
The HSP90 Inhibitor, AUY-922, Protects and Repairs Human Lung Microvascular Endothelial Cells from Hydrochloric Acid-Induced Endothelial Barrier Dysfunction




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture and Cell Viability
2.3. Endothelial Barrier Function
2.4. Protein Isolation and Western Blots
2.5. Immunocytochemistry
2.6. Statistical Analysis
3. Results
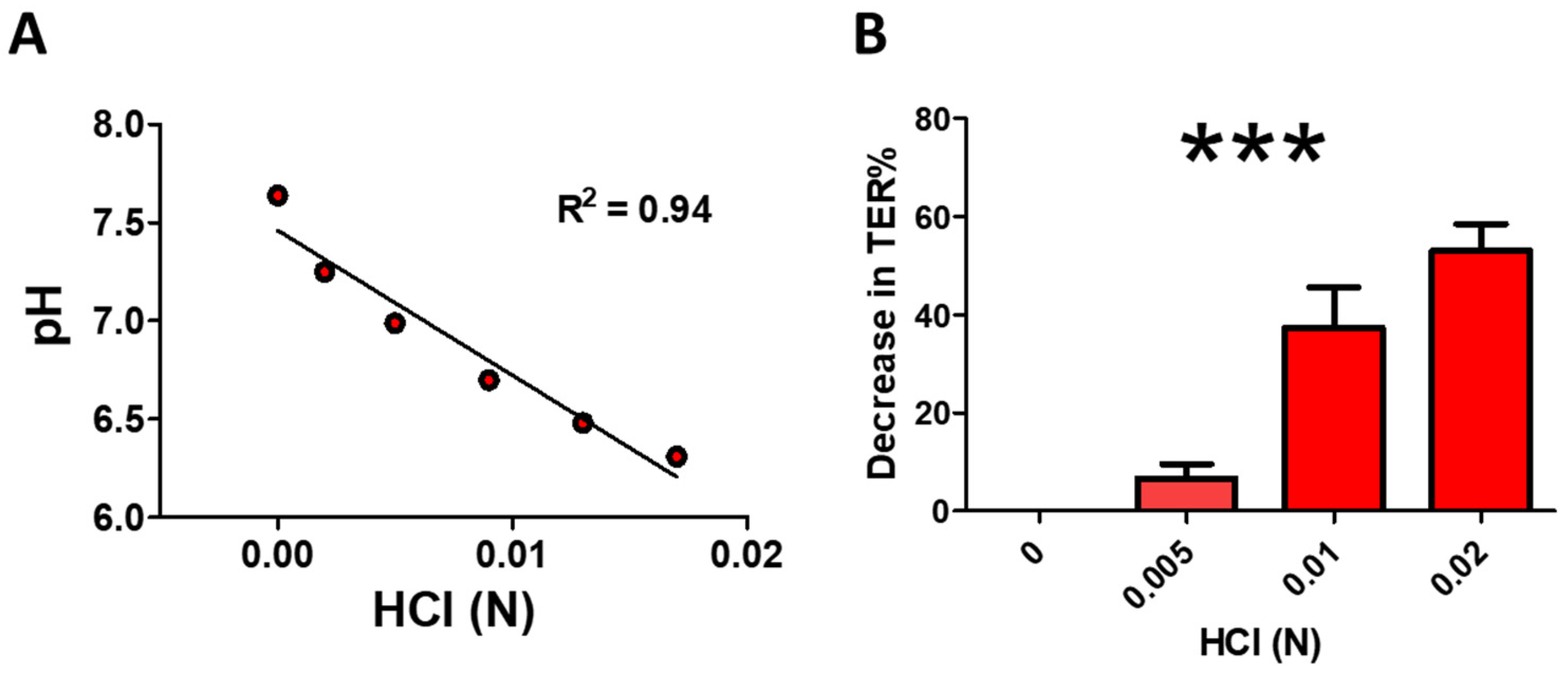
3.1. HCl Elicits a Concentration-Dependent Decrease in TER
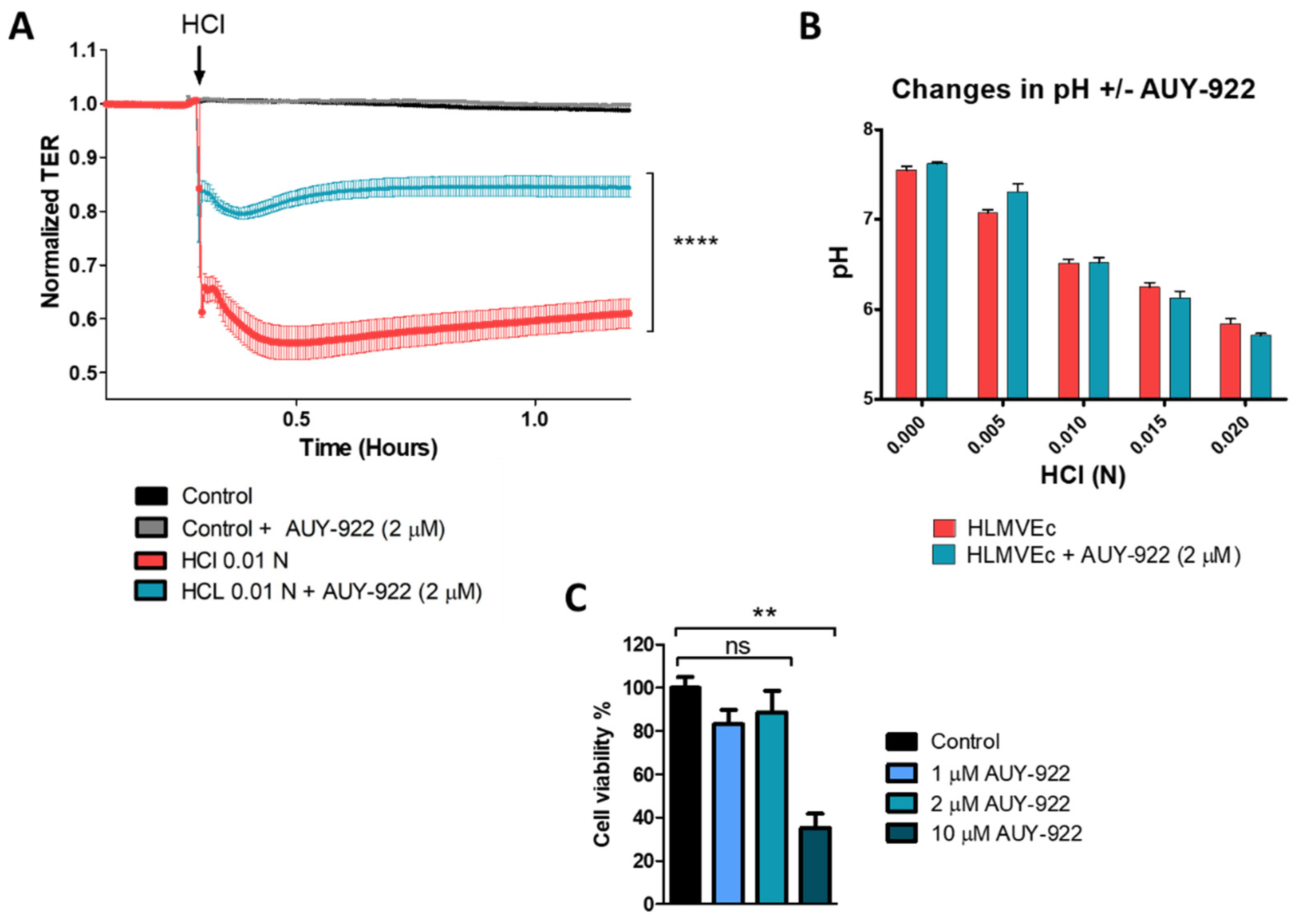
3.2. Pre-Treatment with the HSP90 Inhibitor, AUY-922, Protects HLMVEC from HCl-Induced Endothelial Barrier Dysfunction
3.3. Pre-Treatment with AUY-922 Induces Cytoprotection
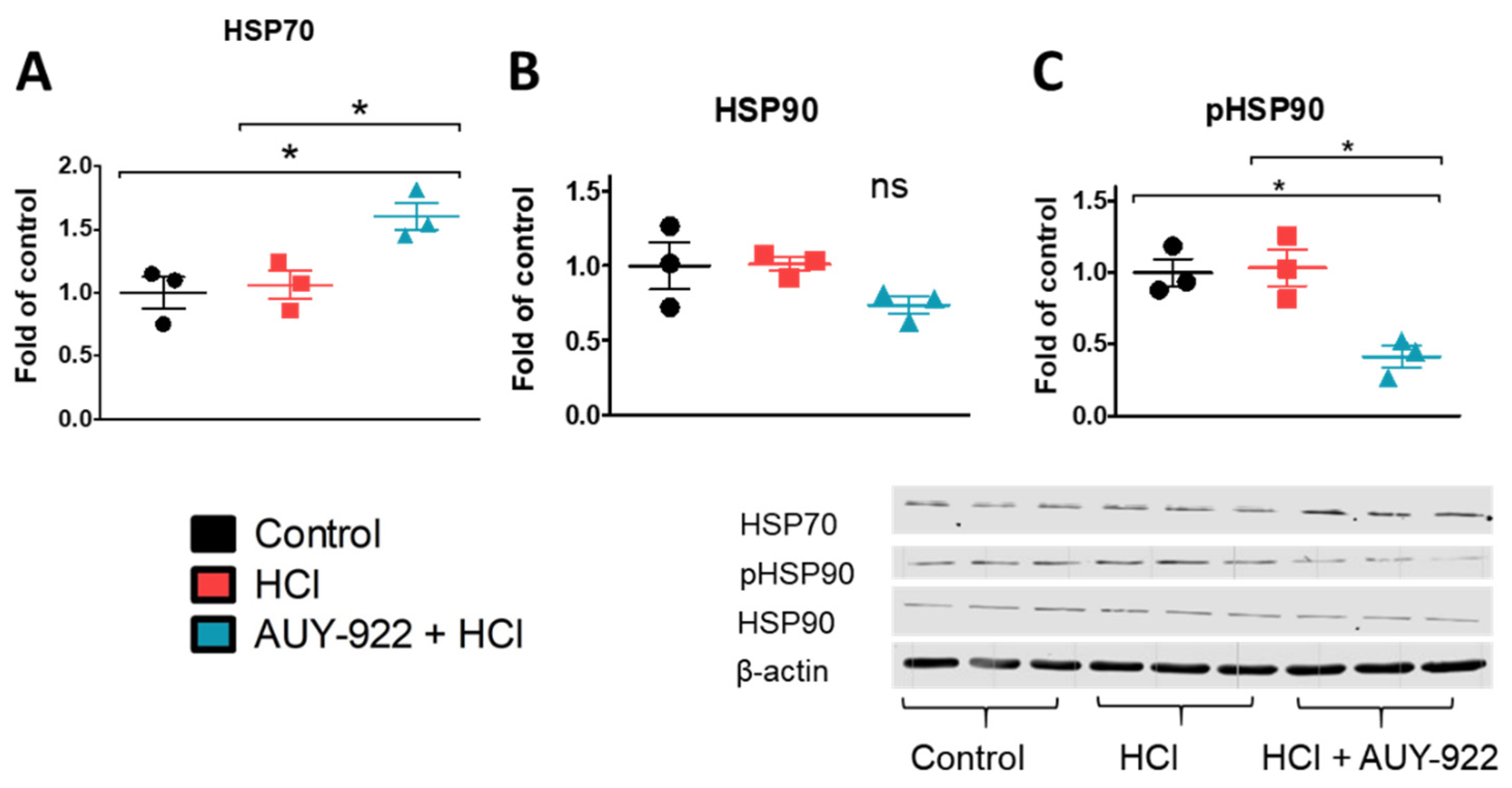
3.4. Pre-Treatment with AUY-922 Prevents the HCl-Induced AKT Phosphorylation
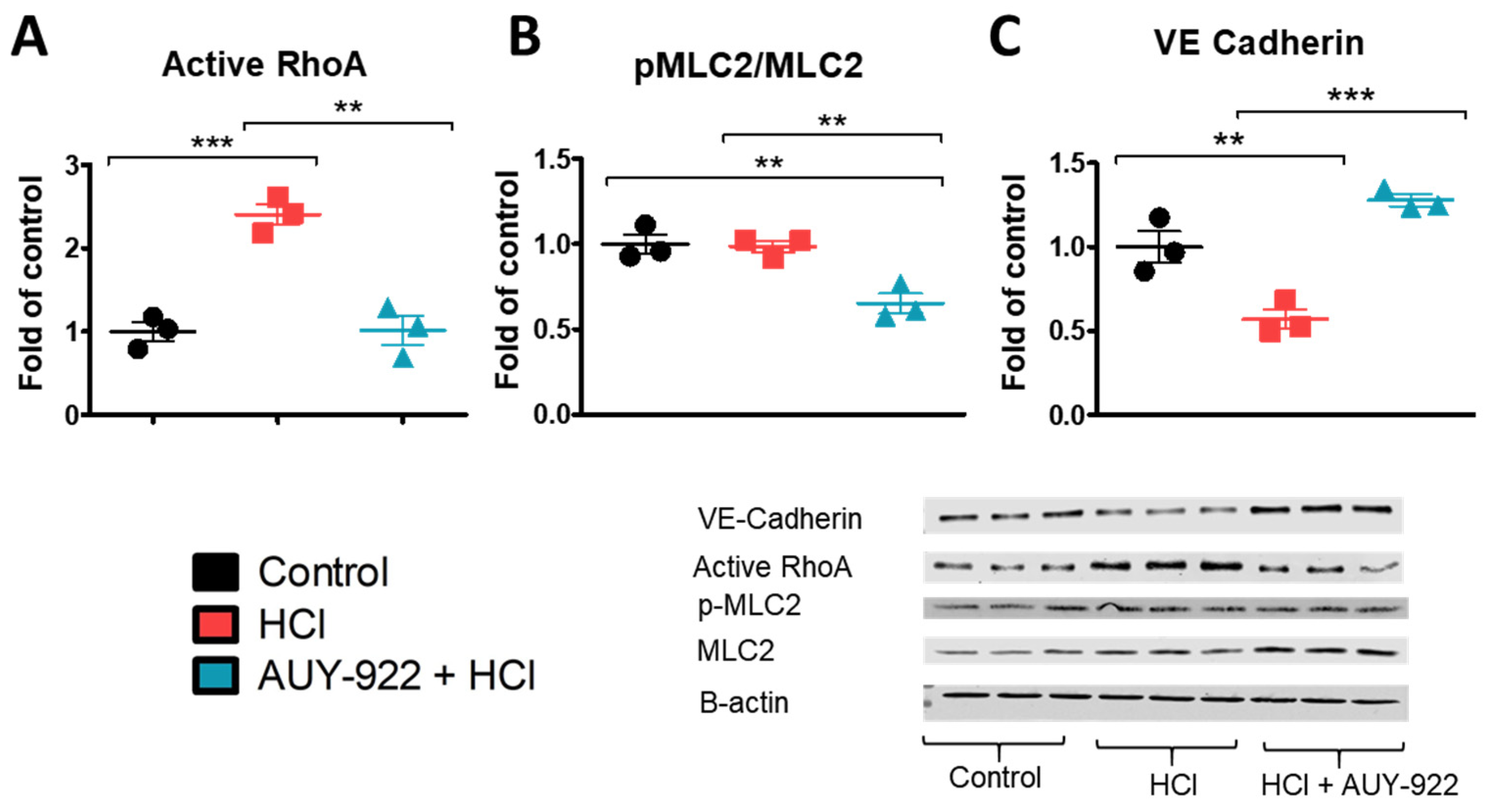
3.5. Pre-Treatment with AUY-922 Modulates the Activation of Cytoskeletal Proteins and the Expression of Tight Junction Proteins
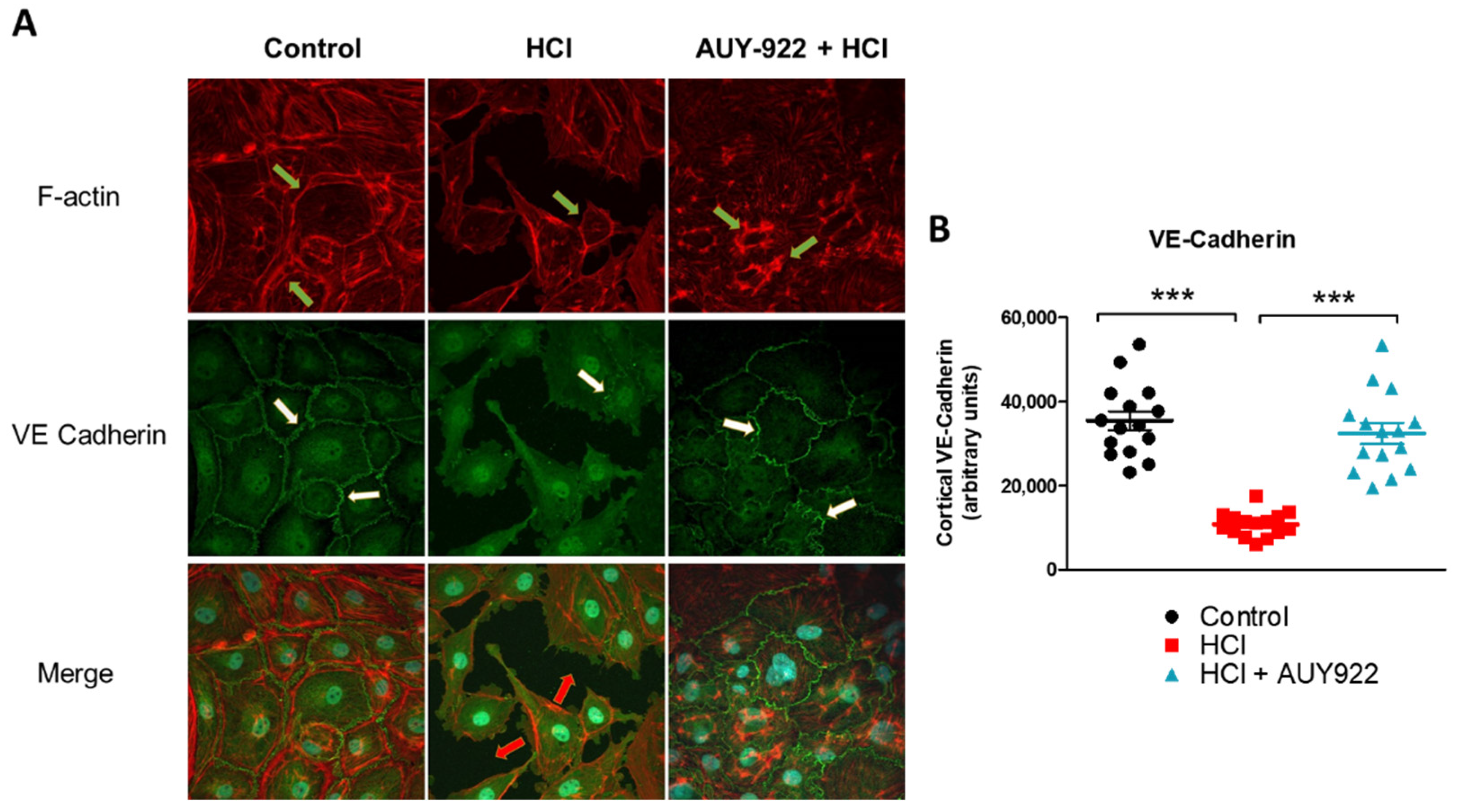
3.6. Pre-Treatment of HLMVEC with AUY-922 for 24 h Prevents the HCl-Induced Loss of Plasmalemmal VE-Cadherin
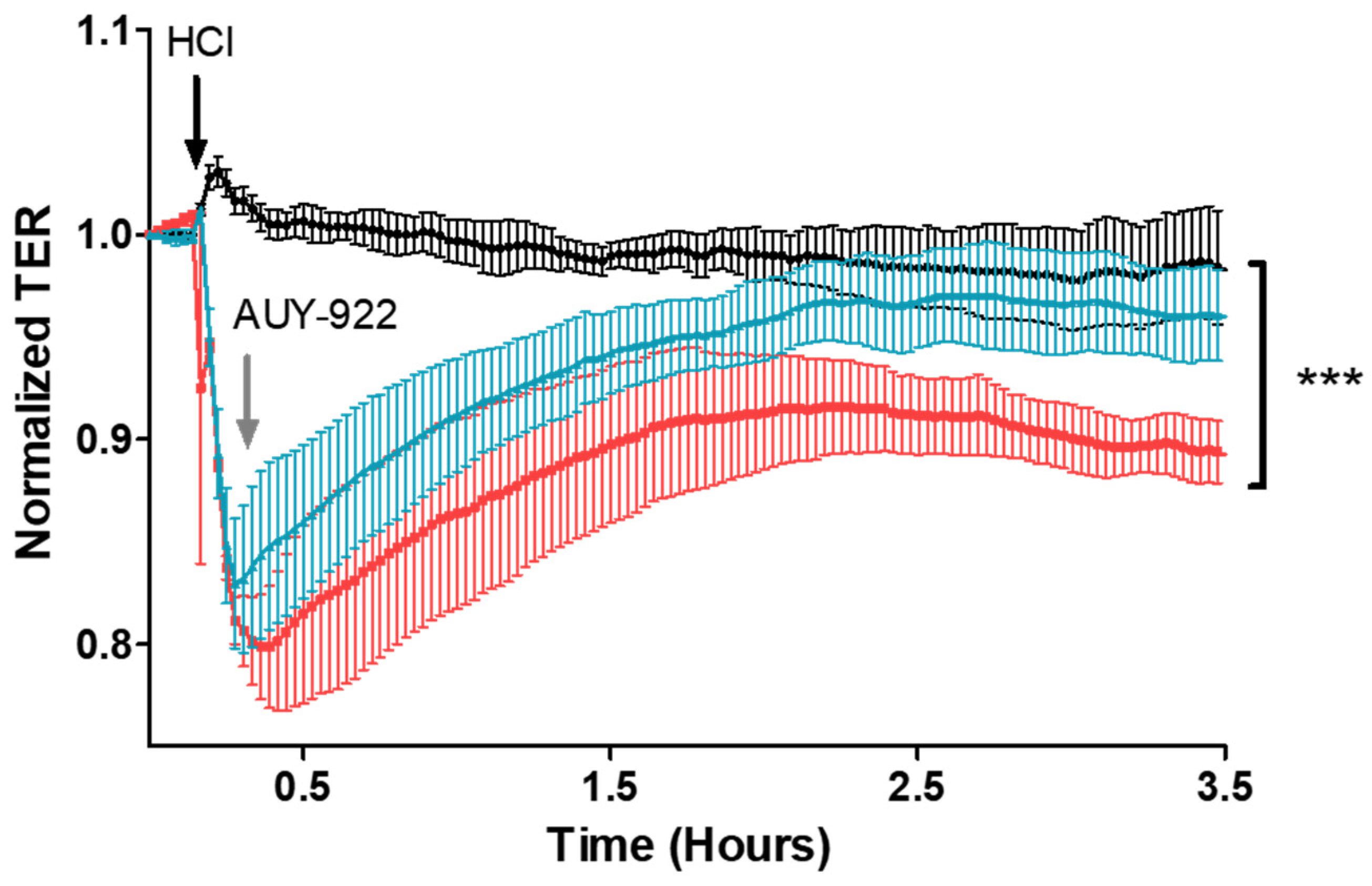
