
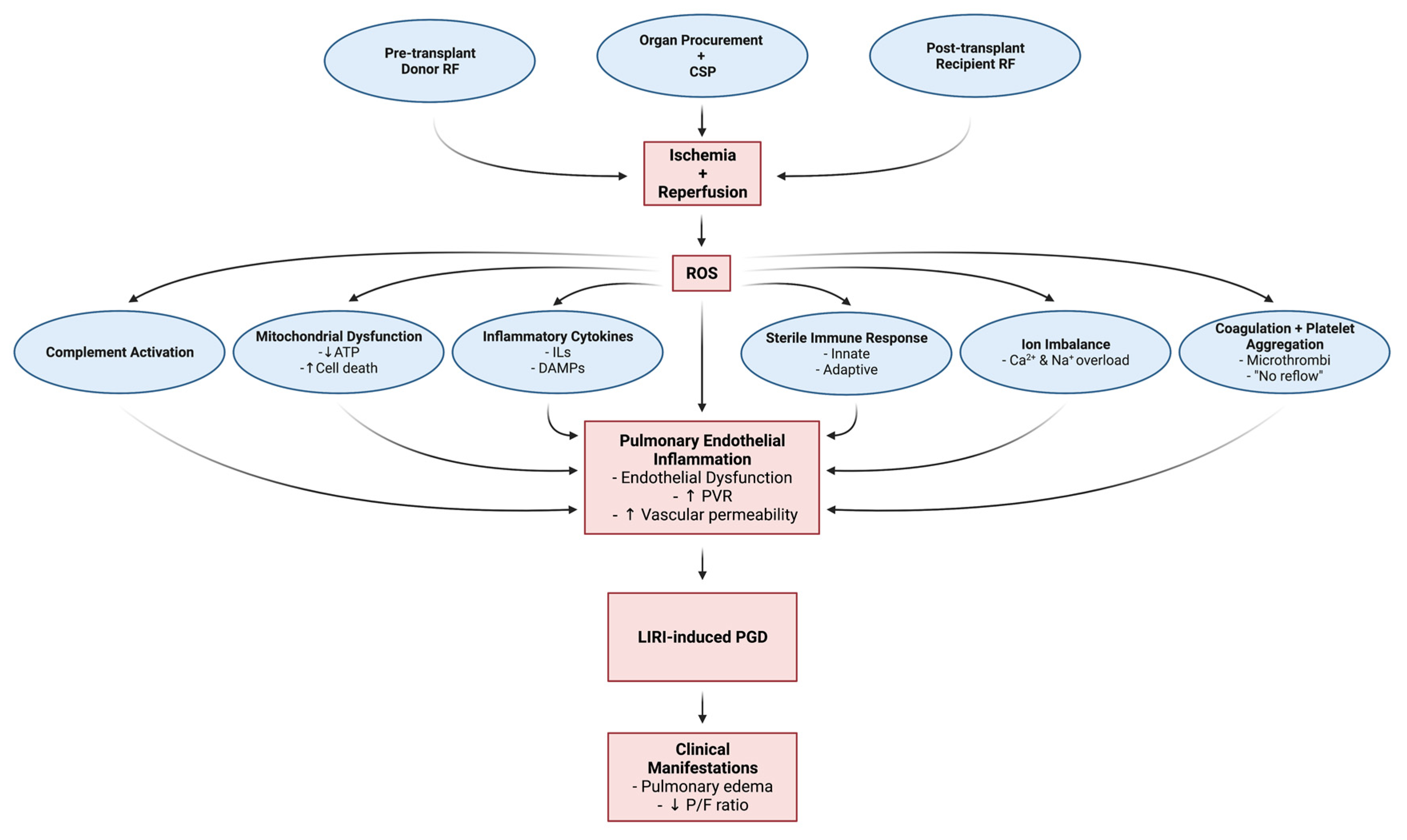
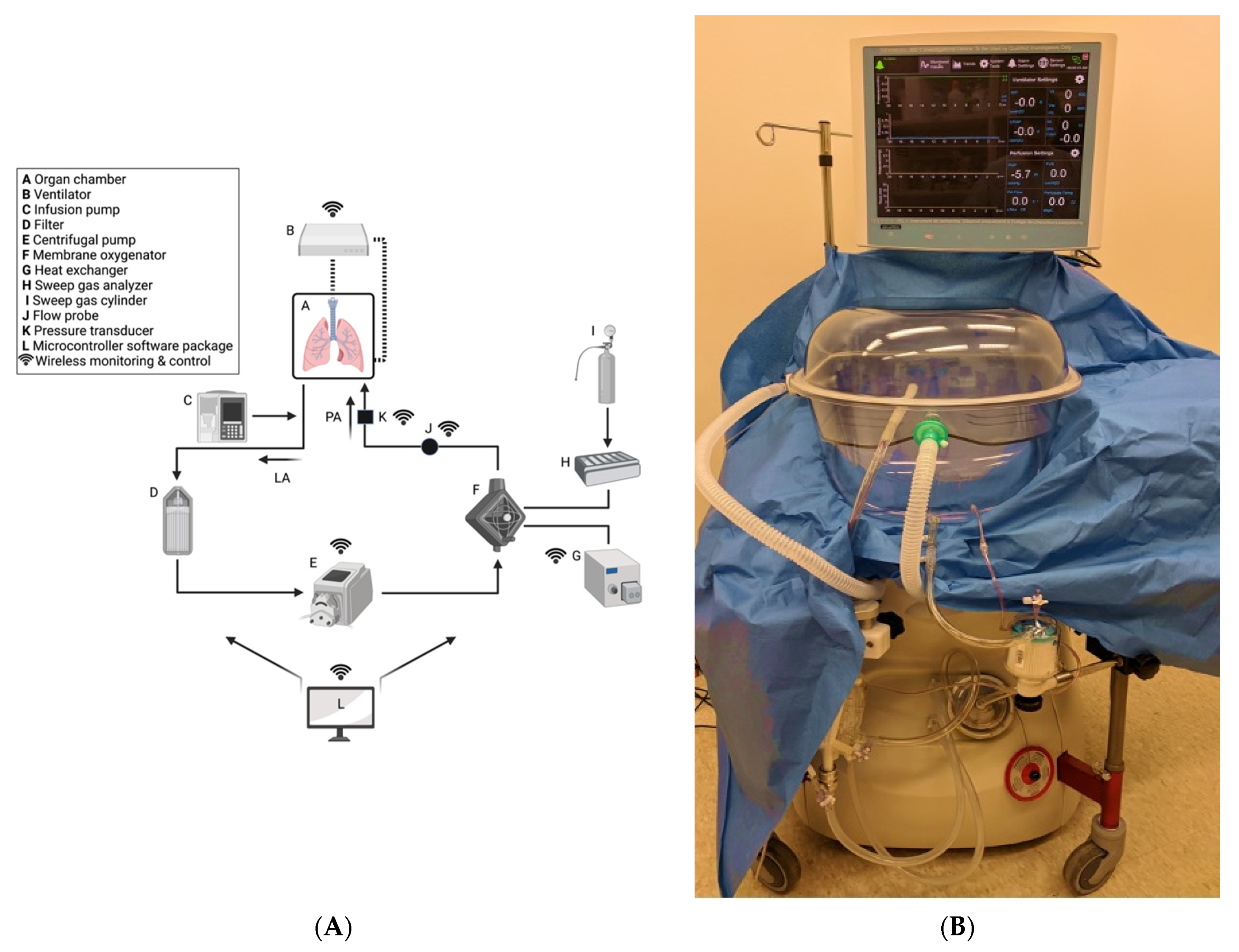
Lung Transplantation, Pulmonary Endothelial Inflammation, and Ex-Situ Lung Perfusion: A Review


{kind=link}
{kind=link}
Abstract
Share and Cite
Forgie, K.A.; Fialka, N.; Freed, D.H.; Nagendran, J. Lung Transplantation, Pulmonary Endothelial Inflammation, and Ex-Situ Lung Perfusion: A Review. Cells 2021, 10, 1417. https://doi.org/10.3390/cells10061417
Forgie KA, Fialka N, Freed DH, Nagendran J. Lung Transplantation, Pulmonary Endothelial Inflammation, and Ex-Situ Lung Perfusion: A Review. Cells. 2021; 10(6):1417. https://doi.org/10.3390/cells10061417
Chicago/Turabian StyleForgie, Keir A., Nicholas Fialka, Darren H. Freed, and Jayan Nagendran. 2021. "Lung Transplantation, Pulmonary Endothelial Inflammation, and Ex-Situ Lung Perfusion: A Review" Cells 10, no. 6: 1417. https://doi.org/10.3390/cells10061417
APA StyleForgie, K. A., Fialka, N., Freed, D. H., & Nagendran, J. (2021). Lung Transplantation, Pulmonary Endothelial Inflammation, and Ex-Situ Lung Perfusion: A Review. Cells, 10(6), 1417. https://doi.org/10.3390/cells10061417