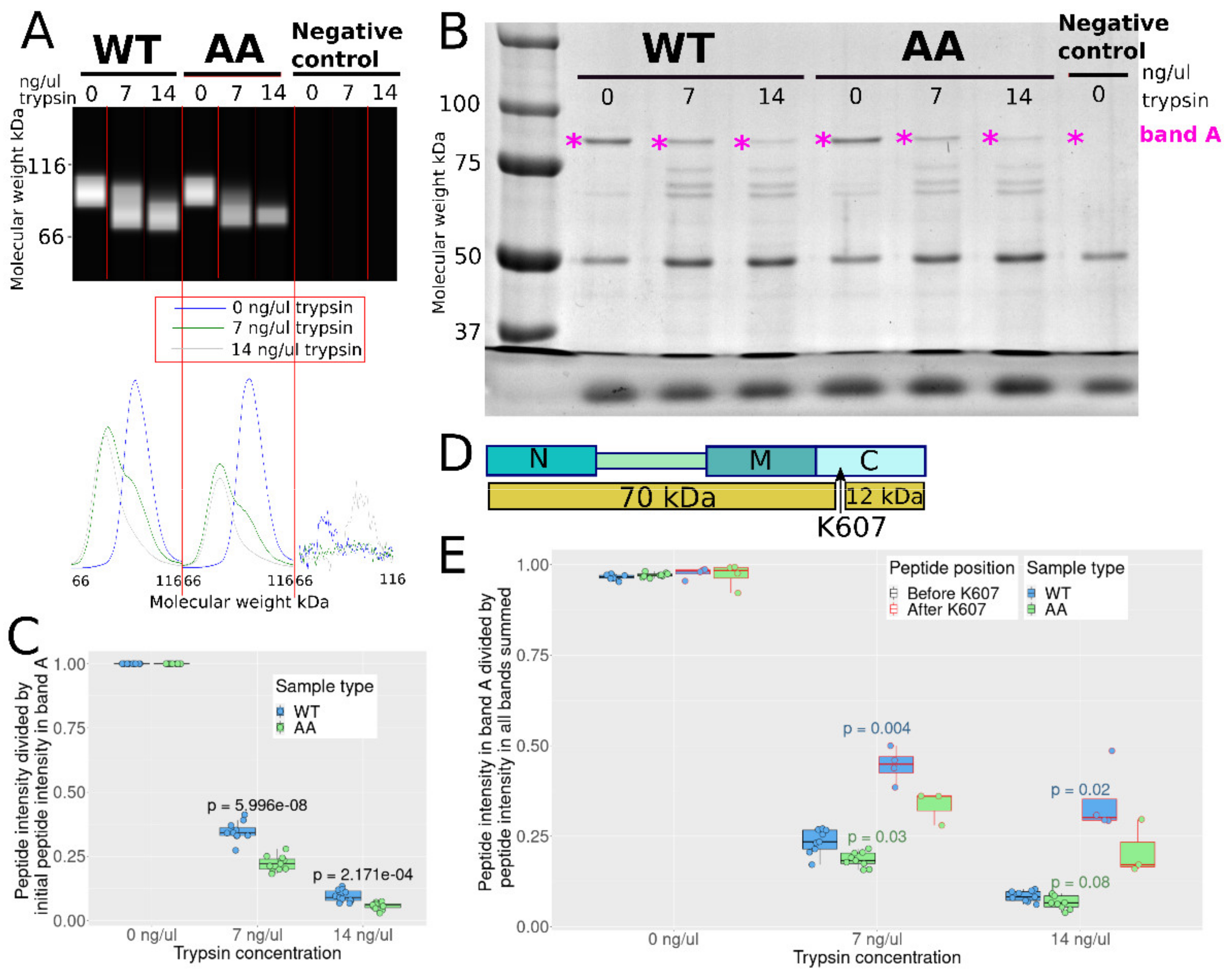
2.10. Limted Proteolysis with Trypsin
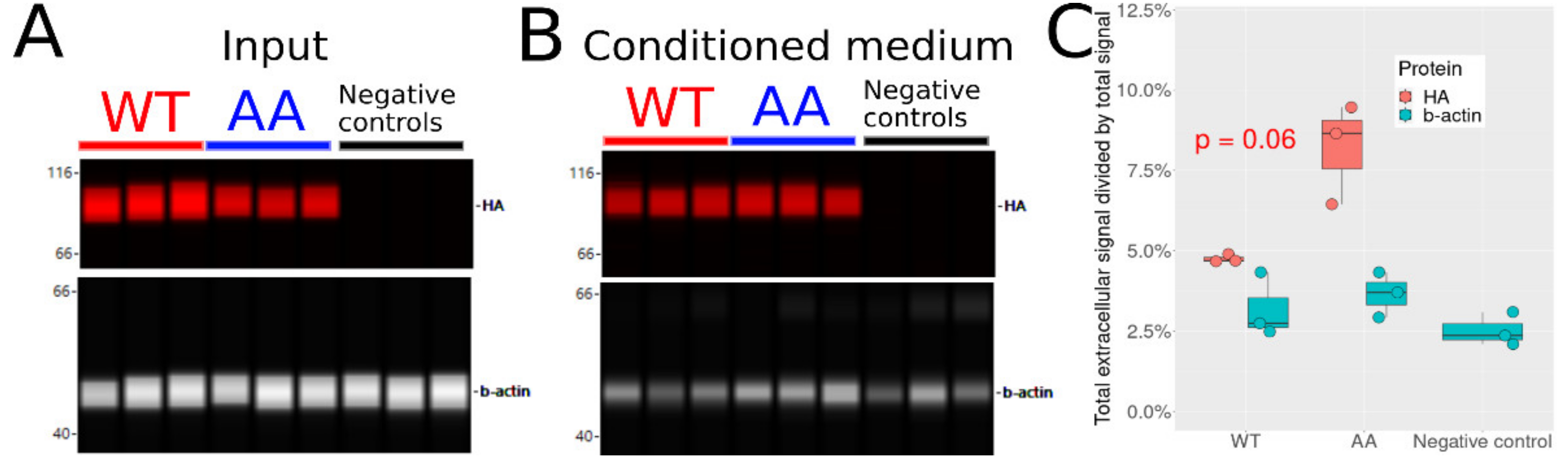
2.11. Analysis of Mutant Secretion
2.12. Cell Treatment and Sample Preparation for Phosphorylation Analysis (TiO2 Enrichment)
2.13. Mutant ATP and ADP Binding Analysis with Active Site Probes
2.14. Raw LC-MS/MS Data Availability
3. Results
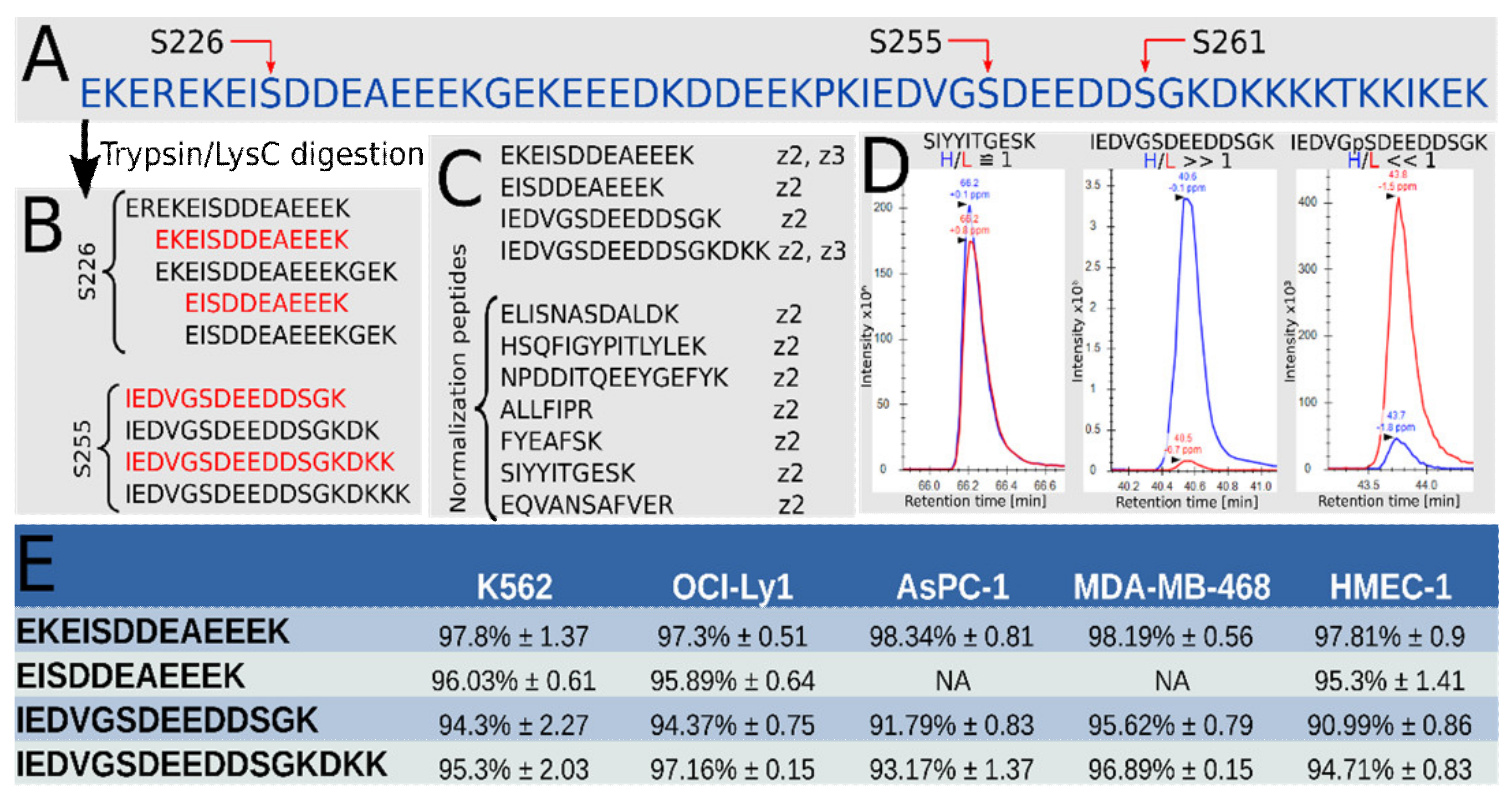
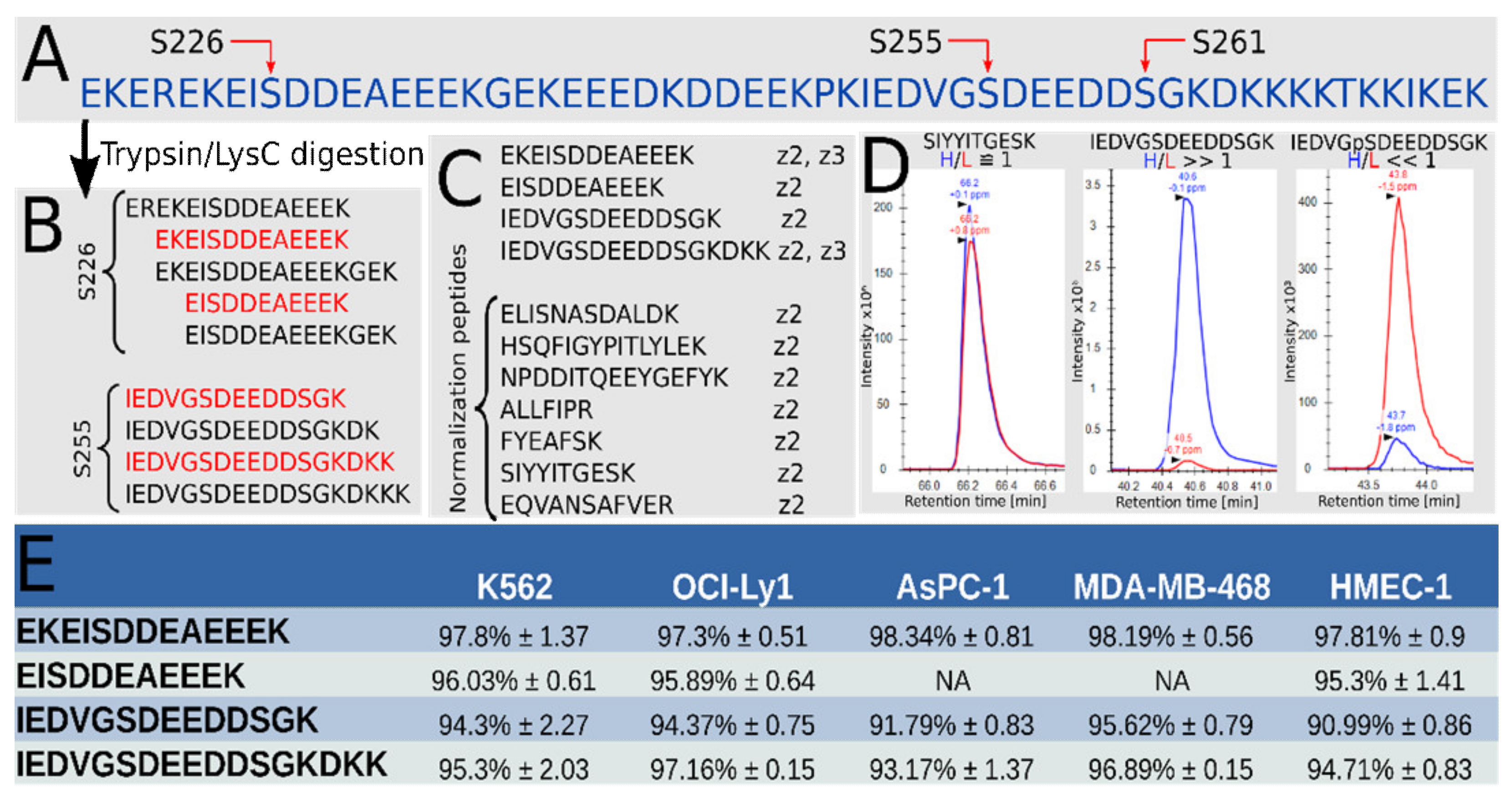
3.1. High Phosphorylation Occupancy for Both S226 and S255 in Hsp90β in a Range of Cell Lines
3.2. Charged Linker Phosphorylation Remains High in Stressed and Heat Shocked Cells
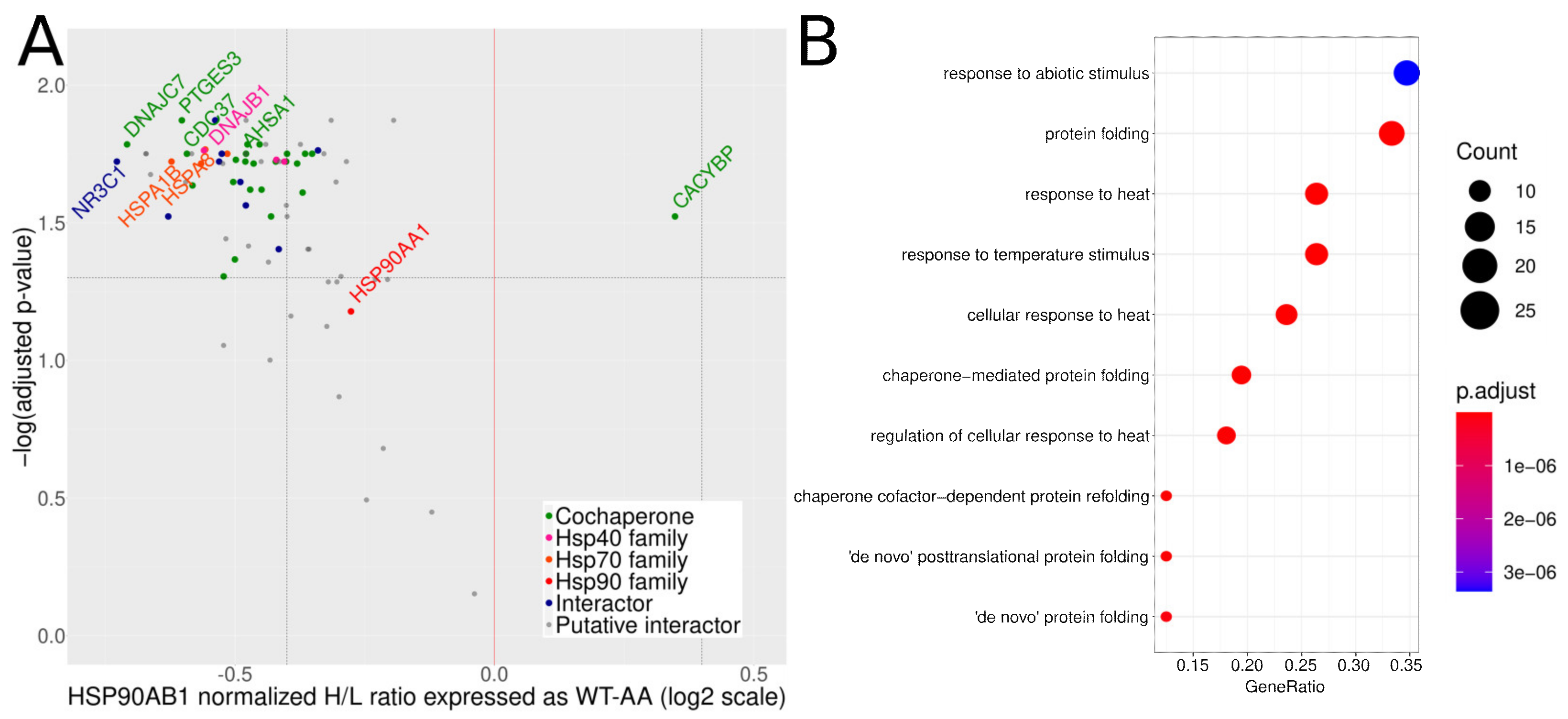
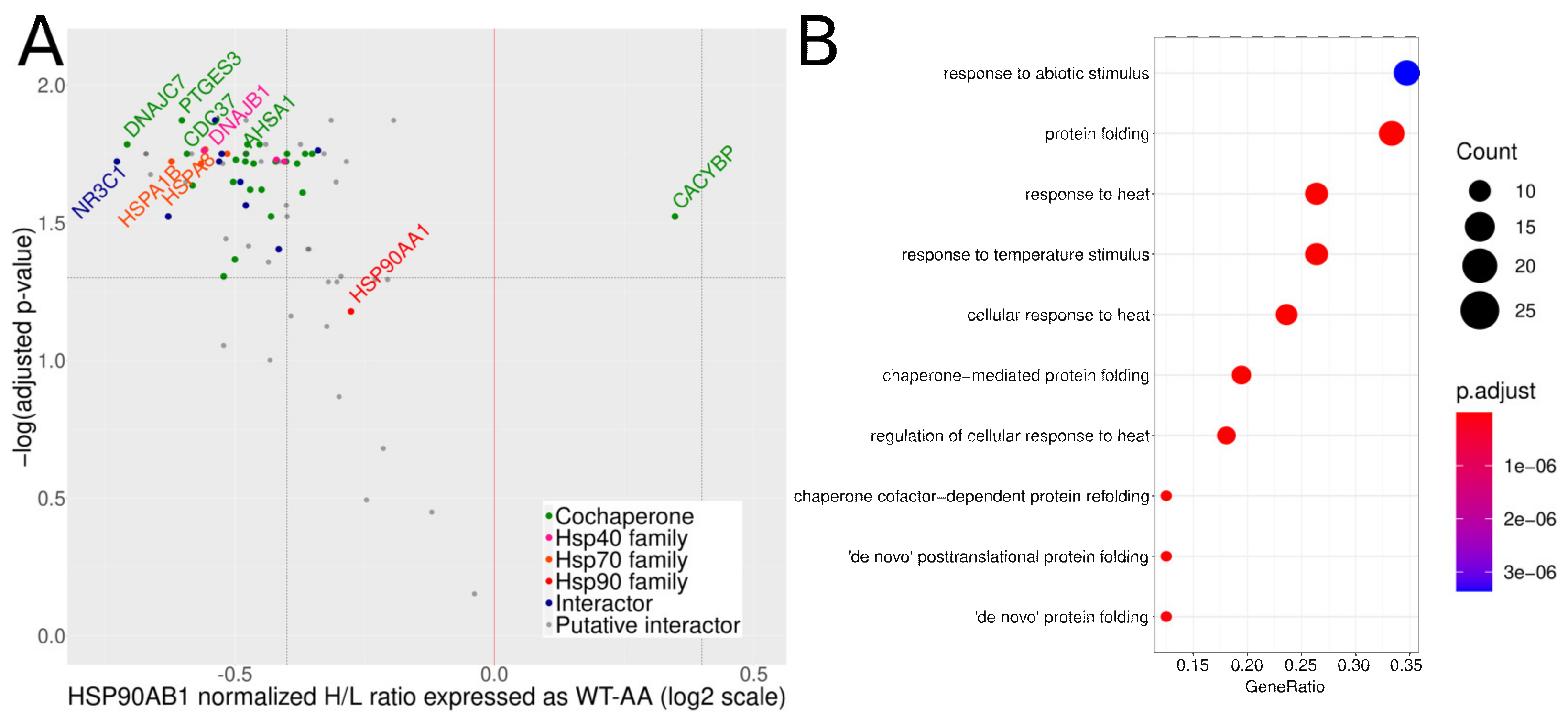
3.3. Mutation of S226 and S255 to Alanines Increases the Amount of Hsp90β Co-Purifying Proteins
3.4. S226 and S255 Mutations to Alanine do Not Alter ATP/ADP Binding
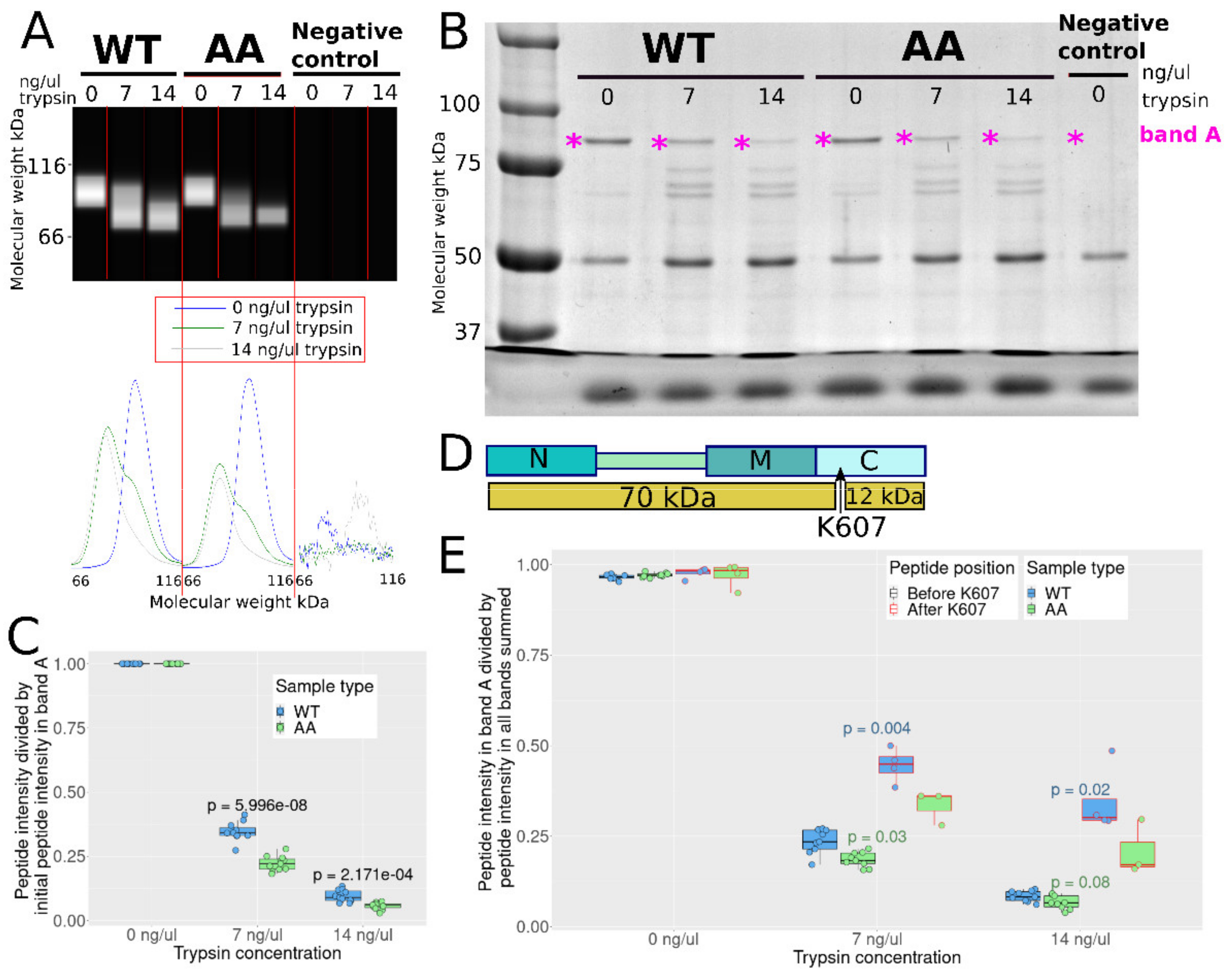
3.5. S226 and S255 Mutation to Alanine Enhances Tryptic Cleavage in the C Domain
3.6. Both Cytosolic and Nuclear Hsp90β Are Highly Phosphorylated on CL
3.7. Extracellular Hsp90β Shows Lower Phosphosite Occupancy
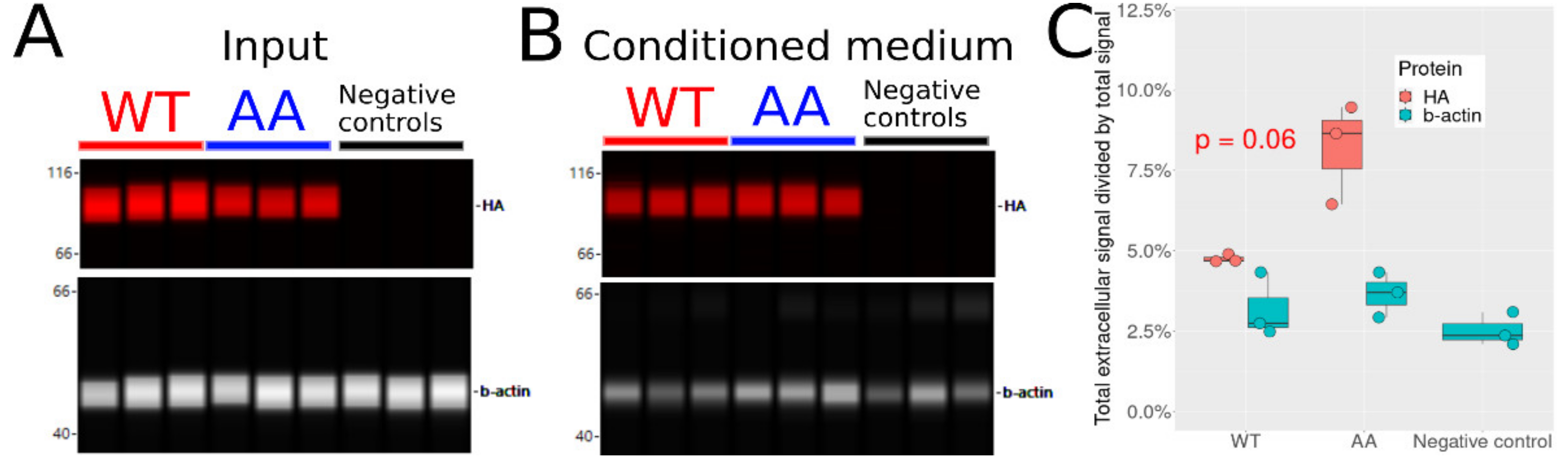
3.8. Non-Phosphorylatable Hsp90β Is Found in Higher Amounts in Conditioned Medium
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pearl, L.H. Review: The HSP90 molecular chaperone-an enigmatic ATPase. Biopolymers 2016, 105, 594–607. [Google Scholar] [CrossRef] [Green Version]
- Prodromou, C. Mechanisms of Hsp90 regulation. Biochem. J. 2016, 473, 2439–2452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef]
- Sima, S.; Richter, K. Regulation of the Hsp90 system. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L. Evolution and function of diverse Hsp90 homologs and cochaperone proteins. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 607–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoter, A.; El-Sabban, M.E.; Naim, H.Y. The HSP90 family: Structure, regulation, function, and implications in health and disease. Int. J. Mol. Sci. 2018, 19, 2560. [Google Scholar] [CrossRef] [Green Version]
- Sreedhar, A.S.; Kalmár, E.; Csermely, P.; Shen, Y.-F. Hsp90 isoforms: Functions, expression and clinical importance. FEBS Lett. 2004, 562, 11–15. [Google Scholar] [CrossRef]
- Cox, M.B.; Johnson, J.L. Evidence for Hsp90 co-chaperones in regulating Hsp90 function and promoting client protein folding. Methods Mol. Biol. 2018, 1709, 397–422. [Google Scholar] [CrossRef]
