
Emerging Roles of Small GTPases in Islet β-Cell Function

Abstract
1. Introduction
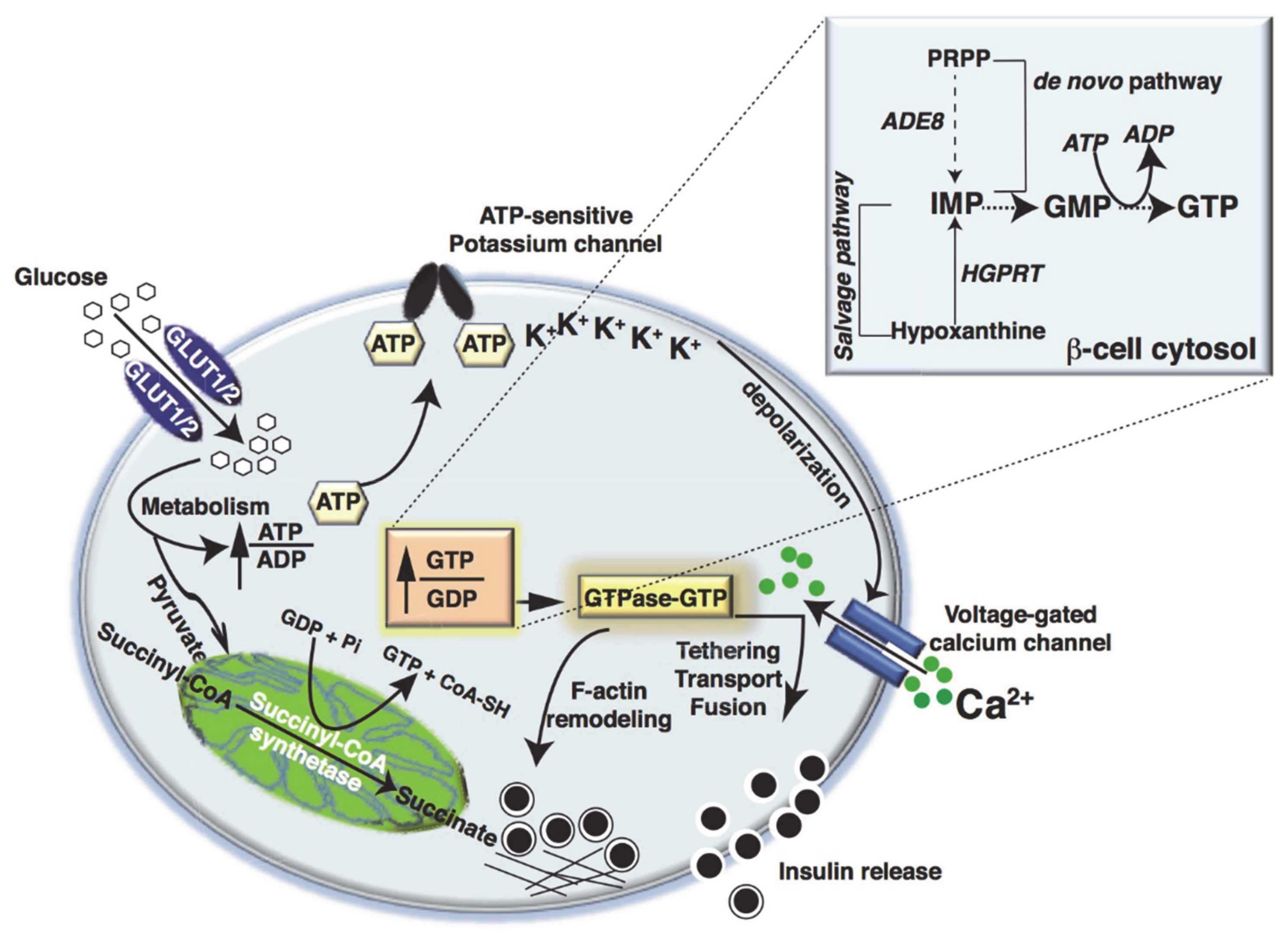
2. Metabolic Fate of Glucose in Islet β-Cells
3. Metabolic Dysfunction and Small GTPase Signaling in Islet β-Cells
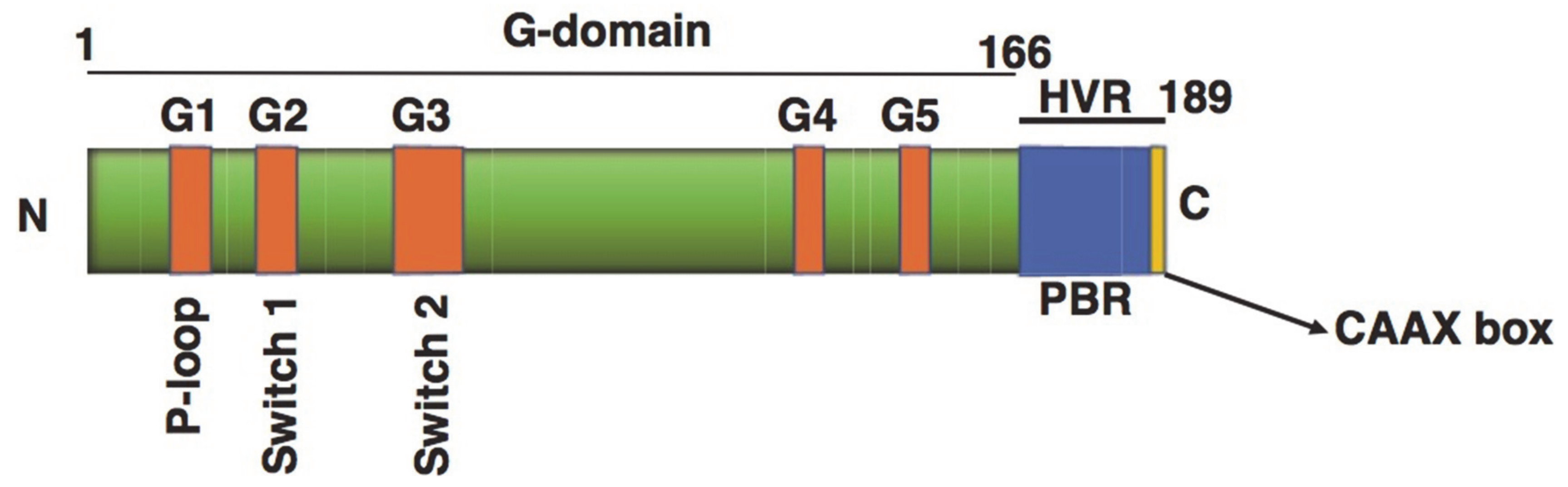
4. Small Monomeric GTPases
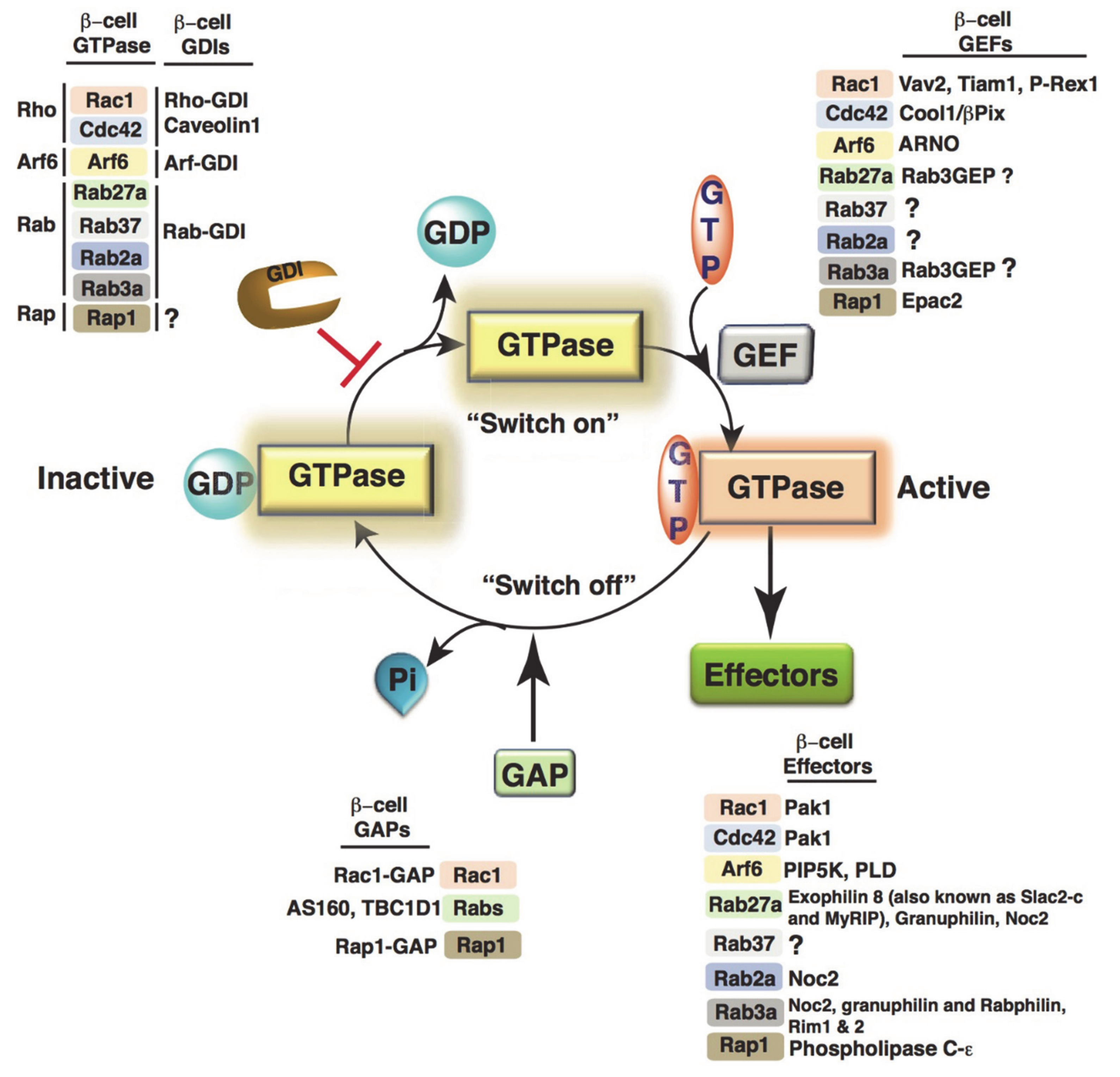
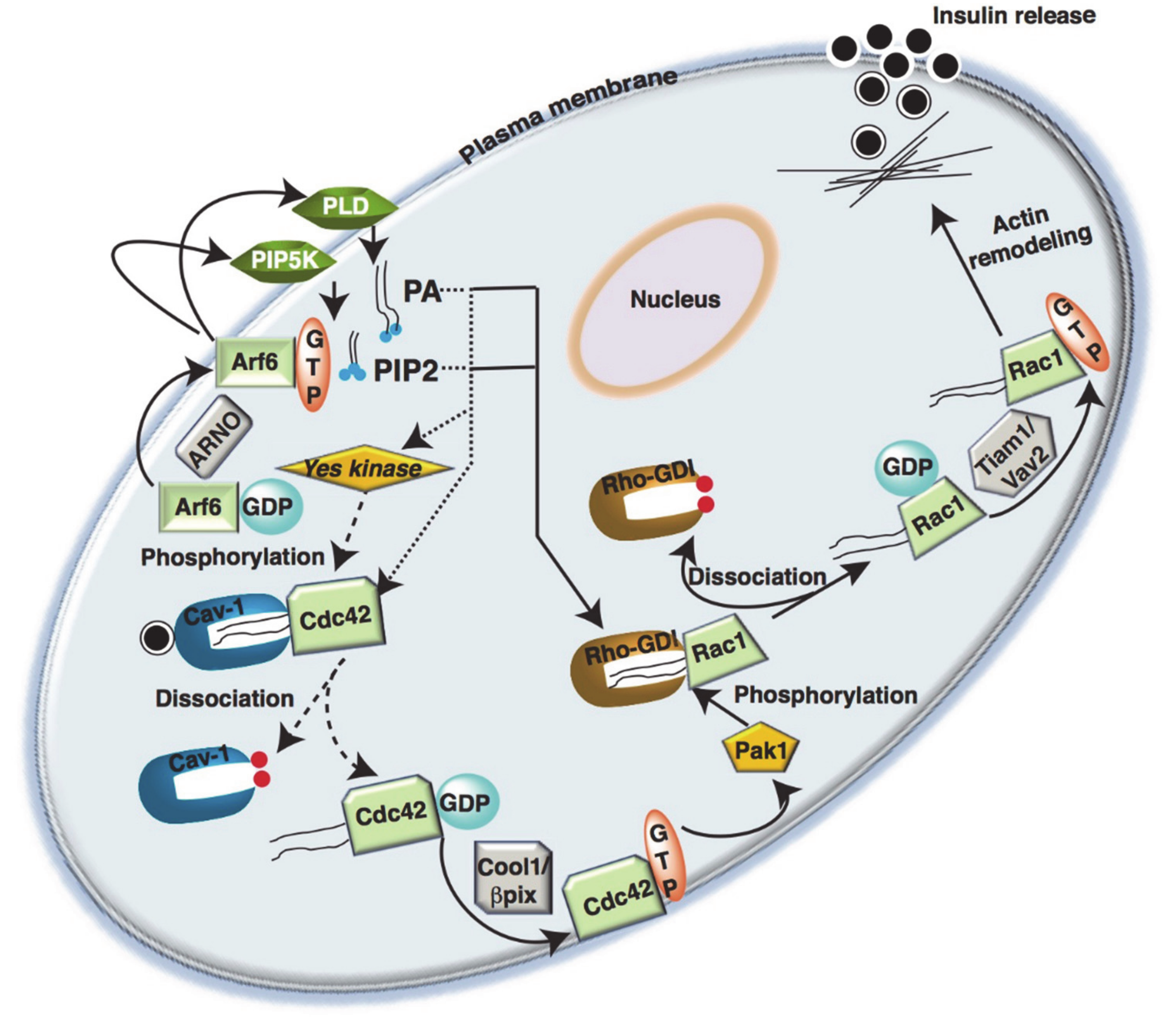
4.1. Small GTPase Regulation in β-Cells
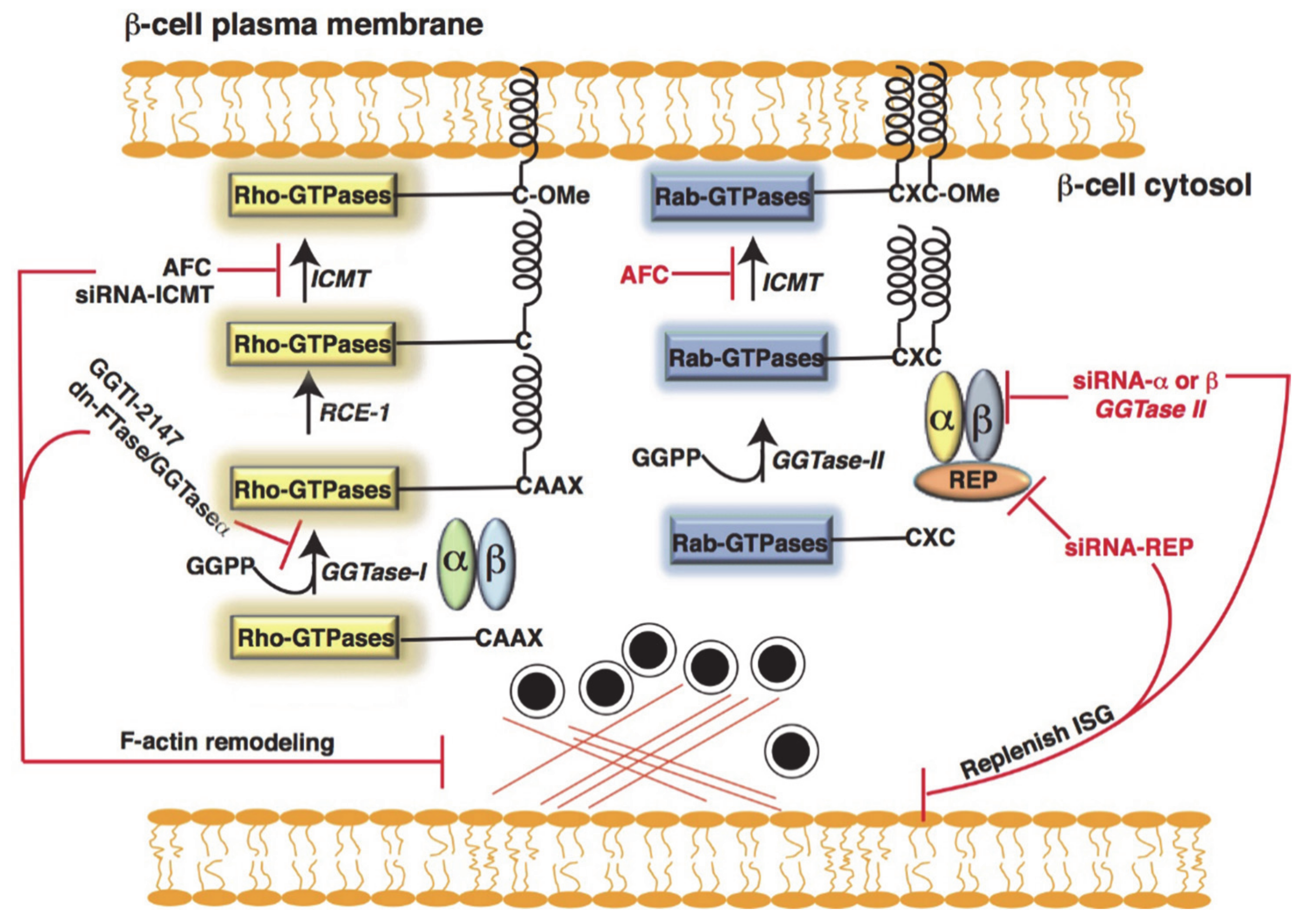
4.2. Post-Translational Modification of Small GTPases in β-Cells
Metabolic Dysfunction and Defective Post-Translational Modification of GTPase in Islet β-Cells
5. Rho-GTPases in Islet β-Cells
5.1. Cdc42
5.2. Rac1
5.3. Rho-GDI
Rac1 and Oxidative Stress in T2D Islet β-Cells
5.4. Arf
5.5. Rab-GTPases
Arf6 and Rab27a Couple Exocytosis and Endocytosis
5.6. Rap-GTPases
6. Identification of GTPase Regulating Proteins as T2D Candidate Genes
7. Conclusions
8. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cerasi, E. Mechanisms of glucose stimulated insulin secretion in health and in diabetes: Some re-evaluations and proposals. Diabetologia 1975, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Cerasi, E. Potentiation of insulin release by glucose in man. II. Role of the insulin response, and enhancement of stimuli other than glucose. Acta Endocrinol. 1975, 79, 502–510. [Google Scholar] [CrossRef]
- Chadt, A.; Al-Hasani, H. Glucose transporters in adipose tissue, liver, and skeletal muscle in metabolic health and disease. Pflug. Arch. Eur. J. Physiol. 2020, 472, 1273–1298. [Google Scholar] [CrossRef]
- Berger, C.; Zdzieblo, D. Glucose transporters in pancreatic islets. Pflug. Arch. Eur. J. Physiol. 2020, 472, 1249–1272. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, F.M.; Proks, P.; Smith, P.A.; Ammala, C.; Bokvist, K.; Rorsman, P. Stimulus-secretion coupling in pancreatic beta cells. J. Cell. Biochem. 1994, 55, 54–65. [Google Scholar] [CrossRef]
- Doliba, N.M.; Qin, W.; Najafi, H.; Liu, C.; Buettger, C.W.; Sotiris, J.; Collins, H.W.; Li, C.; Stanley, C.A.; Wilson, D.F.; et al. Glucokinase activation repairs defective bioenergetics of islets of Langerhans isolated from type 2 diabetics. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E87–E102. [Google Scholar] [CrossRef]
- Gembal, M.; Detimary, P.; Gilon, P.; Gao, Z.Y.; Henquin, J.C. Mechanisms by which glucose can control insulin release independently from its action on adenosine triphosphate-sensitive K+ channels in mouse B cells. J. Clin. Investig. 1993, 91, 871–880. [Google Scholar] [CrossRef]
- Matschinsky, F.M. Banting Lecture 1995. A lesson in metabolic regulation inspired by the glucokinase glucose sensor paradigm. Diabetes 1996, 45, 223–241. [Google Scholar] [CrossRef]
- Prentki, M.; Corkey, B.E.; Madiraju, S.R.M. Lipid-associated metabolic signalling networks in pancreatic beta cell function. Diabetologia 2020, 63, 10–20. [Google Scholar] [CrossRef]
- Rorsman, P.; Braun, M. Regulation of insulin secretion in human pancreatic islets. Annu. Rev. Physiol. 2013, 75, 155–179. [Google Scholar] [CrossRef]
- Seino, S.; Shibasaki, T.; Minami, K. Dynamics of insulin secretion and the clinical implications for obesity and diabetes. J. Clin. Investig. 2011, 121, 2118–2125. [Google Scholar] [CrossRef]
- Henquin, J.C.; Dufrane, D.; Gmyr, V.; Kerr-Conte, J.; Nenquin, M. Pharmacological approach to understanding the control of insulin secretion in human islets. Diabetes Obes. Metab. 2017, 19, 1061–1070. [Google Scholar] [CrossRef]
- Henquin, J.C.; Nenquin, M.; Stiernet, P.; Ahren, B. In vivo and in vitro glucose-induced biphasic insulin secretion in the mouse: Pattern and role of cytoplasmic Ca2+ and amplification signals in beta-cells. Diabetes 2006, 55, 441–451. [Google Scholar] [CrossRef]
- Omar-Hmeadi, M.; Idevall-Hagren, O. Insulin granule biogenesis and exocytosis. Cell. Mol. Life Sci. CMLS 2021, 78, 1957–1970. [Google Scholar] [CrossRef]
- Arous, C.; Halban, P.A. The skeleton in the closet: Actin cytoskeletal remodeling in beta-cell function. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E611–E620. [Google Scholar] [CrossRef]
- Sekine, N.; Cirulli, V.; Regazzi, R.; Brown, L.J.; Gine, E.; Tamarit-Rodriguez, J.; Girotti, M.; Marie, S.; MacDonald, M.J.; Wollheim, C.B.; et al. Low lactate dehydrogenase and high mitochondrial glycerol phosphate dehydrogenase in pancreatic beta-cells. Potential role in nutrient sensing. J. Biol. Chem. 1994, 269, 4895–4902. [Google Scholar] [CrossRef]
- McCommis, K.S.; Hodges, W.T.; Bricker, D.K.; Wisidagama, D.R.; Compan, V.; Remedi, M.S.; Thummel, C.S.; Finck, B.N. An ancestral role for the mitochondrial pyruvate carrier in glucose-stimulated insulin secretion. Mol. Metab. 2016, 5, 602–614. [Google Scholar] [CrossRef]
- Meredith, M.; Rabaglia, M.; Metz, S. Cytosolic biosynthesis of GTP and ATP in normal rat pancreatic islets. Biochim. Biophys. Acta 1995, 1266, 16–22. [Google Scholar] [CrossRef][Green Version]
- Meredith, M.; Rabaglia, M.E.; Metz, S.A. Evidence of a role for GTP in the potentiation of Ca(2+)-induced insulin secretion by glucose in intact rat islets. J. Clin. Investig. 1995, 96, 811–821. [Google Scholar] [CrossRef]
- Metz, S.A.; Meredith, M.; Rabaglia, M.E.; Kowluru, A. Small elevations of glucose concentration redirect and amplify the synthesis of guanosine 5’-triphosphate in rat islets. J. Clin. Investig. 1993, 92, 872–882. [Google Scholar] [CrossRef]
- Stark, R.; Pasquel, F.; Turcu, A.; Pongratz, R.L.; Roden, M.; Cline, G.W.; Shulman, G.I.; Kibbey, R.G. Phosphoenolpyruvate cycling via mitochondrial phosphoenolpyruvate carboxykinase links anaplerosis and mitochondrial GTP with insulin secretion. J. Biol. Chem. 2009, 284, 26578–26590. [Google Scholar] [CrossRef]
- Kibbey, R.G.; Pongratz, R.L.; Romanelli, A.J.; Wollheim, C.B.; Cline, G.W.; Shulman, G.I. Mitochondrial GTP regulates glucose-stimulated insulin secretion. Cell Metab. 2007, 5, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Jesinkey, S.R.; Madiraju, A.K.; Alves, T.C.; Yarborough, O.H.; Cardone, R.L.; Zhao, X.; Parsaei, Y.; Nasiri, A.R.; Butrico, G.; Liu, X.; et al. Mitochondrial GTP Links Nutrient Sensing to beta Cell Health, Mitochondrial Morphology, and Insulin Secretion Independent of OxPhos. Cell Rep. 2019, 28, 759–772.e10. [Google Scholar] [CrossRef] [PubMed]
- Metz, S.A.; Rabaglia, M.E.; Pintar, T.J. Selective inhibitors of GTP synthesis impede exocytotic insulin release from intact rat islets. J. Biol. Chem. 1992, 267, 12517–12527. [Google Scholar] [CrossRef]
- Ottaway, J.H.; McClellan, J.A.; Saunderson, C.L. Succinic thiokinase and metabolic control. Int. J. Biochem. 1981, 13, 401–410. [Google Scholar] [CrossRef]
