
Traumatic Brain Injury: Ultrastructural Features in Neuronal Ferroptosis, Glial Cell Activation and Polarization, and Blood–Brain Barrier Breakdown


Abstract
1. Introduction
2. TBI-Related Ultrastructural Damage to Neurons
2.1. Hydropic Disintegration of Neuronal Body and Process
2.2. Axon Destruction, Demyelination, and Myelination
2.3. Mitochondrial Abnormality
2.4. Endoplasmic Reticulum Dissolution
2.5. Cytoskeleton Destruction
3. Effects of TBI on Glial Cell Ultrastructure
3.1. Structural Changes of Astrocytes
3.2. Structural Changes of Microglia
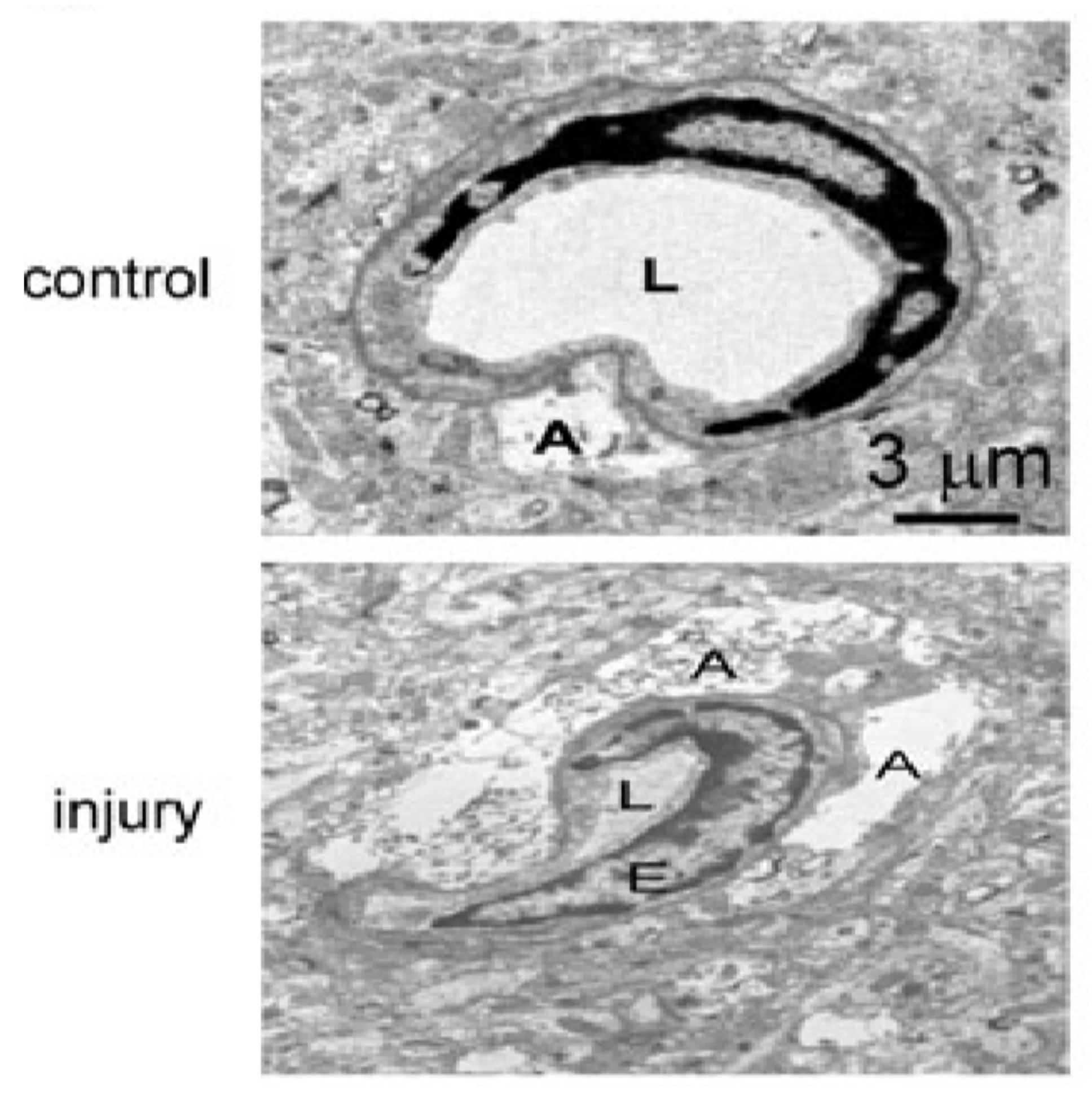
4. TBI-Related Ultrastructural Damage to Blood–Brain Barrier
5. TBI-Induced Ferroptosis
6. Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wiley, C.A.; Bissel, S.J.; Lesniak, A.; Dixon, C.E.; Franks, J.; Beer Stolz, D.; Sun, M.; Wang, G.; Switzer, R.; Kochanek, P.M.; et al. Ultrastructure of Diaschisis Lesions after Traumatic Brain Injury. J. Neurotrauma 2016, 33, 1866–1882. [Google Scholar] [CrossRef] [PubMed]
- Dewan, M.C.; Rattani, A.; Gupta, S.; Baticulon, R.E.; Hung, Y.C.; Punchak, M.; Agrawal, A.; Adeleye, A.O.; Shrime, M.G.; Rubiano, A.M.; et al. Estimating the global incidence of traumatic brain injury. J. Neurosurg. 2018, 1, 1–18. [Google Scholar] [CrossRef]
- Liu, B. Current status and development of traumatic brain injury treatments in China. Chin. J. Traumatol. 2015, 18, 135–136. [Google Scholar] [CrossRef] [PubMed]
- Georges, A.; Das, J.M. Traumatic Brain Injury; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Maas, A.I.R.; Menon, D.K.; Adelson, P.D.; Andelic, N.; Bell, M.J.; Belli, A.; Bragge, P.; Brazinova, A.; Buki, A.; Chesnut, R.M.; et al. Traumatic brain injury: Integrated approaches to improve prevention, clinical care, and research. Lancet Neurol. 2017, 16, 987–1048. [Google Scholar] [CrossRef] [PubMed]
- Janak, J.C.; Pugh, M.J.; Orman, J.A.L. Epidemiology of traumatic brain injury. In Traumatic Brain Injury Rehabilitation Medicine; Future Medicine Ltd.: London, UK, 2015; pp. 6–35. [Google Scholar] [CrossRef]
- Xiong, Y.; Mahmood, A.; Chopp, M. Current understanding of neuroinflammation after traumatic brain injury and cell-based therapeutic opportunities. Chin. J. Traumatol. 2018, 21, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Jha, R.M.; Kochanek, P.M.; Simard, J.M. Pathophysiology and treatment of cerebral edema in traumatic brain injury. Neuropharmacology 2019, 145, 230–246. [Google Scholar] [CrossRef] [PubMed]
- Salehi, A.; Zhang, J.H.; Obenaus, A. Response of the cerebral vasculature following traumatic brain injury. J. Cereb. Blood Flow Metab. 2017, 37, 2320–2339. [Google Scholar] [CrossRef] [PubMed]
- Stokum, J.A.; Gerzanich, V.; Simard, J.M. Molecular pathophysiology of cerebral edema. J. Cereb. Blood Flow Metab. 2016, 36, 513–538. [Google Scholar] [CrossRef]
- Zhang, J.; Pu, H.; Zhang, H.; Wei, Z.; Jiang, X.; Xu, M.; Zhang, L.; Zhang, W.; Liu, J.; Meng, H.; et al. Inhibition of Na(+)-K(+)-2Cl(-) cotransporter attenuates blood-brain-barrier disruption in a mouse model of traumatic brain injury. Neurochem Int. 2017, 111, 23–31. [Google Scholar] [CrossRef]
- Simard, J.M.; Kahle, K.T.; Gerzanich, V. Molecular mechanisms of microvascular failure in central nervous system injury—Synergistic roles of NKCC1 and SUR1/TRPM4. J. Neurosurg. 2010, 113, 622–629. [Google Scholar] [CrossRef]
- Hubbard, J.A.; Szu, J.I.; Binder, D.K. The role of aquaporin-4 in synaptic plasticity, memory and disease. Brain Res. Bull. 2018, 136, 118–129. [Google Scholar] [CrossRef]