- Mollapour, M.; Neckers, L. Post-translational modifications of Hsp90 and their contributions to chaperone regulation. Biochim. Biophys. Acta 2012, 1823, 648–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prodromou, C. The “active life” of Hsp90 complexes. Biochim. Biophys. Acta 2012, 1823, 614–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sager, R.A.; Woodford, M.R.; Neckers, L.; Mollapour, M. Detecting posttranslational modifications of Hsp90. Methods Mol. Biol. 2018, 1709, 209–219. [Google Scholar] [CrossRef]
- Dean, M.E.; Johnson, J.L. Human Hsp90 cochaperones: Perspectives on tissue-specific expression and identification of cochaperones with similar in vivo functions. Cell Stress Chaperones 2021, 26, 3–13. [Google Scholar] [CrossRef]
- Backe, S.J.; Sager, R.A.; Woodford, M.R.; Makedon, A.M.; Mollapour, M. Post-translational modifications of Hsp90 and translating the chaperone code. J. Biol. Chem. 2020, 295, 11099–11117. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.S. Phylogenetic analysis of the 90 kD heat shock family of protein sequences and an examination of the relationship among animals, plants, and fungi species. Mol. Biol. Evol. 1995, 12, 1063–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binart, N.; Chambraud, B.; Levin, J.M.; Garnier, J.; Baulieu, E.E. A highly charged sequence of chick hsp90: A good candidate for interaction with steroid receptors. J. Steroid Biochem. 1989, 34, 369–374. [Google Scholar] [CrossRef]
- Tbarka, N.; Richard-Méreau, C.; Formstecher, P.; Dautrevaux, M. Biochemical and immunological evidence that an acidic domain of hsp 90 is involved in the stabilization of untransformed glucocorticoid receptor complexes. FEBS Lett. 1993, 322, 125–128. [Google Scholar] [CrossRef] [Green Version]
- Hainzl, O.; Lapina, M.C.; Buchner, J.; Richter, K. The charged linker region is an important regulator of Hsp90 function. J. Biol. Chem. 2009, 284, 22559–22567. [Google Scholar] [CrossRef] [Green Version]
- Tsutsumi, S.; Mollapour, M.; Graf, C.; Lee, C.-T.; Scroggins, B.T.; Xu, W.; Haslerova, L.; Hessling, M.; Konstantinova, A.A.; Trepel, J.B.; et al. Hsp90 charged-linker truncation reverses the functional consequences of weakened hydrophobic contacts in the N domain. Nat. Struct. Mol. Biol. 2009, 16, 1141–1147. [Google Scholar] [CrossRef]
- Tsutsumi, S.; Mollapour, M.; Prodromou, C.; Lee, C.-T.; Panaretou, B.; Yoshida, S.; Mayer, M.P.; Neckers, L.M. Charged linker sequence modulates eukaryotic heat shock protein 90 (Hsp90) chaperone activity. Proc. Natl. Acad. Sci. USA 2012, 109, 2937–2942. [Google Scholar] [CrossRef] [Green Version]