- Smith, C.M.; Bryla, J.; Williamson, J.R. Regulation of mitochondrial alpha-ketoglutarate metabolism by product inhibition at alpha-ketoglutarate dehydrogenase. J. Biol. Chem. 1974, 249, 1497–1505. [Google Scholar] [CrossRef]
- Drahota, Z.; Rauchova, H.; Mikova, M.; Kaul, P.; Bass, A. Phosphoenolpyruvate shuttle—Transport of energy from mitochondria to cytosol. FEBS Lett. 1983, 157, 347–349. [Google Scholar] [CrossRef]
- van der Meulen, T.; Mawla, A.M.; DiGruccio, M.R.; Adams, M.W.; Nies, V.; Dolleman, S.; Liu, S.; Ackermann, A.M.; Caceres, E.; Hunter, A.E.; et al. Virgin Beta Cells Persist throughout Life at a Neogenic Niche within Pancreatic Islets. Cell Metab. 2017, 25, 911–926.e6. [Google Scholar] [CrossRef]
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9(th) edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef]
- Holman, R.R.; Clark, A.; Rorsman, P. beta-cell secretory dysfunction: A key cause of type 2 diabetes. Lancet. Diabetes Endocrinol. 2020, 8, 370. [Google Scholar] [CrossRef]
- Hudish, L.I.; Reusch, J.E.; Sussel, L. beta Cell dysfunction during progression of metabolic syndrome to type 2 diabetes. J. Clin. Investig. 2019, 129, 4001–4008. [Google Scholar] [CrossRef]
- Magkos, F.; Hjorth, M.F.; Astrup, A. Diet and exercise in the prevention and treatment of type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2020, 16, 545–555. [Google Scholar] [CrossRef]
- Krentz, N.A.J.; Gloyn, A.L. Insights into pancreatic islet cell dysfunction from type 2 diabetes mellitus genetics. Nat. Rev. Endocrinol. 2020, 16, 202–212. [Google Scholar] [CrossRef]
- Ashcroft, F.M.; Rorsman, P. Diabetes mellitus and the beta cell: The last ten years. Cell 2012, 148, 1160–1171. [Google Scholar] [CrossRef]
- Bray, G.A.; Heisel, W.E.; Afshin, A.; Jensen, M.D.; Dietz, W.H.; Long, M.; Kushner, R.F.; Daniels, S.R.; Wadden, T.A.; Tsai, A.G.; et al. The Science of Obesity Management: An Endocrine Society Scientific Statement. Endocr. Rev. 2018, 39, 79–132. [Google Scholar] [CrossRef]
- Salunkhe, V.A.; Veluthakal, R.; Kahn, S.E.; Thurmond, D.C. Novel approaches to restore beta cell function in prediabetes and type 2 diabetes. Diabetologia 2018, 61, 1895–1901. [Google Scholar] [CrossRef]
- Gandasi, N.R.; Yin, P.; Omar-Hmeadi, M.; Ottosson Laakso, E.; Vikman, P.; Barg, S. Glucose-Dependent Granule Docking Limits Insulin Secretion and Is Decreased in Human Type 2 Diabetes. Cell Metab. 2018, 27, 470–478.e4. [Google Scholar] [CrossRef]
- Katsarou, A.; Gudbjornsdottir, S.; Rawshani, A.; Dabelea, D.; Bonifacio, E.; Anderson, B.J.; Jacobsen, L.M.; Schatz, D.A.; Lernmark, A. Type 1 diabetes mellitus. Nat. Rev. Dis. Primers 2017, 3, 17016. [Google Scholar] [CrossRef]
- Rojas, A.M.; Fuentes, G.; Rausell, A.; Valencia, A. The Ras protein superfamily: Evolutionary tree and role of conserved amino acids. J. Cell Biol. 2012, 196, 189–201. [Google Scholar] [CrossRef]
- Liu, W.N.; Yan, M.; Chan, A.M. A thirty-year quest for a role of R-Ras in cancer: From an oncogene to a multitasking GTPase. Cancer Lett. 2017, 403, 59–65. [Google Scholar] [CrossRef]
- Qu, L.; Pan, C.; He, S.M.; Lang, B.; Gao, G.D.; Wang, X.L.; Wang, Y. The Ras Superfamily of Small GTPases in Non-neoplastic Cerebral Diseases. Front. Mol. Neurosci. 2019, 12, 121. [Google Scholar] [CrossRef] [PubMed]
- Vetter, I.R.; Wittinghofer, A. The guanine nucleotide-binding switch in three dimensions. Science 2001, 294, 1299–1304. [Google Scholar] [CrossRef] [PubMed]
- Bourne, H.R.; Sanders, D.A.; McCormick, F. The GTPase superfamily: Conserved structure and molecular mechanism. Nature 1991, 349, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Wennerberg, K.; Rossman, K.L.; Der, C.J. The Ras superfamily at a glance. J. Cell Sci. 2005, 118, 843–846. [Google Scholar] [CrossRef]
- Karnoub, A.E.; Weinberg, R.A. Ras oncogenes: Split personalities. Nat. Rev. Mol. Cell Biol. 2008, 9, 517–531. [Google Scholar] [CrossRef]
- Toma-Fukai, S.; Shimizu, T. Structural Insights into the Regulation Mechanism of Small GTPases by GEFs. Molecules 2019, 24, 3308. [Google Scholar] [CrossRef]
- Nevins, A.K.; Thurmond, D.C. Glucose regulates the cortical actin network through modulation of Cdc42 cycling to stimulate insulin secretion. Am. J. Physiol. Cell Physiol. 2003, 285, C698–C710. [Google Scholar] [CrossRef]
- Nevins, A.K.; Thurmond, D.C. A direct interaction between Cdc42 and vesicle-associated membrane protein 2 regulates SNARE-dependent insulin exocytosis. J. Biol. Chem. 2005, 280, 1944–1952. [Google Scholar] [CrossRef]
- Nevins, A.K.; Thurmond, D.C. Caveolin-1 functions as a novel Cdc42 guanine nucleotide dissociation inhibitor in pancreatic beta-cells. J. Biol. Chem. 2006, 281, 18961–18972. [Google Scholar] [CrossRef]
- Regazzi, R.; Kikuchi, A.; Takai, Y.; Wollheim, C.B. The small GTP-binding proteins in the cytosol of insulin-secreting cells are complexed to GDP dissociation inhibitor proteins. J. Biol. Chem. 1992, 267, 17512–17519. [Google Scholar] [CrossRef]
- Bravo-Nuevo, A.; Sugimoto, H.; Iyer, S.; Fallon, Z.; Lucas, J.M.; Kazerounian, S.; Prendergast, G.C.; Kalluri, R.; Shapiro, N.I.; Benjamin, L.E. RhoB loss prevents streptozotocin-induced diabetes and ameliorates diabetic complications in mice. Am. J. Pathol. 2011, 178, 245–252. [Google Scholar] [CrossRef]
- Liu, X.; Yan, F.; Yao, H.; Chang, M.; Qin, J.; Li, Y.; Wang, Y.; Pei, X. Involvement of RhoA/ROCK in insulin secretion of pancreatic beta-cells in 3D culture. Cell Tissue Res. 2014, 358, 359–369. [Google Scholar] [CrossRef]
- Kowluru, A.; Li, G.; Rabaglia, M.E.; Segu, V.B.; Hofmann, F.; Aktories, K.; Metz, S.A. Evidence for differential roles of the Rho subfamily of GTP-binding proteins in glucose- and calcium-induced insulin secretion from pancreatic beta cells. Biochem. Pharmacol. 1997, 54, 1097–1108. [Google Scholar] [CrossRef]
- Li, J.; Luo, R.; Kowluru, A.; Li, G. Novel regulation by Rac1 of glucose- and forskolin-induced insulin secretion in INS-1 beta-cells. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E818–E827. [Google Scholar] [CrossRef]
- Kowluru, A.; Rabaglia, M.E.; Muse, K.E.; Metz, S.A. Subcellular localization and kinetic characterization of guanine nucleotide binding proteins in normal rat and human pancreatic islets and transformed beta cells. Biochim. Biophys. Acta 1994, 1222, 348–359. [Google Scholar] [CrossRef]
- Daniel, S.; Noda, M.; Cerione, R.A.; Sharp, G.W. A link between Cdc42 and syntaxin is involved in mastoparan-stimulated insulin release. Biochemistry 2002, 41, 9663–9671. [Google Scholar] [CrossRef]
- Kowluru, A.; Chen, H.Q.; Tannous, M. Novel roles for the rho subfamily of GTP-binding proteins in succinate-induced insulin secretion from betaTC3 cells: Further evidence in support of the succinate mechanism of insulin release. Endocr. Res. 2003, 29, 363–376. [Google Scholar] [CrossRef]