- Patel, A.D.; Gerzanich, V.; Geng, Z.; Simard, J.M. Glibenclamide reduces hippocampal injury and preserves rapid spatial learning in a model of traumatic brain injury. J. Neuropathol. Exp. Neurol. 2010, 69, 1177–1190. [Google Scholar] [CrossRef]
- Simard, J.M.; Kilbourne, M.; Tsymbalyuk, O.; Tosun, C.; Caridi, J.; Ivanova, S.; Keledjian, K.; Bochicchio, G.; Gerzanich, V. Key role of sulfonylurea receptor 1 in progressive secondary hemorrhage after brain contusion. J. Neurotrauma 2009, 26, 2257–2267. [Google Scholar] [CrossRef]
- Winkler, E.A.; Minter, D.; Yue, J.K.; Manley, G.T. Cerebral Edema in Traumatic Brain Injury: Pathophysiology and Prospective Therapeutic Targets. Neurosurg. Clin. N. Am. 2016, 27, 473–488. [Google Scholar] [CrossRef] [PubMed]
- Krieg, S.M.; Trabold, R.; Plesnila, N. Time-Dependent Effects of Arginine-Vasopressin V1 Receptor Inhibition on Secondary Brain Damage after Traumatic Brain Injury. J. Neurotrauma 2017, 34, 1329–1336. [Google Scholar] [CrossRef] [PubMed]
- Filippidis, A.S.; Liang, X.; Wang, W.; Parveen, S.; Baumgarten, C.M.; Marmarou, C.R. Real-time monitoring of changes in brain extracellular sodium and potassium concentrations and intracranial pressure after selective vasopressin-1a receptor inhibition following focal traumatic brain injury in rats. J. Neurotrauma 2014, 31, 1258–1267. [Google Scholar] [CrossRef] [PubMed]
- Pedachenko, E.G.; Biloshytsky, V.V.; Mikhal’sky, S.A.; Gridina, N.Y.; Kvitnitskaya-Ryzhova, T.Y. The effect of gene therapy with the APOE3 Gene on structural and functional manifestations of secondary hippocampal damages in experimental traumatic brain injury. Zh. Vopr. Neirokhir. Im. NN Burdenko 2015, 79, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Mierzwa, A.J.; Marion, C.M.; Sullivan, G.M.; McDaniel, D.P.; Armstrong, R.C. Components of myelin damage and repair in the progression of white matter pathology after mild traumatic brain injury. J. Neuropathol. Exp. Neurol. 2015, 74, 218–232. [Google Scholar] [CrossRef]
- Song, H.; Konan, L.M.; Cui, J.; Johnson, C.E.; Hubler, G.K.; DePalma, R.G.; Gu, Z. Nanometer ultrastructural brain damage following low intensity primary blast wave exposure. Neural. Regen. Res. 2018, 13, 1516–1519. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Konan, L.M.; Cui, J.; Johnson, C.E.; Langenderfer, M.; Grant, D.; Ndam, T.; Simonyi, A.; White, T.; Demirci, U.; et al. Ultrastructural brain abnormalities and associated behavioral changes in mice after low-intensity blast exposure. Behav. Brain Res. 2018, 347, 148–157. [Google Scholar] [CrossRef]
- Dams-O’Connor, K.; Spielman, L.; Singh, A.; Gordon, W.A.; Lingsma, H.F.; Maas, A.I.; Manley, G.T.; Mukherjee, P.; Okonkwo, D.O.; Puccio, A.M.; et al. The impact of previous traumatic brain injury on health and functioning: A TRACK-TBI study. J. Neurotrauma 2013, 30, 2014–2020. [Google Scholar] [CrossRef] [PubMed]
- Donders, J.; Strong, C.A. Clinical utility of the Wechsler Adult Intelligence Scale-Fourth Edition after traumatic brain injury. Assessment 2015, 22, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Clarner, T.; Diederichs, F.; Berger, K.; Denecke, B.; Gan, L.; van der Valk, P.; Beyer, C.; Amor, S.; Kipp, M. Myelin debris regulates inflammatory responses in an experimental demyelination animal model and multiple sclerosis lesions. Glia 2012, 60, 1468–1480. [Google Scholar] [CrossRef]
- Snaidero, N.; Mobius, W.; Czopka, T.; Hekking, L.H.; Mathisen, C.; Verkleij, D.; Goebbels, S.; Edgar, J.; Merkler, D.; Lyons, D.A.; et al. Myelin membrane wrapping of CNS axons by PI(3,4,5)P3-dependent polarized growth at the inner tongue. Cell 2014, 156, 277–290. [Google Scholar] [CrossRef]