3.7. Post-Treatment with AUY-922 Restores HCl-Induced Hyperpermeability
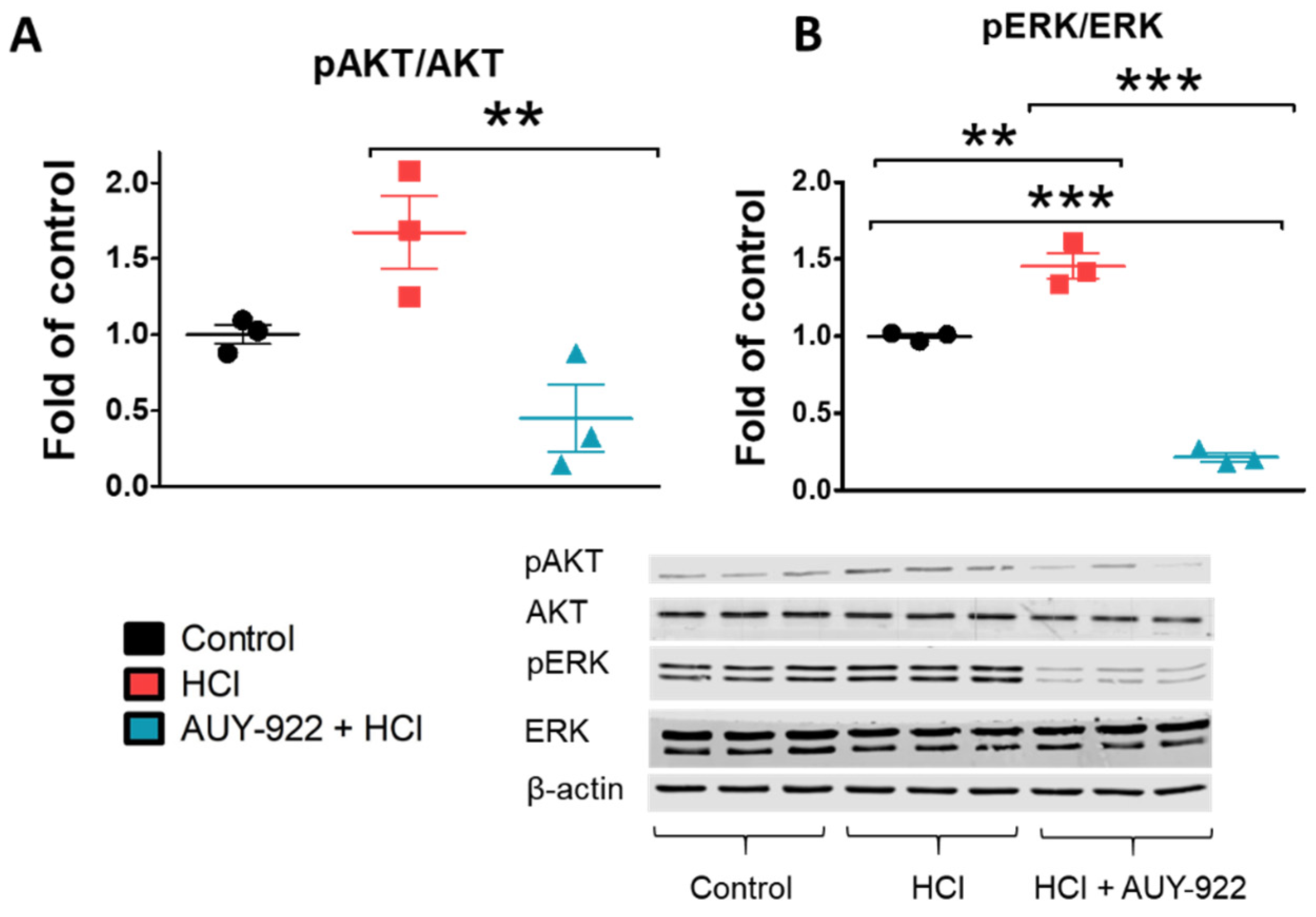
3.8. Post-Treatment with AUY-922 Inhibited the Activation of AKT and ERK
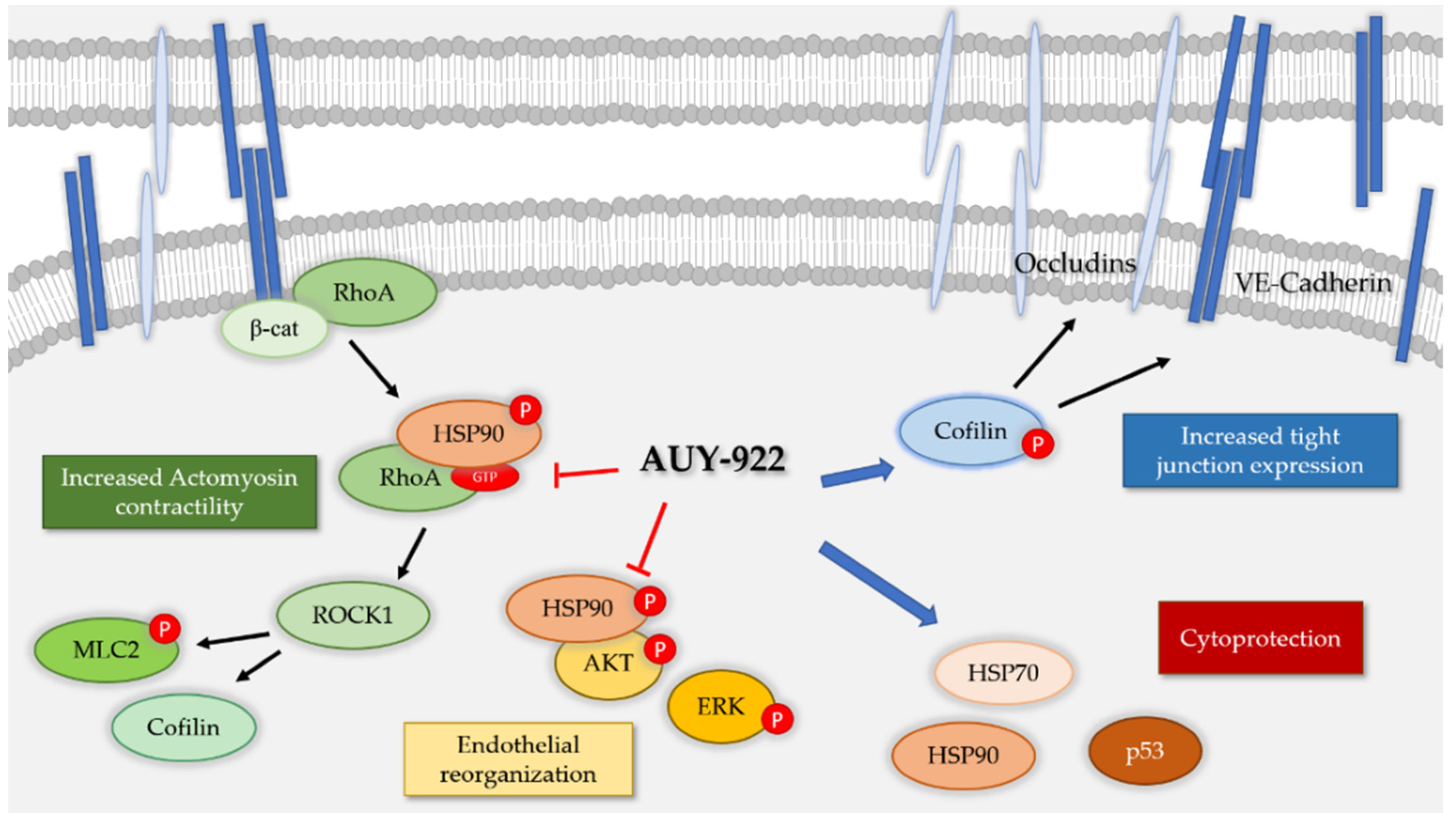
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- National Research Council (US), S.o.R.-E.T. Acute Toxicity of Hydrogen Chloride, Appendix D; National Academies Press (US): Washington, DC, USA, 1998. [Google Scholar]
- Bingham, E.; Cohrssen, B.; Powell, C.H. Patty’s Toxicology, 5th ed.; John Wiley & Sons: New York, NY, USA, 2001; pp. 1–9. [Google Scholar]
- Boulet, L.P. Increases in airway responsiveness following acute exposure to respiratory irritants. Reactive airway dysfunction syndrome or occupational asthma? Chest 1988, 94, 476–481. [Google Scholar] [CrossRef]
- Agabiti, N.; Ancona, C.; Forastiere, F.; Di Napoli, A.; Lo Presti, E.; Corbo, G.M.; D’Orsi, F.; Perucci, C.A. Short term respiratory effects of acute exposure to chlorine due to a swimming pool accident. Occup. Environ. Med. 2001, 58, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Promisloff, R.A.; Lenchner, G.S.; Phan, A.; Cichelli, A.V. Reactive airway dysfunction syndrome in three police officers following a roadside chemical spill. Chest 1990, 98, 928–929. [Google Scholar] [CrossRef] [PubMed]