- Jahn, M.; Rehn, A.; Pelz, B.; Hellenkamp, B.; Richter, K.; Rief, M.; Buchner, J.; Hugel, T. The charged linker of the molecular chaperone Hsp90 modulates domain contacts and biological function. Proc. Natl. Acad. Sci. USA 2014, 111, 17881–17886. [Google Scholar] [CrossRef] [Green Version]
- Suhane, T.; Laskar, S.; Advani, S.; Roy, N.; Varunan, S.; Bhattacharyya, D.; Bhattacharyya, S.; Bhattacharyya, M.K. Both the charged linker region and ATPase domain of Hsp90 are essential for Rad51-dependent DNA repair. Eukaryot. Cell 2015, 14, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Daturpalli, S.; Knieß, R.A.; Lee, C.-T.; Mayer, M.P. Large Rotation of the N-terminal domain of Hsp90 is important for interaction with some but not all client proteins. J. Mol. Biol. 2017, 429, 1406–1423. [Google Scholar] [CrossRef] [PubMed]
- Cadepond, F.; Jibard, N.; Binart, N.; Schweizer-Groyer, G.; Segard-Maurel, I.; Baulieu, E.E. Selective deletions in the 90 kDa heat shock protein (hsp90) impede hetero-oligomeric complex formation with the glucocorticosteroid receptor (GR) or hormone binding by GR. J. Steroid Biochem. Mol. Biol. 1994, 48, 361–367. [Google Scholar] [CrossRef]
- Binart, N.; Lombès, M.; Baulieu, E.E. Distinct functions of the 90 kDa heat-shock protein (hsp90) in oestrogen and mineralocorticosteroid receptor activity: Effects of hsp90 deletion mutants. Biochem. J. 1995, 311 Pt 3, 797–804. [Google Scholar] [CrossRef] [Green Version]
- Dao-Phan, H.P.; Formstecher, P.; Lefebvre, P. Disruption of the glucocorticoid receptor assembly with heat shock protein 90 by a peptidic antiglucocorticoid. Mol. Endocrinol. 1997, 11, 962–972. [Google Scholar] [CrossRef]
- Scheibel, T.; Siegmund, H.I.; Jaenicke, R.; Ganz, P.; Lilie, H.; Buchner, J. The charged region of Hsp90 modulates the function of the N-terminal domain. Proc. Natl. Acad. Sci. USA 1999, 96, 1297–1302. [Google Scholar] [CrossRef] [Green Version]
- Louvion, J.F.; Warth, R.; Picard, D. Two eukaryote-specific regions of Hsp82 are dispensable for its viability and signal transduction functions in yeast. Proc. Natl. Acad. Sci. USA 1996, 93, 13937–13942. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.; Devin, J.; Sullivan, W.P.; Toft, D.; Baulieu, E.E.; Catelli, M.G. Mutational analysis of Hsp90 alpha dimerization and subcellular localization: Dimer disruption does not impede “in vivo’’ interaction with estrogen receptor. J. Cell Sci. 1996, 109 Pt 7, 1677–1687. [Google Scholar] [CrossRef]
- Huai, Q.; Wang, H.; Liu, Y.; Kim, H.Y.; Toft, D.; Ke, H. Structures of the N-terminal and middle domains of E. coli Hsp90 and conformation changes upon ADP binding. Structure 2005, 13, 579–590. [Google Scholar] [CrossRef] [Green Version]