- Sullivan, G.M.; Mierzwa, A.J.; Kijpaisalratana, N.; Tang, H.; Wang, Y.; Song, S.K.; Selwyn, R.; Armstrong, R.C. Oligodendrocyte lineage and subventricular zone response to traumatic axonal injury in the corpus callosum. J. Neuropathol. Exp. Neurol. 2013, 72, 1106–1125. [Google Scholar] [CrossRef]
- Bruce, C.C.; Zhao, C.; Franklin, R.J. Remyelination - An effective means of neuroprotection. Horm. Behav. 2010, 57, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Irvine, K.A.; Blakemore, W.F. Remyelination protects axons from demyelination-associated axon degeneration. Brain 2008, 131, 1464–1477. [Google Scholar] [CrossRef] [PubMed]
- Balan, I.S.; Saladino, A.J.; Aarabi, B.; Castellani, R.J.; Wade, C.; Stein, D.M.; Eisenberg, H.M.; Chen, H.H.; Fiskum, G. Cellular alterations in human traumatic brain injury: Changes in mitochondrial morphology reflect regional levels of injury severity. J. Neurotrauma 2013, 30, 367–381. [Google Scholar] [CrossRef]
- Watson, W.D.; Buonora, J.E.; Yarnell, A.M.; Lucky, J.J.; D’Acchille, M.I.; McMullen, D.C.; Boston, A.G.; Kuczmarski, A.V.; Kean, W.S.; Verma, A.; et al. Impaired cortical mitochondrial function following TBI precedes behavioral changes. Front. Neuroenerg. 2013, 5, 12. [Google Scholar] [CrossRef]
- Wang, Z.; Jiang, H.; Chen, S.; Du, F.; Wang, X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 2012, 148, 228–243. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Wang, Q.; Fan, W.; Cai, Y.; Wu, Q.; Mo, L.; Huang, Z.; Huang, H. Protective effects of taurine in traumatic brain injury via mitochondria and cerebral blood flow. Amino Acids 2016, 48, 2169–2177. [Google Scholar] [CrossRef]
- Wu, Q.; Xia, S.X.; Li, Q.Q.; Gao, Y.; Shen, X.; Ma, L.; Zhang, M.Y.; Wang, T.; Li, Y.S.; Wang, Z.F.; et al. Mitochondrial division inhibitor 1 (Mdivi-1) offers neuroprotection through diminishing cell death and improving functional outcome in a mouse model of traumatic brain injury. Brain Res. 2016, 1630, 134–143. [Google Scholar] [CrossRef]
- Bereiter-Hahn, J.; Jendrach, M. Mitochondrial dynamics. Int. Rev. Cell Mol. Biol. 2010, 284, 1–65. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hajnoczky, G. Altered fusion dynamics underlie unique morphological changes in mitochondria during hypoxia-reoxygenation stress. Cell Death Differ. 2011, 18, 1561–1572. [Google Scholar] [CrossRef] [PubMed]
- Lyamzaev, K.G.; Nepryakhina, O.K.; Saprunova, V.B.; Bakeeva, L.E.; Pletjushkina, O.Y.; Chernyak, B.V.; Skulachev, V.P. Novel mechanism of elimination of malfunctioning mitochondria (mitoptosis): Formation of mitoptotic bodies and extrusion of mitochondrial material from the cell. Biochim. Biophys. Acta 2008, 1777, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Mannella, C.A. The relevance of mitochondrial membrane topology to mitochondrial function. Biochim. Biophys. Acta 2006, 1762, 140–147. [Google Scholar] [CrossRef]
- Zick, M.; Rabl, R.; Reichert, A.S. Cristae formation-linking ultrastructure and function of mitochondria. Biochim. Biophys. Acta 2009, 1793, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Kawane, K.; Motani, K.; Nagata, S. DNA degradation and its defects. Cold Spring Harb. Perspect. Biol. 2014, 6. [Google Scholar] [CrossRef]
- Santucci, R.; Sinibaldi, F.; Cozza, P.; Polticelli, F.; Fiorucci, L. Cytochrome c: An extreme multifunctional protein with a key role in cell fate. Int. J. Biol. Macromol. 2019, 136, 1237–1246. [Google Scholar] [CrossRef] [PubMed]