- Marinova, M.; Solopov, P.; Dimitropoulou, C.; Colunga Biancatelli, R.M.L.; Catravas, J.D. Acute exposure of mice to hydrochloric acid leads to the development of chronic lung injury and pulmonary fibrosis. Inhal. Toxicol. 2019, 31, 147–160. [Google Scholar] [CrossRef]
- Solopov, P.; Colunga Biancatelli, R.M.; Dimitropoulou, C.; Catravas, J.D. Sex-Related Differences in Murine Models of Chemically Induced Pulmonary Fibrosis. Int. J. Mol. Sci. 2021, 22, 5909. [Google Scholar] [CrossRef] [PubMed]
- Davidson, J.T.; Rubin, S.; Eyal, Z.; Polliack, A. A comparison of the pulmonary response to the endotracheal instillation of 0.1 N hydrochloric acid and Hartmann’s solution in the rabbit. Br. J. Anaesth. 1974, 46, 127–132. [Google Scholar] [CrossRef][Green Version]
- Uhlig, S.; Yang, Y.; Waade, J.; Wittenberg, C.; Babendreyer, A.; Kuebler, W.M. Differential Regulation of Lung Endothelial Permeability in vitro and in situ. Cell. Physiol. Biochem. 2014, 34, 1–19. [Google Scholar] [CrossRef]