- Pereira-Leal, J.B.; Seabra, M.C. Evolution of the Rab family of small GTP-binding proteins. J. Mol. Biol. 2001, 313, 889–901. [Google Scholar] [CrossRef]
- Segev, N. Ypt and Rab GTPases: Insight into functions through novel interactions. Curr. Opin. Cell Biol. 2001, 13, 500–511. [Google Scholar] [CrossRef]
- Jackson, C.L.; Bouvet, S. Arfs at a glance. J. Cell Sci. 2014, 127, 4103–4109. [Google Scholar] [CrossRef]
- D’Souza-Schorey, C.; Chavrier, P. ARF proteins: Roles in membrane traffic and beyond. Nat. Rev. Mol. Cell Biol. 2006, 7, 347–358. [Google Scholar] [CrossRef]
- Benarroch, E.E. Nucleocytoplasmic transport: Mechanisms and involvement in neurodegenerative disease. Neurology 2019, 92, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Veluthakal, R.; Kaur, H.; Goalstone, M.; Kowluru, A. Dominant-negative alpha-subunit of farnesyl- and geranyltransferase inhibits glucose-stimulated, but not KCl-stimulated, insulin secretion in INS 832/13 cells. Diabetes 2007, 56, 204–210. [Google Scholar] [CrossRef]
- Veluthakal, R.; Madathilparambil, S.V.; McDonald, P.; Olson, L.K.; Kowluru, A. Regulatory roles for Tiam1, a guanine nucleotide exchange factor for Rac1, in glucose-stimulated insulin secretion in pancreatic beta-cells. Biochem. Pharmacol. 2009, 77, 101–113. [Google Scholar] [CrossRef]
- Wang, Z.; Oh, E.; Thurmond, D.C. Glucose-stimulated Cdc42 signaling is essential for the second phase of insulin secretion. J. Biol. Chem. 2007, 282, 9536–9546. [Google Scholar] [CrossRef]
- Kowluru, A.; Seavey, S.E.; Li, G.; Sorenson, R.L.; Weinhaus, A.J.; Nesher, R.; Rabaglia, M.E.; Vadakekalam, J.; Metz, S.A. Glucose- and GTP-dependent stimulation of the carboxyl methylation of CDC42 in rodent and human pancreatic islets and pure beta cells. Evidence for an essential role of GTP-binding proteins in nutrient-induced insulin secretion. J. Clin. Investig. 1996, 98, 540–555. [Google Scholar] [CrossRef] [PubMed]
- Yi, Z.; Yokota, H.; Torii, S.; Aoki, T.; Hosaka, M.; Zhao, S.; Takata, K.; Takeuchi, T.; Izumi, T. The Rab27a/granuphilin complex regulates the exocytosis of insulin-containing dense-core granules. Mol. Cell Biol. 2002, 22, 1858–1867. [Google Scholar] [CrossRef]
- Ljubicic, S.; Bezzi, P.; Brajkovic, S.; Nesca, V.; Guay, C.; Ohbayashi, N.; Fukuda, M.; Abderrhamani, A.; Regazzi, R. The GTPase Rab37 Participates in the Control of Insulin Exocytosis. PLoS ONE 2013, 8, e68255. [Google Scholar] [CrossRef]
- Matsunaga, K.; Taoka, M.; Isobe, T.; Izumi, T. Rab2a and Rab27a cooperatively regulate the transition from granule maturation to exocytosis through the dual effector Noc2. J. Cell Sci. 2017, 130, 541–550. [Google Scholar] [CrossRef]
- Regazzi, R.; Vallar, L.; Ullrich, S.; Ravazzola, M.; Kikuchi, A.; Takai, Y.; Wollheim, C.B. Characterization of small-molecular-mass guanine-nucleotide-binding regulatory proteins in insulin-secreting cells and PC12 cells. Eur. J. Biochem. 1992, 208, 729–737. [Google Scholar] [CrossRef] [PubMed]
- Shibasaki, T.; Takahashi, H.; Miki, T.; Sunaga, Y.; Matsumura, K.; Yamanaka, M.; Zhang, C.; Tamamoto, A.; Satoh, T.; Miyazaki, J.; et al. Essential role of Epac2/Rap1 signaling in regulation of insulin granule dynamics by cAMP. Proc. Natl. Acad. Sci. USA 2007, 104, 19333–19338. [Google Scholar] [CrossRef]
- Schmidt, A.; Hall, A. Guanine nucleotide exchange factors for Rho GTPases: Turning on the switch. Genes Dev. 2002, 16, 1587–1609. [Google Scholar] [CrossRef]
- Bernards, A.; Settleman, J. GAP control: Regulating the regulators of small GTPases. Trends Cell Biol. 2004, 14, 377–385. [Google Scholar] [CrossRef]
- Bishop, A.L.; Hall, A. Rho GTPases and their effector proteins. Biochem. J. 2000, 348, 241–255. [Google Scholar] [CrossRef]
- Repasky, G.A.; Chenette, E.J.; Der, C.J. Renewing the conspiracy theory debate: Does Raf function alone to mediate Ras oncogenesis? Trends Cell Biol. 2004, 14, 639–647. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Karamouzis, M.V.; Papavassiliou, A.G. Post-translational modifications and regulation of the RAS superfamily of GTPases as anticancer targets. Nat. Rev. Drug Discov. 2007, 6, 541–555. [Google Scholar] [CrossRef]
- Wang, M.; Casey, P.J. Protein prenylation: Unique fats make their mark on biology. Nat. Rev. Mol. Cell Biol. 2016, 17, 110–122. [Google Scholar] [CrossRef]
- Seabra, M.C.; Reiss, Y.; Casey, P.J.; Brown, M.S.; Goldstein, J.L. Protein farnesyltransferase and geranylgeranyltransferase share a common alpha subunit. Cell 1991, 65, 429–434. [Google Scholar] [CrossRef]
- Reid, T.S.; Terry, K.L.; Casey, P.J.; Beese, L.S. Crystallographic analysis of CaaX prenyltransferases complexed with substrates defines rules of protein substrate selectivity. J. Mol. Biol. 2004, 343, 417–433. [Google Scholar] [CrossRef]
- Goalstone, M.; Kamath, V.; Kowluru, A. Glucose activates prenyltransferases in pancreatic islet beta-cells. Biochem. Biophys. Res. Commun. 2010, 391, 895–898. [Google Scholar] [CrossRef]
- Seabra, M.C.; Goldstein, J.L.; Sudhof, T.C.; Brown, M.S. Rab geranylgeranyl transferase. A multisubunit enzyme that prenylates GTP-binding proteins terminating in Cys-X-Cys or Cys-Cys. J. Biol. Chem. 1992, 267, 14497–14503. [Google Scholar] [CrossRef]
- Arora, D.K.; Syed, I.; Machhadieh, B.; McKenna, C.E.; Kowluru, A. Rab-geranylgeranyl transferase regulates glucose-stimulated insulin secretion from pancreatic beta cells. Islets 2012, 4, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.W.; Tan, K.T.; Waldmann, H.; Goody, R.S.; Alexandrov, K. Interaction analysis of prenylated Rab GTPase with Rab escort protein and GDP dissociation inhibitor explains the need for both regulators. Proc. Natl. Acad. Sci. USA 2007, 104, 12294–12299. [Google Scholar] [CrossRef]
- Shinde, S.R.; Maddika, S. Post translational modifications of Rab GTPases. Small GTPases 2018, 9, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Metz, S.A.; Rabaglia, M.E.; Stock, J.B.; Kowluru, A. Modulation of insulin secretion from normal rat islets by inhibitors of the post-translational modifications of GTP-binding proteins. Biochem. J. 1993, 295, 31–40. [Google Scholar] [CrossRef]
- Li, G.; Regazzi, R.; Roche, E.; Wollheim, C.B. Blockade of mevalonate production by lovastatin attenuates bombesin and vasopressin potentiation of nutrient-induced insulin secretion in HIT-T15 cells. Probable involvement of small GTP-binding proteins. Biochem. J. 1993, 289, 379–385. [Google Scholar] [CrossRef]
- Amin, R.; Chen, H.Q.; Tannous, M.; Gibbs, R.; Kowluru, A. Inhibition of glucose- and calcium-induced insulin secretion from betaTC3 cells by novel inhibitors of protein isoprenylation. J. Pharm. Exp. 2002, 303, 82–88. [Google Scholar] [CrossRef]