- Scorrano, L.; Ashiya, M.; Buttle, K.; Weiler, S.; Oakes, S.A.; Mannella, C.A.; Korsmeyer, S.J. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev. Cell 2002, 2, 55–67. [Google Scholar] [CrossRef]
- Estaquier, J.; Vallette, F.; Vayssiere, J.L.; Mignotte, B. The mitochondrial pathways of apoptosis. Adv. Exp. Med. Biol. 2012, 942, 157–183. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, A.; Scorrano, L. Mitochondria: From cell death executioners to regulators of cell differentiation. Trends Cell Biol. 2014, 24, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Frank, S.; Gaume, B.; Bergmann-Leitner, E.S.; Leitner, W.W.; Robert, E.G.; Catez, F.; Smith, C.L.; Youle, R.J. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev. Cell 2001, 1, 515–525. [Google Scholar] [CrossRef]
- Jagasia, R.; Grote, P.; Westermann, B.; Conradt, B. DRP-1-mediated mitochondrial fragmentation during EGL-1-induced cell death in C. elegans. Nature 2005, 433, 754–760. [Google Scholar] [CrossRef]
- Wasiak, S.; Zunino, R.; McBride, H.M. Bax/Bak promote sumoylation of DRP1 and its stable association with mitochondria during apoptotic cell death. J. Cell Biol. 2007, 177, 439–450. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Muneer, P.M.; Chandra, N.; Haorah, J. Interactions of oxidative stress and neurovascular inflammation in the pathogenesis of traumatic brain injury. Mol. Neurobiol. 2015, 51, 966–979. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Hiebert, J.B.; Shen, Q.; Thimmesch, A.R.; Pierce, J.D. Traumatic brain injury and mitochondrial dysfunction. Am. J. Med. Sci. 2015, 350, 132–138. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Khatri, N.; Thakur, M.; Pareek, V.; Kumar, S.; Sharma, S.; Datusalia, A.K. Oxidative Stress: Major Threat in Traumatic Brain Injury. CNS Neurol. Disord. Drug Targets 2018, 17, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, S.; Umeda, M.; Uchida, H.; Kato, H.; Araki, T. Alterations of oxidative stress markers and apoptosis markers in the striatum after transient focal cerebral ischemia in rats. J. Neural. Transm. 2009, 116, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Semple, B.D. Early preservation of mitochondrial bioenergetics supports both structural and functional recovery after neurotrauma. Exp. Neurol. 2014, 261, 291–297. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, H.; Xu, J.; Zhao, D. Neuritin provides neuroprotection against experimental traumatic brain injury in rats. Int. J. Neurosci. 2018, 128, 811–820. [Google Scholar] [CrossRef]
- Chevet, E.; Hetz, C.; Samali, A. Endoplasmic reticulum stress-activated cell reprogramming in oncogenesis. Cancer Discov. 2015, 5, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Banhegyi, G.; Margittai, E.; Szarka, A.; Mandl, J.; Csala, M. Crosstalk and barriers between the electron carriers of the endoplasmic reticulum. Antioxid. Redox Signal 2012, 16, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Bramlett, H.M.; Dietrich, W.D. Progressive damage after brain and spinal cord injury: Pathomechanisms and treatment strategies. Prog. Brain Res. 2007, 161, 125–141. [Google Scholar] [CrossRef]
- Stoica, B.A.; Faden, A.I. Cell death mechanisms and modulation in traumatic brain injury. Neurotherapeutics 2010, 7, 3–12. [Google Scholar] [CrossRef]
- Krebs, J.; Agellon, L.B.; Michalak, M. Ca(2+) homeostasis and endoplasmic reticulum (ER) stress: An integrated view of calcium signaling. Biochem. Biophys. Res. Commun. 2015, 460, 114–121. [Google Scholar] [CrossRef]
- Berridge, M.J. The endoplasmic reticulum: A multifunctional signaling organelle. Cell Calcium 2002, 32, 235–249. [Google Scholar] [CrossRef]