- Joshi, A.D.; Dimitropoulou, C.; Thangjam, G.; Snead, C.; Feldman, S.; Barabutis, N.; Fulton, D.; Hou, Y.; Kumar, S.; Patel, V.; et al. Heat shock protein 90 inhibitors prevent LPS-induced endothelial barrier dysfunction by disrupting RhoA signaling. Am. J. Respir. Cell Mol. Biol. 2014, 50, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Antonov, A.; Snead, C.; Gorshkov, B.; Antonova, G.N.; Verin, A.D.; Catravas, J.D. Heat shock protein 90 inhibitors protect and restore pulmonary endothelial barrier function. Am. J. Respir. Cell Mol. Biol 2008, 39, 551–559. [Google Scholar] [CrossRef]
- Colunga Biancatelli, R.M.; Solopov, P.; Gregory, B.; Catravas, J.D. HSP90 Inhibition and Modulation of the Proteome: Therapeutical Implications for Idiopathic Pulmonary Fibrosis (IPF). Int. J. Mol. Sci. 2020, 21, 5286. [Google Scholar] [CrossRef] [PubMed]
- Marinova, M.; Solopov, P.; Dimitropoulou, C.; Colunga Biancatelli, R.M.L.; Catravas, J.D. Post-treatment with a heat shock protein 90 inhibitor prevents chronic lung injury and pulmonary fibrosis, following acute exposure of mice to HCl. Exp. Lung Res. 2020, 46, 203–216. [Google Scholar] [CrossRef]