- Eisenberg, D.A. Cholesterol lowering in the management of coronary artery disease: The clinical implications of recent trials. Am. J. Med. 1998, 104, 2S–5S. [Google Scholar] [CrossRef]
- Cederberg, H.; Stancakova, A.; Yaluri, N.; Modi, S.; Kuusisto, J.; Laakso, M. Increased risk of diabetes with statin treatment is associated with impaired insulin sensitivity and insulin secretion: A 6 year follow-up study of the METSIM cohort. Diabetologia 2015, 58, 1109–1117. [Google Scholar] [CrossRef]
- Sattar, N.; Preiss, D.; Murray, H.M.; Welsh, P.; Buckley, B.M.; de Craen, A.J.; Seshasai, S.R.; McMurray, J.J.; Freeman, D.J.; Jukema, J.W.; et al. Statins and risk of incident diabetes: A collaborative meta-analysis of randomised statin trials. Lancet 2010, 375, 735–742. [Google Scholar] [CrossRef]
- Preiss, D.; Seshasai, S.R.; Welsh, P.; Murphy, S.A.; Ho, J.E.; Waters, D.D.; DeMicco, D.A.; Barter, P.; Cannon, C.P.; Sabatine, M.S.; et al. Risk of incident diabetes with intensive-dose compared with moderate-dose statin therapy: A meta-analysis. JAMA 2011, 305, 2556–2564. [Google Scholar] [CrossRef]
- Rajpathak, S.N.; Kumbhani, D.J.; Crandall, J.; Barzilai, N.; Alderman, M.; Ridker, P.M. Statin therapy and risk of developing type 2 diabetes: A meta-analysis. Diabetes Care 2009, 32, 1924–1929. [Google Scholar] [CrossRef]
- Ridker, P.M.; Danielson, E.; Fonseca, F.A.; Genest, J.; Gotto, A.M., Jr.; Kastelein, J.J.; Koenig, W.; Libby, P.; Lorenzatti, A.J.; MacFadyen, J.G.; et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N. Engl. J. Med. 2008, 359, 2195–2207. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, J.; Blauw, G.J.; Murphy, M.B.; Bollen, E.L.; Buckley, B.M.; Cobbe, S.M.; Ford, I.; Gaw, A.; Hyland, M.; Jukema, J.W.; et al. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): A randomised controlled trial. Lancet 2002, 360, 1623–1630. [Google Scholar] [CrossRef]
- Crandall, J.P.; Mather, K.; Rajpathak, S.N.; Goldberg, R.B.; Watson, K.; Foo, S.; Ratner, R.; Barrett-Connor, E.; Temprosa, M. Statin use and risk of developing diabetes: Results from the Diabetes Prevention Program. BMJ. Open Diabetes Res. Care 2017, 5, e000438. [Google Scholar] [CrossRef]
- Kowluru, A. Small G proteins in islet beta-cell function. Endocr. Rev. 2010, 31, 52–78. [Google Scholar] [CrossRef]
- Kowluru, A. A lack of ‘glue’ misplaces Rab27A to cause islet dysfunction in diabetes. J. Pathol. 2016, 238, 375–377. [Google Scholar] [CrossRef]
- Jiang, S.; Shen, D.; Jia, W.J.; Han, X.; Shen, N.; Tao, W.; Gao, X.; Xue, B.; Li, C.J. GGPPS-mediated Rab27A geranylgeranylation regulates beta cell dysfunction during type 2 diabetes development by affecting insulin granule docked pool formation. J. Pathol. 2016, 238, 109–119. [Google Scholar] [CrossRef]
- Wang, Z.; Thurmond, D.C. Differential phosphorylation of RhoGDI mediates the distinct cycling of Cdc42 and Rac1 to regulate second-phase insulin secretion. J. Biol. Chem. 2010, 285, 6186–6197. [Google Scholar] [CrossRef]
- Swanston-Flatt, S.K.; Carlsson, L.; Gylfe, E. Actin filament formation in pancreatic beta-cells during glucose stimulation of insulin secretion. FEBS Lett. 1980, 117, 299–302. [Google Scholar] [CrossRef]
- Varadi, A.; Tsuboi, T.; Rutter, G.A. Myosin Va transports dense core secretory vesicles in pancreatic MIN6 beta-cells. Mol. Biol. Cell 2005, 16, 2670–2680. [Google Scholar] [CrossRef] [PubMed]
- Orci, L.; Gabbay, K.H.; Malaisse, W.J. Pancreatic beta-cell web: Its possible role in insulin secretion. Science 1972, 175, 1128–1130. [Google Scholar] [CrossRef] [PubMed]
- Kepner, E.M.; Yoder, S.M.; Oh, E.; Kalwat, M.A.; Wang, Z.; Quilliam, L.A.; Thurmond, D.C. Cool-1/betaPIX functions as a guanine nucleotide exchange factor in the cycling of Cdc42 to regulate insulin secretion. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E1072–E1080. [Google Scholar] [CrossRef] [PubMed]
- Wehinger, S.; Ortiz, R.; Diaz, M.I.; Aguirre, A.; Valenzuela, M.; Llanos, P.; Mc Master, C.; Leyton, L.; Quest, A.F. Phosphorylation of caveolin-1 on tyrosine-14 induced by ROS enhances palmitate-induced death of beta-pancreatic cells. Biochim. Biophys. Acta 2015, 1852, 693–708. [Google Scholar] [CrossRef][Green Version]
- Zeng, W.; Tang, J.; Li, H.; Xu, H.; Lu, H.; Peng, H.; Lin, C.; Gao, R.; Lin, S.; Lin, K.; et al. Caveolin-1 deficiency protects pancreatic beta cells against palmitate-induced dysfunction and apoptosis. Cell Signal. 2018, 47, 65–78. [Google Scholar] [CrossRef]
- Lillo Urzua, P.; Nunez Murillo, O.; Castro-Sepulveda, M.; Torres-Quintana, M.A.; Lladser Caldera, A.; Quest, A.F.G.; Espinoza Robles, C.; Llanos Vidal, P.; Wehinger, S. Loss of Caveolin-1 Is Associated with a Decrease in Beta Cell Death in Mice on a High Fat Diet. Int. J. Mol. Sci. 2020, 21, 5225. [Google Scholar] [CrossRef]
- He, X.Q.; Wang, N.; Zhao, J.J.; Wang, D.; Wang, C.J.; Xie, L.; Zheng, H.Y.; Shi, S.Z.; He, J.; Zhou, J.; et al. Specific deletion of CDC42 in pancreatic beta cells attenuates glucose-induced insulin expression and secretion in mice. Mol. Cell Endocrinol. 2020, 518, 111004. [Google Scholar] [CrossRef]
- Duan, J.; Qian, X.L.; Li, J.; Xiao, X.H.; Lu, X.T.; Lv, L.C.; Huang, Q.Y.; Ding, W.; Zhang, H.Y.; Xiong, L.X. miR-29a Negatively Affects Glucose-Stimulated Insulin Secretion and MIN6 Cell Proliferation via Cdc42/beta-Catenin Signaling. Int. J. Endocrinol. 2019, 2019, 5219782. [Google Scholar] [CrossRef]
- Sun, Y.; Zhou, Y.; Shi, Y.; Zhang, Y.; Liu, K.; Liang, R.; Sun, P.; Chang, X.; Tang, W.; Zhang, Y.; et al. Expression of miRNA-29 in Pancreatic beta Cells Promotes Inflammation and Diabetes via TRAF3. Cell Rep. 2021, 34, 108576. [Google Scholar] [CrossRef]
- Ursino, G.M.; Fu, Y.; Cottle, D.L.; Mukhamedova, N.; Jones, L.K.; Low, H.; Tham, M.S.; Gan, W.J.; Mellett, N.A.; Das, P.P.; et al. ABCA12 regulates insulin secretion from beta-cells. EMBO Rep. 2020, 21, e48692. [Google Scholar] [CrossRef]
- Veluthakal, R.; Chepurny, O.G.; Leech, C.A.; Schwede, F.; Holz, G.G.; Thurmond, D.C. Restoration of Glucose-Stimulated Cdc42-Pak1 Activation and Insulin Secretion by a Selective Epac Activator in Type 2 Diabetic Human Islets. Diabetes 2018, 67, 1999–2011. [Google Scholar] [CrossRef]
- Asahara, S.; Shibutani, Y.; Teruyama, K.; Inoue, H.Y.; Kawada, Y.; Etoh, H.; Matsuda, T.; Kimura-Koyanagi, M.; Hashimoto, N.; Sakahara, M.; et al. Ras-related C3 botulinum toxin substrate 1 (RAC1) regulates glucose-stimulated insulin secretion via modulation of F-actin. Diabetologia 2013, 56, 1088–1097. [Google Scholar] [CrossRef]
- Greiner, T.U.; Kesavan, G.; Stahlberg, A.; Semb, H. Rac1 regulates pancreatic islet morphogenesis. BMC Dev. Biol. 2009, 9, 2. [Google Scholar] [CrossRef]