- Paschen, W. Endoplasmic reticulum: A primary target in various acute disorders and degenerative diseases of the brain. Cell Calcium 2003, 34, 365–383. [Google Scholar] [CrossRef]
- Heath-Engel, H.M.; Wang, B.; Shore, G.C. Bcl2 at the endoplasmic reticulum protects against a Bax/Bak-independent paraptosis-like cell death pathway initiated via p20Bap31. Biochim. Biophys. Acta 2012, 1823, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Oakes, S.A.; Papa, F.R. The role of endoplasmic reticulum stress in human pathology. Annu. Rev. Pathol. 2015, 10, 173–194. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Kaufman, R.J. The impact of the unfolded protein response on human disease. J. Cell Biol. 2012, 197, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Sakaki, K.; Yoshina, S.; Shen, X.; Han, J.; DeSantis, M.R.; Xiong, M.; Mitani, S.; Kaufman, R.J. RNA surveillance is required for endoplasmic reticulum homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 8079–8084. [Google Scholar] [CrossRef] [PubMed]
- DeGracia, D.J.; Montie, H.L. Cerebral ischemia and the unfolded protein response. J. Neurochem. 2004, 91, 1–8. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef]
- Banhegyi, G.; Baumeister, P.; Benedetti, A.; Dong, D.; Fu, Y.; Lee, A.S.; Li, J.; Mao, C.; Margittai, E.; Ni, M.; et al. Endoplasmic reticulum stress. Ann. N. Y. Acad. Sci. 2007, 1113, 58–71. [Google Scholar] [CrossRef]
- Truettner, J.S.; Hu, B.; Alonso, O.F.; Bramlett, H.M.; Kokame, K.; Dietrich, W.D. Subcellular stress response after traumatic brain injury. J. Neurotrauma 2007, 24, 599–612. [Google Scholar] [CrossRef]
- Chen, X.; Kintner, D.B.; Luo, J.; Baba, A.; Matsuda, T.; Sun, D. Endoplasmic reticulum Ca2+ dysregulation and endoplasmic reticulum stress following in vitro neuronal ischemia: Role of Na+-K+-Cl- cotransporter. J. Neurochem. 2008, 106, 1563–1576. [Google Scholar] [CrossRef]
- Hohmann, T.; Dehghani, F. The Cytoskeleton-A Complex Interacting Meshwork. Cells 2019, 8, 362. [Google Scholar] [CrossRef]
- Ogawa, Y.; Schafer, D.P.; Horresh, I.; Bar, V.; Hales, K.; Yang, Y.; Susuki, K.; Peles, E.; Stankewich, M.C.; Rasband, M.N. Spectrins and ankyrinB constitute a specialized paranodal cytoskeleton. J. Neurosci. 2006, 26, 5230–5239. [Google Scholar] [CrossRef]
- Schafer, D.P.; Jha, S.; Liu, F.; Akella, T.; McCullough, L.D.; Rasband, M.N. Disruption of the axon initial segment cytoskeleton is a new mechanism for neuronal injury. J. Neurosci. 2009, 29, 13242–13254. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.T.; Barres, B.A. Axon degeneration: Where the Wlds things are. Curr. Biol. 2012, 22, R221–R223. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vargas, M.E.; Barres, B.A. Why is Wallerian degeneration in the CNS so slow? Annu. Rev. Neurosci. 2007, 30, 153–179. [Google Scholar] [CrossRef] [PubMed]
- Christman, C.W.; Salvant, J.B.; Walker, S.A.; Povlishock, J.T. Characterization of a prolonged regenerative attempt by diffusely injured axons following traumatic brain injury in adult cat: A light and electron microscopic immunocytochemical study. Acta Neuropathol. 1997, 94, 329–337. [Google Scholar] [CrossRef]
- Saatman, K.E.; Abai, B.; Grosvenor, A.; Vorwerk, C.K.; Smith, D.H.; Meaney, D.F. Traumatic axonal injury results in biphasic calpain activation and retrograde transport impairment in mice. J. Cereb. Blood Flow Metab. 2003, 23, 34–42. [Google Scholar] [CrossRef]
- Blennow, K.; Brody, D.L.; Kochanek, P.M.; Levin, H.; McKee, A.; Ribbers, G.M.; Yaffe, K.; Zetterberg, H. Traumatic brain injuries. Nat. Rev. Dis. Primers 2016, 2, 16084. [Google Scholar] [CrossRef]