- Sibinska, Z.; Tian, X.; Korfei, M.; Kojonazarov, B.; Kolb, J.S.; Klepetko, W.; Kosanovic, D.; Wygrecka, M.; Ghofrani, H.A.; Weissmann, N.; et al. Amplified canonical transforming growth factor-β signalling viaheat shock protein 90 in pulmonary fibrosis. Eur. Respir. J. 2017, 49, 1501941. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Takahashi, T.; Obata, Y.; Nishida, T.; Ohkubo, S.; Nakagawa, F.; Serada, S.; Fujimoto, M.; Ohkawara, T.; Nishigaki, T.; et al. TAS-116 inhibits oncogenic KIT signalling on the Golgi in both imatinib-naïve and imatinib-resistant gastrointestinal stromal tumours. Br. J. Cancer 2020, 122, 658–667. [Google Scholar] [CrossRef]
- Bohush, A.; Bieganowski, P.; Filipek, A. Hsp90 and Its Co-Chaperones in Neurodegenerative Diseases. Int J. Mol. Sci 2019, 20, 4976. [Google Scholar] [CrossRef]
- Luo, Q.; Boczek, E.E.; Wang, Q.; Buchner, J.; Kaila, V.R.I. Hsp90 dependence of a kinase is determined by its conformational landscape. Sci. Rep. 2017, 7, 43996. [Google Scholar] [CrossRef] [PubMed]
- Costa, T.E.M.M.; Raghavendra, N.M.; Penido, C. Natural heat shock protein 90 inhibitors in cancer and inflammation. Eur. J. Med. Chem. 2020, 189, 112063. [Google Scholar] [CrossRef]