- Veluthakal, R.; Tunduguru, R.; Arora, D.K.; Sidarala, V.; Syeda, K.; Vlaar, C.P.; Thurmond, D.C.; Kowluru, A. VAV2, a guanine nucleotide exchange factor for Rac1, regulates glucose-stimulated insulin secretion in pancreatic beta cells. Diabetologia 2015, 58, 2573–2581. [Google Scholar] [CrossRef]
- Gao, Y.; Dickerson, J.B.; Guo, F.; Zheng, J.; Zheng, Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc. Natl. Acad. Sci. USA 2004, 101, 7618–7623. [Google Scholar] [CrossRef]
- Crespo, P.; Schuebel, K.E.; Ostrom, A.A.; Gutkind, J.S.; Bustelo, X.R. Phosphotyrosine-dependent activation of Rac-1 GDP/GTP exchange by the vav proto-oncogene product. Nature 1997, 385, 169–172. [Google Scholar] [CrossRef]
- Schuebel, K.E.; Movilla, N.; Rosa, J.L.; Bustelo, X.R. Phosphorylation-dependent and constitutive activation of Rho proteins by wild-type and oncogenic Vav-2. EMBO J. 1998, 17, 6608–6621. [Google Scholar] [CrossRef]
- Han, J.; Das, B.; Wei, W.; Van Aelst, L.; Mosteller, R.D.; Khosravi-Far, R.; Westwick, J.K.; Der, C.J.; Broek, D. Lck regulates Vav activation of members of the Rho family of GTPases. Mol. Cell Biol. 1997, 17, 1346–1353. [Google Scholar] [CrossRef]
- Michel, F.; Grimaud, L.; Tuosto, L.; Acuto, O. Fyn and ZAP-70 are required for Vav phosphorylation in T cells stimulated by antigen-presenting cells. J. Biol. Chem. 1998, 273, 31932–31938. [Google Scholar] [CrossRef]
- Deckert, M.; Tartare-Deckert, S.; Couture, C.; Mustelin, T.; Altman, A. Functional and physical interactions of Syk family kinases with the Vav proto-oncogene product. Immunity 1996, 5, 591–604. [Google Scholar] [CrossRef]
- Bustelo, X.R.; Barbacid, M. Tyrosine phosphorylation of the vav proto-oncogene product in activated B cells. Science 1992, 256, 1196–1199. [Google Scholar] [CrossRef]
- Bustelo, X.R.; Ledbetter, J.A.; Barbacid, M. Product of vav proto-oncogene defines a new class of tyrosine protein kinase substrates. Nature 1992, 356, 68–71. [Google Scholar] [CrossRef] [PubMed]
- Margolis, B.; Hu, P.; Katzav, S.; Li, W.; Oliver, J.M.; Ullrich, A.; Weiss, A.; Schlessinger, J. Tyrosine phosphorylation of vav proto-oncogene product containing SH2 domain and transcription factor motifs. Nature 1992, 356, 71–74. [Google Scholar] [CrossRef]
- Yoder, S.M.; Dineen, S.L.; Wang, Z.; Thurmond, D.C. YES, a Src family kinase, is a proximal glucose-specific activator of cell division cycle control protein 42 (Cdc42) in pancreatic islet beta cells. J. Biol. Chem. 2014, 289, 11476–11487. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, A.; Veluthakal, R. Rho guanosine diphosphate-dissociation inhibitor plays a negative modulatory role in glucose-stimulated insulin secretion. Diabetes 2005, 54, 3523–3529. [Google Scholar] [CrossRef]
- Thamilselvan, V.; Kowluru, A. Paradoxical regulation of glucose-induced Rac1 activation and insulin secretion by RhoGDIbeta in pancreatic beta-cells. Small GTPases 2021, 12, 114–121. [Google Scholar] [CrossRef]
- Hodge, R.G.; Ridley, A.J. Regulating Rho GTPases and their regulators. Nat. Rev. Mol. Cell Biol. 2016, 17, 496–510. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Mata, R.; Boulter, E.; Burridge, K. The ’invisible hand’: Regulation of RHO GTPases by RHOGDIs. Nat. Rev. Mol. Cell Biol. 2011, 12, 493–504. [Google Scholar] [CrossRef] [PubMed]
- DerMardirossian, C.; Rocklin, G.; Seo, J.Y.; Bokoch, G.M. Phosphorylation of RhoGDI by Src regulates Rho GTPase binding and cytosol-membrane cycling. Mol. Biol. Cell 2006, 17, 4760–4768. [Google Scholar] [CrossRef]
- Dovas, A.; Choi, Y.; Yoneda, A.; Multhaupt, H.A.; Kwon, S.H.; Kang, D.; Oh, E.S.; Couchman, J.R. Serine 34 phosphorylation of rho guanine dissociation inhibitor (RhoGDIalpha) links signaling from conventional protein kinase C to RhoGTPase in cell adhesion. J. Biol. Chem. 2010, 285, 23296–23308. [Google Scholar] [CrossRef] [PubMed]
- Sabbatini, M.E.; Williams, J.A. Cholecystokinin-mediated RhoGDI phosphorylation via PKCalpha promotes both RhoA and Rac1 signaling. PLoS ONE 2013, 8, e66029. [Google Scholar] [CrossRef]
- DerMardirossian, C.M.; Bokoch, G.M. Phosphorylation of RhoGDI by p21-activated kinase 1. Methods Enzym. 2006, 406, 80–90. [Google Scholar] [CrossRef]
- Fei, F.; Kweon, S.M.; Haataja, L.; De Sepulveda, P.; Groffen, J.; Heisterkamp, N. The Fer tyrosine kinase regulates interactions of Rho GDP-Dissociation Inhibitor alpha with the small GTPase Rac. BMC Biochem. 2010, 11, 48. [Google Scholar] [CrossRef]
- Oishi, A.; Makita, N.; Sato, J.; Iiri, T. Regulation of RhoA signaling by the cAMP-dependent phosphorylation of RhoGDIalpha. J. Biol. Chem. 2012, 287, 38705–38715. [Google Scholar] [CrossRef]
- Tkachenko, E.; Sabouri-Ghomi, M.; Pertz, O.; Kim, C.; Gutierrez, E.; Machacek, M.; Groisman, A.; Danuser, G.; Ginsberg, M.H. Protein kinase A governs a RhoA-RhoGDI protrusion-retraction pacemaker in migrating cells. Nat. Cell Biol. 2011, 13, 660–667. [Google Scholar] [CrossRef]
- Cho, H.J.; Kim, J.T.; Lee, S.J.; Hwang, Y.S.; Park, S.Y.; Kim, B.Y.; Yoo, J.; Hong, K.S.; Min, J.K.; Lee, C.H.; et al. Protein phosphatase 1B dephosphorylates Rho guanine nucleotide dissociation inhibitor 1 and suppresses cancer cell migration and invasion. Cancer Lett. 2018, 417, 141–151. [Google Scholar] [CrossRef]
- Cho, H.J.; Kim, J.T.; Baek, K.E.; Kim, B.Y.; Lee, H.G. Regulation of Rho GTPases by RhoGDIs in Human Cancers. Cells 2019, 8, 1037. [Google Scholar] [CrossRef]
- Oude Weernink, P.A.; Lopez de Jesus, M.; Schmidt, M. Phospholipase D signaling: Orchestration by PIP2 and small GTPases. Naunyn Schmiedebergs Arch. Pharm. 2007, 374, 399–411. [Google Scholar] [CrossRef]
- Chuang, T.H.; Bohl, B.P.; Bokoch, G.M. Biologically active lipids are regulators of Rac.GDI complexation. J. Biol. Chem. 1993, 268, 26206–26211. [Google Scholar] [CrossRef]
- Faure, J.; Vignais, P.V.; Dagher, M.C. Phosphoinositide-dependent activation of Rho A involves partial opening of the RhoA/Rho-GDI complex. Eur. J. Biochem. 1999, 262, 879–889. [Google Scholar] [CrossRef]
- Fleming, I.N.; Elliott, C.M.; Collard, J.G.; Exton, J.H. Lysophosphatidic acid induces threonine phosphorylation of Tiam1 in Swiss 3T3 fibroblasts via activation of protein kinase C. J. Biol. Chem. 1997, 272, 33105–33110. [Google Scholar] [CrossRef] [PubMed]
- McDonald, P.; Veluthakal, R.; Kaur, H.; Kowluru, A. Biologically active lipids promote trafficking and membrane association of Rac1 in insulin-secreting INS 832/13 cells. Am. J. Physiol. Cell Physiol. 2007, 292, C1216–C1220. [Google Scholar] [CrossRef]
- del Pozo, M.A.; Alderson, N.B.; Kiosses, W.B.; Chiang, H.H.; Anderson, R.G.; Schwartz, M.A. Integrins regulate Rac targeting by internalization of membrane domains. Science 2004, 303, 839–842. [Google Scholar] [CrossRef] [PubMed]