- Buki, A.; Povlishock, J.T. All roads lead to disconnection?—Traumatic axonal injury revisited. Acta Neurochir. 2006, 148, 181–193. [Google Scholar] [CrossRef]
- Smith, D.H.; Meaney, D.F.; Shull, W.H. Diffuse axonal injury in head trauma. J. Head Trauma Rehabil. 2003, 18, 307–316. [Google Scholar] [CrossRef]
- Ma, M. Role of calpains in the injury-induced dysfunction and degeneration of the mammalian axon. Neurobiol. Dis. 2013, 60, 61–79. [Google Scholar] [CrossRef] [PubMed]
- Perrot, R.; Berges, R.; Bocquet, A.; Eyer, J. Review of the multiple aspects of neurofilament functions, and their possible contribution to neurodegeneration. Mol. Neurobiol. 2008, 38, 27–65. [Google Scholar] [CrossRef]
- Yuan, A.; Rao, M.V.; Veeranna; Nixon, R.A. Neurofilaments at a glance. J. Cell Sci. 2012, 125, 3257–3263. [Google Scholar] [CrossRef] [PubMed]
- Siedler, D.G.; Chuah, M.I.; Kirkcaldie, M.T.; Vickers, J.C.; King, A.E. Diffuse axonal injury in brain trauma: Insights from alterations in neurofilaments. Front. Cell Neurosci. 2014, 8, 429. [Google Scholar] [CrossRef]
- Ma, M.; Ferguson, T.A.; Schoch, K.M.; Li, J.; Qian, Y.; Shofer, F.S.; Saatman, K.E.; Neumar, R.W. Calpains mediate axonal cytoskeleton disintegration during Wallerian degeneration. Neurobiol. Dis. 2013, 56, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Villapol, S.; Byrnes, K.R.; Symes, A.J. Temporal dynamics of cerebral blood flow, cortical damage, apoptosis, astrocyte-vasculature interaction and astrogliosis in the pericontusional region after traumatic brain injury. Front. Neurol. 2014, 5, 82. [Google Scholar] [CrossRef]
- Susarla, B.T.; Villapol, S.; Yi, J.H.; Geller, H.M.; Symes, A.J. Temporal patterns of cortical proliferation of glial cell populations after traumatic brain injury in mice. ASN Neuro 2014, 6, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Uchida, K.; Papadopoulos, M.C.; Zador, Z.; Manley, G.T.; Verkman, A.S. Mildly Reduced Brain Swelling and Improved Neurological Outcome in Aquaporin-4 Knockout Mice following Controlled Cortical Impact Brain Injury. J. Neurotrauma 2015, 32, 1458–1464. [Google Scholar] [CrossRef]
- Shitaka, Y.; Tran, H.T.; Bennett, R.E.; Sanchez, L.; Levy, M.A.; Dikranian, K.; Brody, D.L. Repetitive closed-skull traumatic brain injury in mice causes persistent multifocal axonal injury and microglial reactivity. J. Neuropathol. Exp. Neurol. 2011, 70, 551–567. [Google Scholar] [CrossRef]
- Burda, J.E.; Sofroniew, M.V. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 2014, 81, 229–248. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Swanson, R.A. Astrocytes and brain injury. J. Cereb. Blood Flow Metab. 2003, 23, 137–149. [Google Scholar] [CrossRef]
- Abbott, N.J.; Ronnback, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Burda, J.E.; Bernstein, A.M.; Sofroniew, M.V. Astrocyte roles in traumatic brain injury. Exp. Neurol. 2016, 275 Pt 3, 305–315. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Astrocyte barriers to neurotoxic inflammation. Nat. Rev. Neurosci. 2015, 16, 249–263. [Google Scholar] [CrossRef]
- Wang, J.; Dore, S. Inflammation after intracerebral hemorrhage. J. Cereb. Blood Flow Metab. 2007, 27, 894–908. [Google Scholar] [CrossRef]
- Zhu, W.; Gao, Y.; Wan, J.; Lan, X.; Han, X.; Zhu, S.; Zang, W.; Chen, X.; Ziai, W.; Hanley, D.F.; et al. Changes in motor function, cognition, and emotion-related behavior after right hemispheric intracerebral hemorrhage in various brain regions of mouse. Brain Behav. Immun. 2018, 69, 568–581. [Google Scholar] [CrossRef]