- Catravas, J.D.; Snead, C.; Dimitropoulou, C.; Chang, A.S.Y.; Lucas, R.; Verin, A.D.; Black, S.M. Harvesting, identification and barrier function of human lung microvascular endothelial cells. Vasc. Pharmacol. 2010, 52, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Barabutis, N.; Marinova, M.; Solopov, P.; Uddin, M.A.; Croston, G.E.; Reinheimer, T.M.; Catravas, J.D. Protective mechanism of the selective vasopressin V1A receptor agonist selepressin against endothelial barrier dysfunction. J. Pharmacol. Exp. Ther. 2020. [Google Scholar] [CrossRef] [PubMed]
- Karar, J.; Maity, A. PI3K/AKT/mTOR Pathway in Angiogenesis. Front. Mol. Neurosci. 2011, 4, 51. [Google Scholar] [CrossRef]
- Iaccarino, G.; Ciccarelli, M.; Sorriento, D.; Cipolletta, E.; Cerullo, V.; Iovino, G.L.; Paudice, A.; Elia, A.; Santulli, G.; Campanile, A.; et al. AKT participates in endothelial dysfunction in hypertension. Circulation 2004, 109, 2587–2593. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa-Masuda, A.; Terai, K. ERK activation in endothelial cells is a novel marker during neovasculogenesis. Genes Cells 2016, 21, 1164–1175. [Google Scholar] [CrossRef] [PubMed]
- Kawana, K.; Miyamoto, Y.; Tanonaka, K.; Han-no, Y.; Yoshida, H.; Takahashi, M.; Takeo, S. Cytoprotective mechanism of heat shock protein 70 against hypoxia/reoxygenation injury. J. Mol. Cell Cardiol 2000, 32, 2229–2237. [Google Scholar] [CrossRef]