- Binart, N.; Chambraud, B.; Dumas, B.; Rowlands, D.A.; Bigogne, C.; Levin, J.M.; Garnier, J.; Baulieu, E.E.; Catelli, M.G. The cDNA-derived amino acid sequence of chick heat shock protein Mr 90,000 (HSP 90) reveals a “DNA like” structure: Potential site of interaction with steroid receptors. Biochem. Biophys. Res. Commun. 1989, 159, 140–147. [Google Scholar] [CrossRef]
- López, A.; Elimelech, A.R.; Klimm, K.; Sattler, M. The charged linker modulates conformations and molecular interactions of Hsp90. ChemBioChem 2020. [Google Scholar] [CrossRef]
- Lees-Miller, S.P.; Anderson, C.W. Two human 90-kDa heat shock proteins are phosphorylated in vivo at conserved serines that are phosphorylated in vitro by casein kinase II. J. Biol. Chem. 1989, 264, 2431–2437. [Google Scholar] [CrossRef]
- Kelley, P.M.; Schlesinger, M.J. Antibodies to two major chicken heat shock proteins cross-react with similar proteins in widely divergent species. Mol. Cell. Biol. 1982, 2, 267–274. [Google Scholar] [CrossRef] [Green Version]
- Iannotti, A.M.; Rabideau, D.A.; Dougherty, J.J. Characterization of purified avian 90,000-Da heat shock protein. Arch. Biochem. Biophys. 1988, 264, 54–60. [Google Scholar] [CrossRef]
- Lundby, A.; Secher, A.; Lage, K.; Nordsborg, N.B.; Dmytriyev, A.; Lundby, C.; Olsen, J. V Quantitative maps of protein phosphorylation sites across 14 different rat organs and tissues. Nat. Commun. 2012, 3, 876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garnier, C.; Lafitte, D.; Jorgensen, T.J.D.; Jensen, O.N.; Briand, C.; Peyrot, V. Phosphorylation and oligomerization states of native pig brain HSP90 studied by mass spectrometry. Eur. J. Biochem. 2001, 268, 2402–2407. [Google Scholar] [CrossRef] [PubMed]
- Franchin, C.; Cesaro, L.; Salvi, M.; Millioni, R.; Iori, E.; Cifani, P.; James, P.; Arrigoni, G.; Pinna, L. Quantitative analysis of a phosphoproteome readily altered by the protein kinase CK2 inhibitor quinalizarin in HEK-293T cells. Biochim. Biophys. Acta 2015, 1854, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Franchin, C.; Borgo, C.; Cesaro, L.; Zaramella, S.; Vilardell, J.; Salvi, M.; Arrigoni, G.; Pinna, L.A. Re-evaluation of protein kinase CK2 pleiotropy: New insights provided by a phosphoproteomics analysis of CK2 knockout cells. Cell. Mol. Life Sci. 2018, 75, 2011–2026. [Google Scholar] [CrossRef]