- Del Pozo, M.A.; Kiosses, W.B.; Alderson, N.B.; Meller, N.; Hahn, K.M.; Schwartz, M.A. Integrins regulate GTP-Rac localized effector interactions through dissociation of Rho-GDI. Nat. Cell Biol. 2002, 4, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Tiedge, M.; Lortz, S.; Drinkgern, J.; Lenzen, S. Relation between antioxidant enzyme gene expression and antioxidative defense status of insulin-producing cells. Diabetes 1997, 46, 1733–1742. [Google Scholar] [CrossRef]
- Matsuoka, T.; Kajimoto, Y.; Watada, H.; Kaneto, H.; Kishimoto, M.; Umayahara, Y.; Fujitani, Y.; Kamada, T.; Kawamori, R.; Yamasaki, Y. Glycation-dependent, reactive oxygen species-mediated suppression of the insulin gene promoter activity in HIT cells. J. Clin. Investig. 1997, 99, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Matsumoto, K.; Nishikawa, T.; Suefuji, M.; Nakamaru, K.; Hirashima, Y.; Kawashima, J.; Shirotani, T.; Ichinose, K.; Brownlee, M.; et al. Mitochondrial reactive oxygen species reduce insulin secretion by pancreatic beta-cells. Biochem. Biophys. Res. Commun. 2003, 300, 216–222. [Google Scholar] [CrossRef]
- Kaneto, H.; Xu, G.; Song, K.H.; Suzuma, K.; Bonner-Weir, S.; Sharma, A.; Weir, G.C. Activation of the hexosamine pathway leads to deterioration of pancreatic beta-cell function through the induction of oxidative stress. J. Biol. Chem. 2001, 276, 31099–31104. [Google Scholar] [CrossRef]
- Oliveira, H.R.; Verlengia, R.; Carvalho, C.R.; Britto, L.R.; Curi, R.; Carpinelli, A.R. Pancreatic beta-cells express phagocyte-like NAD(P)H oxidase. Diabetes 2003, 52, 1457–1463. [Google Scholar] [CrossRef]
- Morgan, D.; Oliveira-Emilio, H.R.; Keane, D.; Hirata, A.E.; Santos da Rocha, M.; Bordin, S.; Curi, R.; Newsholme, P.; Carpinelli, A.R. Glucose, palmitate and pro-inflammatory cytokines modulate production and activity of a phagocyte-like NADPH oxidase in rat pancreatic islets and a clonal beta cell line. Diabetologia 2007, 50, 359–369. [Google Scholar] [CrossRef]
- Rastogi, R.; Geng, X.; Li, F.; Ding, Y. NOX Activation by Subunit Interaction and Underlying Mechanisms in Disease. Front. Cell. Neurosci. 2016, 10, 301. [Google Scholar] [CrossRef]
- Abo, A.; Pick, E.; Hall, A.; Totty, N.; Teahan, C.G.; Segal, A.W. Activation of the NADPH oxidase involves the small GTP-binding protein p21rac1. Nature 1991, 353, 668–670. [Google Scholar] [CrossRef]
- Syed, I.; Kyathanahalli, C.N.; Jayaram, B.; Govind, S.; Rhodes, C.J.; Kowluru, R.A.; Kowluru, A. Increased phagocyte-like NADPH oxidase and ROS generation in type 2 diabetic ZDF rat and human islets: Role of Rac1-JNK1/2 signaling pathway in mitochondrial dysregulation in the diabetic islet. Diabetes 2011, 60, 2843–2852. [Google Scholar] [CrossRef]
- Syed, I.; Kyathanahalli, C.N.; Kowluru, A. Phagocyte-like NADPH oxidase generates ROS in INS 832/13 cells and rat islets: Role of protein prenylation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R756–R762. [Google Scholar] [CrossRef]
- Sidarala, V.; Veluthakal, R.; Syeda, K.; Vlaar, C.; Newsholme, P.; Kowluru, A. Phagocyte-like NADPH oxidase (Nox2) promotes activation of p38MAPK in pancreatic beta-cells under glucotoxic conditions: Evidence for a requisite role of Ras-related C3 botulinum toxin substrate 1 (Rac1). Biochem. Pharmacol. 2015, 95, 301–310. [Google Scholar] [CrossRef]
- Veluthakal, R.; Sidarala, V.; Kowluru, A. NSC23766, a Known Inhibitor of Tiam1-Rac1 Signaling Module, Prevents the Onset of Type 1 Diabetes in the NOD Mouse Model. Cell Physiol. Biochem. 2016, 39, 760–767. [Google Scholar] [CrossRef]
- Baidwan, S.; Chekuri, A.; Hynds, D.L.; Kowluru, A. Glucotoxicity promotes aberrant activation and mislocalization of Ras-related C3 botulinum toxin substrate 1 [Rac1] and metabolic dysfunction in pancreatic islet beta-cells: Reversal of such metabolic defects by metformin. Apoptosis 2017, 22, 1380–1393. [Google Scholar] [CrossRef]
- Gaschet, J.; Hsu, V.W. Distribution of ARF6 between membrane and cytosol is regulated by its GTPase cycle. J. Biol. Chem. 1999, 274, 20040–20045. [Google Scholar] [CrossRef]
- Honda, A.; Nogami, M.; Yokozeki, T.; Yamazaki, M.; Nakamura, H.; Watanabe, H.; Kawamoto, K.; Nakayama, K.; Morris, A.J.; Frohman, M.A.; et al. Phosphatidylinositol 4-phosphate 5-kinase alpha is a downstream effector of the small G protein ARF6 in membrane ruffle formation. Cell 1999, 99, 521–532. [Google Scholar] [CrossRef]
- Shome, K.; Nie, Y.; Romero, G. ADP-ribosylation factor proteins mediate agonist-induced activation of phospholipase D. J. Biol. Chem. 1998, 273, 30836–30841. [Google Scholar] [CrossRef]
- Cockcroft, S.; De Matteis, M.A. Inositol lipids as spatial regulators of membrane traffic. J. Membr. Biol. 2001, 180, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, J.T.; Birnbaum, M.J. ADP-ribosylation factor 6 regulates insulin secretion through plasma membrane phosphatidylinositol 4,5-bisphosphate. Proc. Natl. Acad. Sci. USA 2003, 100, 13320–13325. [Google Scholar] [CrossRef] [PubMed]
- Jayaram, B.; Syed, I.; Kyathanahalli, C.N.; Rhodes, C.J.; Kowluru, A. Arf nucleotide binding site opener [ARNO] promotes sequential activation of Arf6, Cdc42 and Rac1 and insulin secretion in INS 832/13 beta-cells and rat islets. Biochem. Pharmacol. 2011, 81, 1016–1027. [Google Scholar] [CrossRef] [PubMed]
- Aramata, S.; Han, S.I.; Yasuda, K.; Kataoka, K. Synergistic activation of the insulin gene promoter by the beta-cell enriched transcription factors MafA, Beta2, and Pdx1. Biochim. Biophys. Acta 2005, 1730, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Docherty, H.M.; Hay, C.W.; Ferguson, L.A.; Barrow, J.; Durward, E.; Docherty, K. Relative contribution of PDX-1, MafA and E47/beta2 to the regulation of the human insulin promoter. Biochem. J. 2005, 389, 813–820. [Google Scholar] [CrossRef]
- Ma, W.N.; Park, S.Y.; Han, J.S. Role of phospholipase D1 in glucose-induced insulin secretion in pancreatic Beta cells. Exp. Mol. Med. 2010, 42, 456–464. [Google Scholar] [CrossRef]
- Stenmark, H. Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 2009, 10, 513–525. [Google Scholar] [CrossRef]
- Wandinger-Ness, A.; Zerial, M. Rab proteins and the compartmentalization of the endosomal system. Cold Spring Harb. Perspect. Biol. 2014, 6, a022616. [Google Scholar] [CrossRef]
- Pfeffer, S.R. Rab GTPase regulation of membrane identity. Curr. Opin. Cell Biol. 2013, 25, 414–419. [Google Scholar] [CrossRef]
- Chavrier, P.; Gorvel, J.P.; Stelzer, E.; Simons, K.; Gruenberg, J.; Zerial, M. Hypervariable C-terminal domain of rab proteins acts as a targeting signal. Nature 1991, 353, 769–772. [Google Scholar] [CrossRef]