- Lan, X.; Han, X.; Liu, X.; Wang, J. Inflammatory responses after intracerebral hemorrhage: From cellular function to therapeutic targets. J. Cereb. Blood Flow Metab. 2019, 39, 184–186. [Google Scholar] [CrossRef]
- Wang, J. Preclinical and clinical research on inflammation after intracerebral hemorrhage. Prog. Neurobiol. 2010, 92, 463–477. [Google Scholar] [CrossRef]
- Karve, I.P.; Taylor, J.M.; Crack, P.J. The contribution of astrocytes and microglia to traumatic brain injury. Br. J. Pharmacol. 2016, 173, 692–702. [Google Scholar] [CrossRef]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef]
- Gorina, R.; Font-Nieves, M.; Marquez-Kisinousky, L.; Santalucia, T.; Planas, A.M. Astrocyte TLR4 activation induces a proinflammatory environment through the interplay between MyD88-dependent NFkappaB signaling, MAPK, and Jak1/Stat1 pathways. Glia 2011, 59, 242–255. [Google Scholar] [CrossRef]
- Paintlia, M.K.; Paintlia, A.S.; Singh, A.K.; Singh, I. S-nitrosoglutathione induces ciliary neurotrophic factor expression in astrocytes, which has implications to protect the central nervous system under pathological conditions. J. Biol. Chem. 2013, 288, 3831–3843. [Google Scholar] [CrossRef]
- Zamanian, J.L.; Xu, L.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic analysis of reactive astrogliosis. J. Neurosci. 2012, 32, 6391–6410. [Google Scholar] [CrossRef]
- Pan, L.N.; Zhu, W.; Li, Y.; Xu, X.L.; Guo, L.J.; Lu, Q.; Wang, J. Astrocytic Toll-like receptor 3 is associated with ischemic preconditioning-induced protection against brain ischemia in rodents. PLoS One 2014, 9, e99526. [Google Scholar] [CrossRef]
- Wu, T.; Wu, H.; Wang, J.; Wang, J. Expression and cellular localization of cyclooxygenases and prostaglandin E synthases in the hemorrhagic brain. J Neuroinflamm. 2011, 8, 22. [Google Scholar] [CrossRef]
- Lan, X.; Han, X.; Li, Q.; Wang, J. (-)-Epicatechin, a Natural Flavonoid Compound, Protects Astrocytes Against Hemoglobin Toxicity via Nrf2 and AP-1 Signaling Pathways. Mol. Neurobiol. 2017, 54, 7898–7907. [Google Scholar] [CrossRef]
- Jayakumar, A.R.; Tong, X.Y.; Ruiz-Cordero, R.; Bregy, A.; Bethea, J.R.; Bramlett, H.M.; Norenberg, M.D. Activation of NF-kappaB mediates astrocyte swelling and brain edema in traumatic brain injury. J. Neurotrauma 2014, 31, 1249–1257. [Google Scholar] [CrossRef]
- Unterberg, A.W.; Stover, J.; Kress, B.; Kiening, K.L. Edema and brain trauma. Neuroscience 2004, 129, 1021–1029. [Google Scholar] [CrossRef]
- Portella, G.; Beaumont, A.; Corwin, F.; Fatouros, P.; Marmarou, A. Characterizing edema associated with cortical contusion and secondary insult using magnetic resonance spectroscopy. Acta Neurochir. Suppl. 2000, 76, 273–275. [Google Scholar] [CrossRef]
- Beaumont, A.; Fatouros, P.; Gennarelli, T.; Corwin, F.; Marmarou, A. Bolus tracer delivery measured by MRI confirms edema without blood-brain barrier permeability in diffuse traumatic brain injury. Acta Neurochir. Suppl. 2006, 96, 171–174. [Google Scholar] [CrossRef]
- Laird, M.D.; Shields, J.S.; Sukumari-Ramesh, S.; Kimbler, D.E.; Fessler, R.D.; Shakir, B.; Youssef, P.; Yanasak, N.; Vender, J.R.; Dhandapani, K.M. High mobility group box protein-1 promotes cerebral edema after traumatic brain injury via activation of toll-like receptor 4. Glia 2014, 62, 26–38. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Astrogliosis. Cold Spring Harb. Perspect. Biol. 2014, 7, a020420. [Google Scholar] [CrossRef]