- Kurokawa, M.; Zhao, C.; Reya, T.; Kornbluth, S. Inhibition of apoptosome formation by suppression of Hsp90beta phosphorylation in tyrosine kinase-induced leukemias. Mol. Cell. Biol. 2008, 28, 5494–5506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.F.; Griffiths, S.; Rodríguez-Suárez, E.; Pierce, A.; Unwin, R.D.; Jaworska, E.; Evans, C.A.; Gaskell, S.J.; Whetton, A.D. Assessment of downstream effectors of BCR/ABL protein tyrosine kinase using combined proteomic approaches. Proteomics 2010, 10, 3321–3342. [Google Scholar] [CrossRef] [PubMed]
- Unwin, R.D.; Sternberg, D.W.; Lu, Y.; Pierce, A.; Gilliland, D.G.; Whetton, A.D. Global effects of BCR/ABL and TEL/PDGFRbeta expression on the proteome and phosphoproteome: Identification of the Rho pathway as a target of BCR/ABL. J. Biol. Chem. 2005, 280, 6316–6326. [Google Scholar] [CrossRef] [Green Version]
- Legagneux, V.; Morange, M.; Bensaude, O. Heat shock increases turnover of 90 kDa heat shock protein phosphate groups in HeLa cells. FEBS Lett. 1991, 291, 359–362. [Google Scholar] [CrossRef] [Green Version]
- Weeraphan, C.; Phongdara, A.; Chaiyawat, P.; Diskul-Na-Ayudthaya, P.; Chokchaichamnankit, D.; Verathamjamras, C.; Netsirisawan, P.; Yingchutrakul, Y.; Roytrakul, S.; Champattanachai, V.; et al. Phosphoproteome Profiling of isogenic cancer cell-derived exosome reveals HSP90 as a potential marker for human cholangiocarcinoma. Proteomics 2019, 19, e1800159. [Google Scholar] [CrossRef]
- Ogiso, H.; Kagi, N.; Matsumoto, E.; Nishimoto, M.; Arai, R.; Shirouzu, M.; Mimura, J.; Fujii-Kuriyama, Y.; Yokoyama, S. Phosphorylation analysis of 90 kDa heat shock protein within the cytosolic arylhydrocarbon receptor complex. Biochemistry 2004, 43, 15510–15519. [Google Scholar] [CrossRef]
- Kim, S.W.; Hasanuzzaman, M.; Cho, M.; Heo, Y.R.; Ryu, M.-J.; Ha, N.-Y.; Park, H.J.; Park, H.-Y.; Shin, J.-G. Casein Kinase 2 (CK2)-mediated Phosphorylation of Hsp90β as a Novel Mechanism of Rifampin-induced MDR1 Expression. J. Biol. Chem. 2015, 290, 17029–17040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olesen, S.H.; Ingles, D.J.; Zhu, J.Y.; Martin, M.P.; Betzi, S.; Georg, G.I.; Tash, J.S.; Schönbrunn, E. Stability of the human Hsp90-p50Cdc37 chaperone complex against nucleotides and Hsp90 inhibitors, and the influence of phosphorylation by casein kinase 2. Molecules 2015, 20, 1643–1660. [Google Scholar] [CrossRef] [Green Version]