- Ferro-Novick, S.; Novick, P. The role of GTP-binding proteins in transport along the exocytic pathway. Annu. Rev. Cell Biol. 1993, 9, 575–599. [Google Scholar] [CrossRef]
- Merrins, M.J.; Stuenkel, E.L. Kinetics of Rab27a-dependent actions on vesicle docking and priming in pancreatic beta-cells. J. Physiol. 2008, 586, 5367–5381. [Google Scholar] [CrossRef]
- Haddad, E.K.; Wu, X.; Hammer, J.A., 3rd; Henkart, P.A. Defective granule exocytosis in Rab27a-deficient lymphocytes from Ashen mice. J. Cell Biol. 2001, 152, 835–842. [Google Scholar] [CrossRef]
- Kasai, K.; Ohara-Imaizumi, M.; Takahashi, N.; Mizutani, S.; Zhao, S.; Kikuta, T.; Kasai, H.; Nagamatsu, S.; Gomi, H.; Izumi, T. Rab27a mediates the tight docking of insulin granules onto the plasma membrane during glucose stimulation. J. Clin. Investig. 2005, 115, 388–396. [Google Scholar] [CrossRef]
- Regazzi, R.; Ravazzola, M.; Iezzi, M.; Lang, J.; Zahraoui, A.; Andereggen, E.; Morel, P.; Takai, Y.; Wollheim, C.B. Expression, localization and functional role of small GTPases of the Rab3 family in insulin-secreting cells. J. Cell Sci. 1996, 109, 2265–2273. [Google Scholar] [CrossRef]
- Coppola, T.; Perret-Menoud, V.; Luthi, S.; Farnsworth, C.C.; Glomset, J.A.; Regazzi, R. Disruption of Rab3-calmodulin interaction, but not other effector interactions, prevents Rab3 inhibition of exocytosis. EMBO J. 1999, 18, 5885–5891. [Google Scholar] [CrossRef]
- Zhao, S.; Torii, S.; Yokota-Hashimoto, H.; Takeuchi, T.; Izumi, T. Involvement of Rab27b in the regulated secretion of pituitary hormones. Endocrinology 2002, 143, 1817–1824. [Google Scholar] [CrossRef]
- Lam, A.D.; Ismail, S.; Wu, R.; Yizhar, O.; Passmore, D.R.; Ernst, S.A.; Stuenkel, E.L. Mapping dynamic protein interactions to insulin secretory granule behavior with TIRF-FRET. Biophys. J. 2010, 99, 1311–1320. [Google Scholar] [CrossRef]
- Waselle, L.; Coppola, T.; Fukuda, M.; Iezzi, M.; El-Amraoui, A.; Petit, C.; Regazzi, R. Involvement of the Rab27 binding protein Slac2c/MyRIP in insulin exocytosis. Mol. Biol. Cell 2003, 14, 4103–4113. [Google Scholar] [CrossRef]
- Yaekura, K.; Julyan, R.; Wicksteed, B.L.; Hays, L.B.; Alarcon, C.; Sommers, S.; Poitout, V.; Baskin, D.G.; Wang, Y.; Philipson, L.H.; et al. Insulin secretory deficiency and glucose intolerance in Rab3A null mice. J. Biol. Chem. 2003, 278, 9715–9721. [Google Scholar] [CrossRef]
- Coppola, T.; Frantz, C.; Perret-Menoud, V.; Gattesco, S.; Hirling, H.; Regazzi, R. Pancreatic beta-cell protein granuphilin binds Rab3 and Munc-18 and controls exocytosis. Mol. Biol. Cell 2002, 13, 1906–1915. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, T.S.; Fukuda, M.; Ariga, H.; Mikoshiba, K. Synaptotagmin-like protein 5: A novel Rab27A effector with C-terminal tandem C2 domains. Biochem. Biophys. Res. Commun. 2002, 293, 899–906. [Google Scholar] [CrossRef]
- Kuroda, T.S.; Fukuda, M.; Ariga, H.; Mikoshiba, K. The Slp homology domain of synaptotagmin-like proteins 1-4 and Slac2 functions as a novel Rab27A binding domain. J. Biol. Chem. 2002, 277, 9212–9218. [Google Scholar] [CrossRef] [PubMed]
- Stermann, T.; Menzel, F.; Weidlich, C.; Jeruschke, K.; Weiss, J.; Altenhofen, D.; Benninghoff, T.; Pujol, A.; Bosch, F.; Rustenbeck, I.; et al. Deletion of the RabGAP TBC1D1 Leads to Enhanced Insulin Secretion and Fatty Acid Oxidation in Islets From Male Mice. Endocrinology 2018, 159, 1748–1761. [Google Scholar] [CrossRef]
- Orci, L.; Malaisse-Lagae, F.; Ravazzola, M.; Amherdt, M.; Renold, A.E. Exocytosis-endocytosis coupling in the pancreatic beta cell. Science 1973, 181, 561–562. [Google Scholar] [CrossRef]
- MacDonald, P.E.; Rorsman, P. The ins and outs of secretion from pancreatic beta-cells: Control of single-vesicle exo- and endocytosis. Physiology 2007, 22, 113–121. [Google Scholar] [CrossRef]
- Cousin, M.A. Synaptic vesicle endocytosis: Calcium works overtime in the nerve terminal. Mol. Neurobiol. 2000, 22, 115–128. [Google Scholar] [CrossRef]
- Ryan, T.A. A pre-synaptic to-do list for coupling exocytosis to endocytosis. Curr. Opin. Cell Biol. 2006, 18, 416–421. [Google Scholar] [CrossRef]
- Takei, K.; Yoshida, Y.; Yamada, H. Regulatory mechanisms of dynamin-dependent endocytosis. J. Biochem. 2005, 137, 243–247. [Google Scholar] [CrossRef]
- Yamaoka, M.; Ando, T.; Terabayashi, T.; Okamoto, M.; Takei, M.; Nishioka, T.; Kaibuchi, K.; Matsunaga, K.; Ishizaki, R.; Izumi, T.; et al. PI3K regulates endocytosis after insulin secretion by mediating signaling crosstalk between Arf6 and Rab27a. J. Cell Sci. 2016, 129, 637–649. [Google Scholar] [CrossRef]
- Paleotti, O.; Macia, E.; Luton, F.; Klein, S.; Partisani, M.; Chardin, P.; Kirchhausen, T.; Franco, M. The small G-protein Arf6GTP recruits the AP-2 adaptor complex to membranes. J. Biol. Chem. 2005, 280, 21661–21666. [Google Scholar] [CrossRef]
- Wang, H.; Ishizaki, R.; Xu, J.; Kasai, K.; Kobayashi, E.; Gomi, H.; Izumi, T. The Rab27a effector exophilin7 promotes fusion of secretory granules that have not been docked to the plasma membrane. Mol. Biol. Cell 2013, 24, 319–330. [Google Scholar] [CrossRef]
- Yamaoka, M.; Ishizaki, T.; Kimura, T. Interplay between Rab27a effectors in pancreatic beta-cells. World J. Diabetes 2015, 6, 508–516. [Google Scholar] [CrossRef]
- Janoueix-Lerosey, I.; Pasheva, E.; de Tand, M.F.; Tavitian, A.; de Gunzburg, J. Identification of a specific effector of the small GTP-binding protein Rap2. Eur. J. Biochem. 1998, 252, 290–298. [Google Scholar] [CrossRef]
- Holz, G.G. Epac: A new cAMP-binding protein in support of glucagon-like peptide-1 receptor-mediated signal transduction in the pancreatic beta-cell. Diabetes 2004, 53, 5–13. [Google Scholar] [CrossRef]
- Kang, G.; Chepurny, O.G.; Holz, G.G. cAMP-regulated guanine nucleotide exchange factor II (Epac2) mediates Ca2+-induced Ca2+ release in INS-1 pancreatic beta-cells. J. Physiol. 2001, 536, 375–385. [Google Scholar] [CrossRef]
- McAvoy, T.; Zhou, M.M.; Greengard, P.; Nairn, A.C. Phosphorylation of Rap1GAP, a striatally enriched protein, by protein kinase A controls Rap1 activity and dendritic spine morphology. Proc. Natl. Acad. Sci. USA 2009, 106, 3531–3536. [Google Scholar] [CrossRef]
- Takahashi, M.; Dillon, T.J.; Liu, C.; Kariya, Y.; Wang, Z.; Stork, P.J. Protein kinase A-dependent phosphorylation of Rap1 regulates its membrane localization and cell migration. J. Biol. Chem. 2013, 288, 27712–27723. [Google Scholar] [CrossRef]
- Leech, C.A.; Chepurny, O.G.; Holz, G.G. Epac2-dependent rap1 activation and the control of islet insulin secretion by glucagon-like peptide-1. Vitam. Horm. 2010, 84, 279–302. [Google Scholar] [CrossRef]