- Thangjam, G.S.; Dimitropoulou, C.; Joshi, A.D.; Barabutis, N.; Shaw, M.C.; Kovalenkov, Y.; Wallace, C.M.; Fulton, D.J.; Patel, V.; Catravas, J.D. Novel mechanism of attenuation of LPS-induced NF-κB activation by the heat shock protein 90 inhibitor, 17-N-allylamino-17-demethoxygeldanamycin, in human lung microvascular endothelial cells. Am. J. Respir. Cell Mol. Biol. 2014, 50, 942–952. [Google Scholar] [CrossRef] [PubMed]
- Thangjam, G.S.; Birmpas, C.; Barabutis, N.; Gregory, B.W.; Clemens, M.A.; Newton, J.R.; Fulton, D.; Catravas, J.D. Hsp90 inhibition suppresses NF-κB transcriptional activation via Sirt-2 in human lung microvascular endothelial cells. Am. J. Physiol. -Lung Cell. Mol. Physiol. 2016, 310, L964–L974. [Google Scholar] [CrossRef]
- Solopov, P.; Biancatelli, R.M.; Marinova, M.; Dimitropoulou, C.; Catravas, J.D. The HSP90 Inhibitor, AUY-922, Ameliorates the Development of Nitrogen Mustard-Induced Pulmonary Fibrosis and Lung Dysfunction in Mice. Int. J. Mol. Sci. 2020, 21, 4740. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colunga Biancatelli, R.M.L.; Solopov, P.; Gregory, B.; Catravas, J.D. The HSP90 Inhibitor, AUY-922, Protects and Repairs Human Lung Microvascular Endothelial Cells from Hydrochloric Acid-Induced Endothelial Barrier Dysfunction. Cells 2021, 10, 1489. https://doi.org/10.3390/cells10061489
Colunga Biancatelli RML, Solopov P, Gregory B, Catravas JD. The HSP90 Inhibitor, AUY-922, Protects and Repairs Human Lung Microvascular Endothelial Cells from Hydrochloric Acid-Induced Endothelial Barrier Dysfunction. Cells. 2021; 10(6):1489. https://doi.org/10.3390/cells10061489
Chicago/Turabian StyleColunga Biancatelli, Ruben M. L., Pavel Solopov, Betsy Gregory, and John D. Catravas. 2021. "The HSP90 Inhibitor, AUY-922, Protects and Repairs Human Lung Microvascular Endothelial Cells from Hydrochloric Acid-Induced Endothelial Barrier Dysfunction" Cells 10, no. 6: 1489. https://doi.org/10.3390/cells10061489
APA StyleColunga Biancatelli, R. M. L., Solopov, P., Gregory, B., & Catravas, J. D. (2021). The HSP90 Inhibitor, AUY-922, Protects and Repairs Human Lung Microvascular Endothelial Cells from Hydrochloric Acid-Induced Endothelial Barrier Dysfunction. Cells, 10(6), 1489. https://doi.org/10.3390/cells10061489