- Woo, S.H.; An, S.; Lee, H.-C.; Jin, H.-O.; Seo, S.-K.; Yoo, D.-H.; Lee, K.-H.; Rhee, C.H.; Choi, E.-J.; Hong, S.-I.; et al. A truncated form of p23 down-regulates telomerase activity via disruption of Hsp90 function. J. Biol. Chem. 2009, 284, 30871–30880. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, K.; Weidenauer, L.; Luengo, T.M.; Pieters, E.C.; Echeverría, P.C.; Bernasconi, L.; Wider, D.; Sadian, Y.; Koopman, M.B.; Villemin, M.; et al. The Hsp70-Hsp90 co-chaperone Hop/Stip1 shifts the proteostatic balance from folding towards degradation. Nat. Commun. 2020, 11, 5975. [Google Scholar] [CrossRef]
- Ong, S.E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteom. 2002, 1, 376–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Okayama, H. High-efficiency transformation of mammalian cells by plasmid DNA. Mol. Cell. Biol. 1987, 7, 2745–2752. [Google Scholar] [CrossRef] [PubMed]
- Kulak, N.A.; Pichler, G.; Paron, I.; Nagaraj, N.; Mann, M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods 2014, 11, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteom. 2014, 13, 2513–2526. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020; Available online: https://www.R-project.org/ (accessed on 17 May 2021).
- Wickham, H. The split-apply-combine strategy for data analysis. J. Stat. Softw. 2011, 40, 1–29. [Google Scholar] [CrossRef] [Green Version]
- Sievert, C. Interactive Web-Based Data Visualization with R, Plotly, and Shiny; Chapman and Hall/CRC: Boca Raton, FL, USA, 2020; ISBN 9781138331457. [Google Scholar]
- Thevenot, E.A. Ropls: PCA, PLS(-DA) and OPLS(-DA) for Multivariate Analysis and Feature Selection of Omics Data. R package version 1.20.0. 2020. Available online: https://bioconductor.statistik.tu-dortmund.de/packages/3.6/bioc/vignettes/ropls/inst/doc/ropls-vignette.pdf (accessed on 17 May 2021).
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Thévenot, E.A.; Roux, A.; Xu, Y.; Ezan, E.; Junot, C. Analysis of the human adult urinary metabolome variations with age, body mass index, and gender by implementing a comprehensive workflow for univariate and OPLS statistical analyses. J. Proteome Res. 2015, 14, 3322–3335. [Google Scholar] [CrossRef]
- Xie, Y.; Allaire, J.J.; Grolemund, G. R Markdown: The Definitive Guide; Chapman and Hall/CRC: Boca Raton, FL, USA, 2018. [Google Scholar]
- Xie, Y. TinyTeX: A lightweight, cross-platform, and easy-to-maintain LaTeX distribution based on TeX Live. TUGboat 2019, 40, 30–32. [Google Scholar]
- Wickham, H.; Chang, W.; Henry, L.; Pedersen, T.L.; Takahashi, K.; Wilke, C.; Woo, K.; Yutani, H.; Dunnington, D. ggplot2; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Xie, Y. knitr: A General-Purpose Package for Dynamic Report Generation in R; R package version 1.29. 2020. Available online: https://CRAN.R-project.org/package=knitr (accessed on 17 May 2021).