- Nakazaki, M.; Crane, A.; Hu, M.; Seghers, V.; Ullrich, S.; Aguilar-Bryan, L.; Bryan, J. cAMP-activated protein kinase-independent potentiation of insulin secretion by cAMP is impaired in SUR1 null islets. Diabetes 2002, 51, 3440–3449. [Google Scholar] [CrossRef]
- Kelly, P.; Bailey, C.L.; Fueger, P.T.; Newgard, C.B.; Casey, P.J.; Kimple, M.E. Rap1 promotes multiple pancreatic islet cell functions and signals through mammalian target of rapamycin complex 1 to enhance proliferation. J. Biol. Chem. 2010, 285, 15777–15785. [Google Scholar] [CrossRef]
- Zhang, Y.; Parajuli, K.R.; Fava, G.E.; Gupta, R.; Xu, W.; Nguyen, L.U.; Zakaria, A.F.; Fonseca, V.A.; Wang, H.; Mauvais-Jarvis, F.; et al. GLP-1 Receptor in Pancreatic alpha-Cells Regulates Glucagon Secretion in a Glucose-Dependent Bidirectional Manner. Diabetes 2019, 68, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Richards, P.; Parker, H.E.; Adriaenssens, A.E.; Hodgson, J.M.; Cork, S.C.; Trapp, S.; Gribble, F.M.; Reimann, F. Identification and characterization of GLP-1 receptor-expressing cells using a new transgenic mouse model. Diabetes 2014, 63, 1224–1233. [Google Scholar] [CrossRef] [PubMed]
- Moens, K.; Heimberg, H.; Flamez, D.; Huypens, P.; Quartier, E.; Ling, Z.; Pipeleers, D.; Gremlich, S.; Thorens, B.; Schuit, F. Expression and functional activity of glucagon, glucagon-like peptide I, and glucose-dependent insulinotropic peptide receptors in rat pancreatic islet cells. Diabetes 1996, 45, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J. The biology of incretin hormones. Cell Metab. 2006, 3, 153–165. [Google Scholar] [CrossRef]
- Drucker, D.J.; Nauck, M.A. The incretin system: Glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet 2006, 368, 1696–1705. [Google Scholar] [CrossRef]
- Gheni, G.; Ogura, M.; Iwasaki, M.; Yokoi, N.; Minami, K.; Nakayama, Y.; Harada, K.; Hastoy, B.; Wu, X.; Takahashi, H.; et al. Glutamate acts as a key signal linking glucose metabolism to incretin/cAMP action to amplify insulin secretion. Cell Rep. 2014, 9, 661–673. [Google Scholar] [CrossRef]
- Dzhura, I.; Chepurny, O.G.; Kelley, G.G.; Leech, C.A.; Roe, M.W.; Dzhura, E.; Afshari, P.; Malik, S.; Rindler, M.J.; Xu, X.; et al. Epac2-dependent mobilization of intracellular Ca(2)+ by glucagon-like peptide-1 receptor agonist exendin-4 is disrupted in beta-cells of phospholipase C-epsilon knockout mice. J. Physiol. 2010, 588, 4871–4889. [Google Scholar] [CrossRef]
- Dzhura, I.; Chepurny, O.G.; Leech, C.A.; Roe, M.W.; Dzhura, E.; Xu, X.; Lu, Y.; Schwede, F.; Genieser, H.G.; Smrcka, A.V.; et al. Phospholipase C-epsilon links Epac2 activation to the potentiation of glucose-stimulated insulin secretion from mouse islets of Langerhans. Islets 2011, 3, 121–128. [Google Scholar] [CrossRef]
- Chundru, S. Novel Regulatory Roles Of Rhog And Iqgaps In Pancreatic Islet Beta Cell Function. Ph.D. Thesis, Wayne State University, Detroit, MI, USA, 2020. [Google Scholar]
- Mercader, J.M.; Puiggros, M.; Segre, A.V.; Planet, E.; Sorianello, E.; Sebastian, D.; Rodriguez-Cuenca, S.; Ribas, V.; Bonas-Guarch, S.; Draghici, S.; et al. Identification of novel type 2 diabetes candidate genes involved in the crosstalk between the mitochondrial and the insulin signaling systems. PLoS Genet. 2012, 8, e1003046. [Google Scholar] [CrossRef]
- Thamilselvan, V.; Gamage, S.; Harajli, A.; Chundru, S.A.; Kowluru, A. P-Rex1 Mediates Glucose-Stimulated Rac1 Activation and Insulin Secretion in Pancreatic beta-Cells. Cell Physiol. Biochem. 2020, 54, 1218–1230. [Google Scholar] [CrossRef]
- Lewis, J.P.; Palmer, N.D.; Ellington, J.B.; Divers, J.; Ng, M.C.; Lu, L.; Langefeld, C.D.; Freedman, B.I.; Bowden, D.W. Analysis of candidate genes on chromosome 20q12-13.1 reveals evidence for BMI mediated association of PREX1 with type 2 diabetes in European Americans. Genomics 2010, 96, 211–219. [Google Scholar] [CrossRef]
- Moltke, I.; Grarup, N.; Jorgensen, M.E.; Bjerregaard, P.; Treebak, J.T.; Fumagalli, M.; Korneliussen, T.S.; Andersen, M.A.; Nielsen, T.S.; Krarup, N.T.; et al. A common Greenlandic TBC1D4 variant confers muscle insulin resistance and type 2 diabetes. Nature 2014, 512, 190–193. [Google Scholar] [CrossRef]
- Bouzakri, K.; Ribaux, P.; Tomas, A.; Parnaud, G.; Rickenbach, K.; Halban, P.A. Rab GTPase-activating protein AS160 is a major downstream effector of protein kinase B/Akt signaling in pancreatic beta-cells. Diabetes 2008, 57, 1195–1204. [Google Scholar] [CrossRef]
- Ndiaye, F.K.; Ortalli, A.; Canouil, M.; Huyvaert, M.; Salazar-Cardozo, C.; Lecoeur, C.; Verbanck, M.; Pawlowski, V.; Boutry, R.; Durand, E.; et al. Expression and functional assessment of candidate type 2 diabetes susceptibility genes identify four new genes contributing to human insulin secretion. Mol. Metab. 2017, 6, 459–470. [Google Scholar] [CrossRef]
- Bottcher, Y.; Schleinitz, D.; Tonjes, A.; Bluher, M.; Stumvoll, M.; Kovacs, P. R1467H variant in the rho guanine nucleotide exchange factor 11 (ARHGEF11) is associated with impaired glucose tolerance and type 2 diabetes in German Caucasians. J. Hum. Genet. 2008, 53, 365–367. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GTPases in β-Cells | Location | Function | References |
|---|---|---|---|
| Rac1 | Cytosol (GDP-loaded), PM (GTP-loaded) | Cytoskeletal rearrangement | [63,64] |
| Cdc42 | Cytosol/Insulin-containing secretory granules (GDP-loaded), PM (GTP-loaded) | Cytoskeletal rearrangement/ Vesicle fusion | [47,48,49,65,66] |
| Arf6 | Cytosol (GDP loaded), PM (GTP loaded) | Vesicle fusion | [50] |
| Rab27a | Insulin-containing secretory granules (GDP and GTP loaded) | Docking/priming | [67] |
| Rab37 | Cytosol (GDP loaded) Insulin-containing secretory granules (GTP loaded) | Docking/priming | [68] |
| Rab2a | Cytosol (GDP loaded) Perinuclear immature granules (GTP loaded) | Docking/priming | [69] |
| Rab3a | Cytosol (GDP loaded) Insulin-containing secretory granules (GTP loaded) | Docking/priming | [70] |
| Rap1 | Colocalized with insulin granules (GDP loaded), PM (GTP loaded) | Docking/priming | [71] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Veluthakal, R.; Thurmond, D.C. Emerging Roles of Small GTPases in Islet β-Cell Function. Cells 2021, 10, 1503. https://doi.org/10.3390/cells10061503
Veluthakal R, Thurmond DC. Emerging Roles of Small GTPases in Islet β-Cell Function. Cells. 2021; 10(6):1503. https://doi.org/10.3390/cells10061503
Chicago/Turabian StyleVeluthakal, Rajakrishnan, and Debbie C. Thurmond. 2021. "Emerging Roles of Small GTPases in Islet β-Cell Function" Cells 10, no. 6: 1503. https://doi.org/10.3390/cells10061503
APA StyleVeluthakal, R., & Thurmond, D. C. (2021). Emerging Roles of Small GTPases in Islet β-Cell Function. Cells, 10(6), 1503. https://doi.org/10.3390/cells10061503