- Allaire, J.J.; Xie, Y.; McPherson, J.; Luraschi, J.; Ushey, K.; Atkins, A.; Wickham, H.; Cheng, J.; Chang, W.; Iannone, R. Rmarkdown: Dynamic Documents for R, R package version 2.3; R-Studio: Boston, MA, USA, 2020; Available online: https://rmarkdown.rstudio.com (accessed on 17 May 2021).
- Pagès, H.; Carlson, M.; Falcon, S.; Li, N. AnnotationDbi: Manipulation of SQLite-Based Annotations in Bioconductor. R package version 1.50.3. 2020. Available online: https://bioconductor.org/packages/release/bioc/html/AnnotationDbi.html (accessed on 17 May 2021).
- Auguie, B. gridExtra: Miscellaneous Functions for “Grid” Graphics, R package version 2.3. 2017. Available online: https://CRAN.R-project.org/package=gridExtra (accessed on 17 May 2021).
- Oehlschlägel, J.; Ripley, B. bit: Classes and Methods for Fast Memory-Efficient Boolean Selections; R package version 4.0.4. 2020. Available online: https://CRAN.R-project.org/package=bit (accessed on 17 May 2021).
- Yu, G. DOSE: Disease Ontology Semantic and Enrichment Analysis. R package version 3.14.0. 2020. Available online: https://guangchuangyu.github.io/software/DOSE (accessed on 17 May 2021).
- Yu, G.; Wang, L.-G.; Yan, G.-R.; He, Q.-Y. DOSE: An R/Bioconductor package for disease ontology semantic and enrichment analysis. Bioinformatics 2015, 31, 608–609. [Google Scholar] [CrossRef] [Green Version]
- Yu, G. ReactomePA: Reactome Pathway Analysis. R package version 1.32.0. 2020. Available online: https://guangchuangyu.github.io/software/ReactomePA (accessed on 17 May 2021).
- Inkscape Project. Inkscape: Open Source Scalable Vector Graphics Editor. Version 0.92.5. 2020. Available online: https://inkscape.org/ (accessed on 17 May 2021).
- Yu, G.; He, Q.-Y. ReactomePA: An R/Bioconductor package for reactome pathway analysis and visualization. Mol. Biosyst. 2016, 12, 477–479. [Google Scholar] [CrossRef]
- Li, J. DO.db: A Set of Annotation Maps Describing the Entire Disease Ontology. R package version 2.9. 2015. Available online: https://bioconductor.org/packages/release/data/annotation/html/DO.db.html (accessed on 17 May 2021).
- Acker, D. gg3D: 3D Perspective Plots for ggplot2. R package version 0.0.0.9. 2020. Available online: https://github.com/AckerDWM/gg3D (accessed on 17 May 2021).
- Campitelli, E. ggnewscale: Multiple Fill and Colour Scales in “ggplot2”. R package version 0.4.5. 2021. Available online: https://CRAN.R-project.org/package=ggnewscale (accessed on 17 May 2021).
- Zhu, H. kableExtra: Construct Complex Table with “kable” and Pipe Syntax. R package version 1.1.0. 2019. Available online: https://CRAN.R-project.org/package=kableExtra (accessed on 17 May 2021).
- Kassambara, A. ggpubr: “ggplot2” Based Publication Ready Plots. R package version 0.4.0. 2020. Available online: https://CRAN.R-project.org/package=ggpubr (accessed on 17 May 2021).
- Pedersen, T.L. ggraph: An Implementation of Grammar of Graphics for Graphs and Networks. R package version 2.0.3. 2020. Available online: https://CRAN.R-project.org/package=ggraph (accessed on 17 May 2021).
- Wickham, H. stringr: Simple, Consistent Wrappers for Common String Operations. R package version 1.4.0. 2019. Available online: https://CRAN.R-project.org/package=stringr (accessed on 17 May 2021).
- Bache, S.M.; Wickham, H. magrittr: A Forward-Pipe Operator for R. R package version 1.5. 2014. Available online: https://CRAN.R-project.org/package=magrittr (accessed on 17 May 2021).
- Carlson, M. org.Hs.eg.db: Genome wide annotation for Human. R package version 3.11.14. 2020. Available online: https://bioconductor.org/packages/release/data/annotation/html/org.Hs.eg.db.html (accessed on 17 May 2021).
- Luo; Weijun; Brouwer; Cory Pathview: An R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics 2013, 29, 1830–1831. [CrossRef] [Green Version]
- Luo, W. Pathview: A Tool Set for Pathway Based Data Integration and Visualization. R package version 1.28.1. 2020. Available online: https://pathview.uncc.edu (accessed on 17 May 2021).
- Sievert, C.; Parmer, C.; Hocking, T.; Chamberlain, S.; Ram, K.; Corvellec, M.; Despouy, P. plotly: Create Interactive Web Graphics via “plotly.js”. R package version 4.9.2.1. 2020. Available online: https://CRAN.R-project.org/package=plotly (accessed on 17 May 2021).
- Perkins, D.N.; Pappin, D.J.; Creasy, D.M.; Cottrell, J.S. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 1999, 20, 3551–3567. [Google Scholar] [CrossRef]
- Pino, L.K.; Searle, B.C.; Bollinger, J.G.; Nunn, B.; MacLean, B.; MacCoss, M.J. The Skyline ecosystem: Informatics for quantitative mass spectrometry proteomics. Mass Spectrom. Rev. 2020, 39, 229–244. [Google Scholar] [CrossRef]
- Schilling, B.; Rardin, M.J.; MacLean, B.X.; Zawadzka, A.M.; Frewen, B.E.; Cusack, M.P.; Sorensen, D.J.; Bereman, M.S.; Jing, E.; Wu, C.C.; et al. Platform-independent and label-free quantitation of proteomic data using MS1 extracted ion chromatograms in skyline: Application to protein acetylation and phosphorylation. Mol. Cell. Proteom. 2012, 11, 202–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsen, J.V.; Vermeulen, M.; Santamaria, A.; Kumar, C.; Miller, M.L.; Jensen, L.J.; Gnad, F.; Cox, J.; Jensen, T.S.; Nigg, E.A.; et al. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 2010, 3, ra3. [Google Scholar] [CrossRef]
- Shevchenko, A.; Tomas, H.; Havliš, J.; Olsen, J.V.; Mann, M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 2007, 1, 2856–2860. [Google Scholar] [CrossRef] [PubMed]
- Cortes, S.; Baker-Williams, A.J.; Mollapour, M.; Bourboulia, D. Detection and Analysis of Extracellular Hsp90 (eHsp90). In Rules of Thumb for Chemical Engineers; Elsevier: Amsterdam, The Netherlands, 2018; Volume 1709, pp. 321–329. [Google Scholar]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]
- Deutsch, E.W.; Bandeira, N.; Sharma, V.; Perez-Riverol, Y.; Carver, J.J.; Kundu, D.J.; García-Seisdedos, D.; Jarnuczak, A.F.; Hewapathirana, S.; Pullman, B.S.; et al. The ProteomeXchange consortium in 2020: Enabling “big data” approaches in proteomics. Nucleic Acids Res. 2020, 48, D1145–D1152. [Google Scholar] [CrossRef] [Green Version]
- Šlechtová, T.; Gilar, M.; Kalíková, K.; Tesařová, E. Insight into trypsin miscleavage: Comparison of kinetic constants of problematic peptide sequences. Anal. Chem. 2015, 87, 7636–7643. [Google Scholar] [CrossRef]
- Nordin, B.E.; Liu, Y.; Aban, A.; Brown, H.E.; Wu, J.; Hainley, A.K.; Rosenblum, J.S.; Nomanbhoy, T.K.; Kozarich, J.W. ATP acyl phosphate reactivity reveals native conformations of Hsp90 paralogs and inhibitor target engagement. Biochemistry 2015, 54, 3024–3036. [Google Scholar] [CrossRef]
- Beebe, K.; Mollapour, M.; Scroggins, B.; Prodromou, C.; Xu, W.; Tokita, M.; Taldone, T.; Pullen, L.; Zierer, B.K.; Lee, J.; et al. Post-translational modification and conformational state of Heat Shock Protein 90 differentially affect binding of chemically diverse small molecule inhibitors. Oncotarget 2013, 4, 1065–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Chang, C.; Li, W. The role of secreted heat shock protein-90 (Hsp90) in wound healing—how could it shape future therapeutics? Expert Rev. Proteom. 2017, 14. [Google Scholar] [CrossRef]
- Wong, D.S.; Jay, D.G. Emerging roles of extracellular Hsp90 in cancer. Adv. Cancer Res. 2016, 129, 141–163. [Google Scholar] [CrossRef]
- Tsutsumi, S.; Neckers, L. Extracellular heat shock protein 90: A role for a molecular chaperone in cell motility and cancer metastasis. Cancer Sci. 2007, 98, 1536–1539. [Google Scholar] [CrossRef]
- Li, W.; Sahu, D.; Tsen, F. Secreted heat shock protein-90 (Hsp90) in wound healing and cancer. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 730–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, C.F.; Sahu, D.; Tsen, F.; Zhao, Z.; Fan, J.; Kim, R.; Wang, X.; O’Brien, K.; Li, Y.; Kuang, Y.; et al. A fragment of secreted Hsp90α carries properties that enable it to accelerate effectively both acute and diabetic wound healing in mice. J. Clin. Investig. 2011, 121, 4348–4361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, C.; Wang, Y.; Yen, H.; Tsou, C.; Ku, W.; Lin, P.; Chen, H.-Y.; Nesvizhskii, A.I.; Ishihama, Y.; Chen, Y. Large-scale determination of absolute phosphorylation stoichiometries in human cells by motif-targeting quantitative proteomics. Nat. Commun. 2015, 6, 6622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Góral, A.; Bieganowski, P.; Prus, W.; Krzemieñ-Ojak, Ł.; Dziołka, B.K.; Fabczak, H.; Filipek, A. Calcyclin binding protein/siah-1 interacting protein is a hsp90 binding chaperone. PLoS ONE 2016, 11, 1–18. [Google Scholar] [CrossRef]
- Prodromou, C.; Roe, S.M.; O’Brien, R.; Ladbury, J.E.; Piper, P.W.; Pearl, L.H. Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell 1997, 90, 65–75. [Google Scholar] [CrossRef] [Green Version]
- McLaughlin, S.H.; Ventouras, L.-A.; Lobbezoo, B.; Jackson, S.E. Independent ATPase activity of Hsp90 subunits creates a flexible assembly platform. J. Mol. Biol. 2004, 344, 813–826. [Google Scholar] [CrossRef]
- Mimnaugh, E.G.; Worland, P.J.; Whitesell, L.; Neckers, L.M. Possible role for serine/threonine phosphorylation in the regulation of the heteroprotein complex between the hsp90 stress protein and the pp60v-src tyrosine kinase. J. Biol. Chem. 1995, 270, 28654–28659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.G.; Gilmore, R.; Leone, G.; Coffey, M.C.; Weber, B.; Lee, P.W.K. Hsp90 phosphorylation is linked to its chaperoning function: Assembly of the reovirus cell attachment protein. J. Biol. Chem. 2001, 276, 32822–32827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csermely, P.; Kahn, C.R. The 90-kDa heat shock protein (hsp-90) possesses an ATP binding site and autophosphorylating activity. J. Biol. Chem. 1991, 266, 4943–4950. [Google Scholar] [CrossRef]
- Wang, X.; Song, X.; Zhuo, W.; Fu, Y.; Shi, H.; Liang, Y.; Tong, M.; Chang, G.; Luo, Y. The regulatory mechanism of Hsp90alpha secretion and its function in tumor malignancy. Proc. Natl. Acad. Sci. USA 2009, 106, 21288–21293. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.M.U.; Roe, S.M.; Vaughan, C.K.; Meyer, P.; Panaretou, B.; Piper, P.W.; Prodromou, C.; Pearl, L.H. Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature 2006, 440, 1013–1017. [Google Scholar] [CrossRef] [Green Version]
- Verba, K.A.; Wang, R.Y.-R.; Arakawa, A.; Liu, Y.; Shirouzu, M.; Yokoyama, S.; Agard, D.A. Atomic structure of Hsp90-Cdc37-Cdk4 reveals that Hsp90 traps and stabilizes an unfolded kinase. Science 2016, 352, 1542–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Primer Sequence (5′-3′) |
|---|---|
| A1 | AAGGAAATTGCTGATGATGAGGCAG |
| A2 (BGH_Reverse) | TAGAAGGCACAGTCGAGG |
| A3 (T7_forward) | TAATACGACTCACTATAGGG |
| A4 | CATCATCAGCAATTTCCTTCTCTCG |
| A5 | ATGTGGGTGCAGATGAGGAGG |
| A6 | TCATCTGCACCCACATCTTCG |
| Sample | Average ± s.d. | p-Value |
|---|---|---|
| WT + ATP | 0.46 ± 0.03 | |
| AA + ATP | 0.53 ± 0.05 | ATP:0.12 |
| WT + ADP | 0.34 ± 0.04 | ADP:0.8 |
| AA + ADP | 0.35 ± 0.06 |
| Peptide | Average Occupancy ± s.d. | Adjusted p-Value |
|---|---|---|
| EKEIpSDDEAEEEK | 93.3% ± 0.8 | 0.01 |
| EIpSDDEAEEEK | 94.5% ± 0.4 | NA |
| IEDVGpSDEEDDSGK | 85.0% ± 2.2 | 0.01 |
| IEDVGpSDEEDDSGKDKK | 81.3% ± 2.8 | 0.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weidenauer, L.; Quadroni, M. Phosphorylation in the Charged Linker Modulates Interactions and Secretion of Hsp90β. Cells 2021, 10, 1701. https://doi.org/10.3390/cells10071701
Weidenauer L, Quadroni M. Phosphorylation in the Charged Linker Modulates Interactions and Secretion of Hsp90β. Cells. 2021; 10(7):1701. https://doi.org/10.3390/cells10071701
Chicago/Turabian StyleWeidenauer, Lorenz, and Manfredo Quadroni. 2021. "Phosphorylation in the Charged Linker Modulates Interactions and Secretion of Hsp90β" Cells 10, no. 7: 1701. https://doi.org/10.3390/cells10071701
APA StyleWeidenauer, L., & Quadroni, M. (2021). Phosphorylation in the Charged Linker Modulates Interactions and Secretion of Hsp90β. Cells, 10(7), 1701. https://doi.org/10.3390/cells10071701