- Vajtr, D.; Benada, O.; Kukacka, J.; Prusa, R.; Houstava, L.; Toupalik, P.; Kizek, R. Correlation of ultrastructural changes of endothelial cells and astrocytes occurring during blood brain barrier damage after traumatic brain injury with biochemical markers of BBB leakage and inflammatory response. Physiol. Res. 2009, 58, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Stoica, B.A.; Loane, D.J.; Yang, M.; Abulwerdi, G.; Khan, N.; Kumar, A.; Thom, S.R.; Faden, A.I. Microglial-derived microparticles mediate neuroinflammation after traumatic brain injury. J. Neuroinflamm. 2017, 14, 47. [Google Scholar] [CrossRef]
- Roth, T.L.; Nayak, D.; Atanasijevic, T.; Koretsky, A.P.; Latour, L.L.; McGavern, D.B. Transcranial amelioration of inflammation and cell death after brain injury. Nature 2014, 505, 223–228. [Google Scholar] [CrossRef]
- Corps, K.N.; Roth, T.L.; McGavern, D.B. Inflammation and neuroprotection in traumatic brain injury. JAMA Neurol. 2015, 72, 355–362. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Z.; Lu, H.; Yang, Q.; Wu, H.; Wang, J. Microglial Polarization and Inflammatory Mediators After Intracerebral Hemorrhage. Mol. Neurobiol. 2017, 54, 1874–1886. [Google Scholar] [CrossRef]
- Ren, H.; Han, R.; Chen, X.; Liu, X.; Wan, J.; Wang, L.; Yang, X.; Wang, J. Potential therapeutic targets for intracerebral hemorrhage-associated inflammation: An update. J. Cereb. Blood Flow Metab. 2020, 40, 1752–1768. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef]
- Donat, C.K.; Scott, G.; Gentleman, S.M.; Sastre, M. Microglial Activation in Traumatic Brain Injury. Front. Aging Neurosci. 2017, 9, 208. [Google Scholar] [CrossRef]
- Lan, X.; Han, X.; Li, Q.; Yang, Q.W.; Wang, J. Modulators of microglial activation and polarization after intracerebral haemorrhage. Nat. Rev. Neurol. 2017, 13, 420–433. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.F.; Wan, J.; Li, Q.; Renfroe, S.C.; Heller, N.M.; Wang, J. Alternative activation-skewed microglia/macrophages promote hematoma resolution in experimental intracerebral hemorrhage. Neurobiol. Dis. 2017, 103, 54–69. [Google Scholar] [CrossRef]
- Loane, D.J.; Kumar, A. Microglia in the TBI brain: The good, the bad, and the dysregulated. Exp. Neurol. 2016, 275 Pt 3, 316–327. [Google Scholar] [CrossRef]
- Boche, D.; Perry, V.H.; Nicoll, J.A. Review: Activation patterns of microglia and their identification in the human brain. Neuropathol. Appl. Neurobiol. 2013, 39, 3–18. [Google Scholar] [CrossRef]
- Hu, X.; Leak, R.K.; Shi, Y.; Suenaga, J.; Gao, Y.; Zheng, P.; Chen, J. Microglial and macrophage polarization-new prospects for brain repair. Nat. Rev. Neurol. 2015, 11, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Kigerl, K.A.; Gensel, J.C.; Ankeny, D.P.; Alexander, J.K.; Donnelly, D.J.; Popovich, P.G. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J. Neurosci. 2009, 29, 13435–13444. [Google Scholar] [CrossRef] [PubMed]
- Lan, X.; Han, X.; Li, Q.; Li, Q.; Gao, Y.; Cheng, T.; Wan, J.; Zhu, W.; Wang, J. Pinocembrin protects hemorrhagic brain primarily by inhibiting toll-like receptor 4 and reducing M1 phenotype microglia. Brain Behav. Immun. 2017, 61, 326–339. [Google Scholar] [CrossRef]
- Sica, A.; Schioppa, T.; Mantovani, A.; Allavena, P. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: Potential targets of anti-cancer therapy. Eur. J. Cancer 2006, 42, 717–727. [Google Scholar] [CrossRef]
- Zhu, H.; Wang, Z.; Yu, J.; Yang, X.; He, F.; Liu, Z.; Che, F.; Chen, X.; Ren, H.; Hong, M.; et al. Role and mechanisms of cytokines in the secondary brain injury after intracerebral hemorrhage. Prog. Neurobiol. 2019, 178, 101610. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, H.; Lee, D.H.; Yu, J.; Cheng, T.; Hong, M.; Jiang, S.; Fan, H.; Huang, X.; Zhou, J.; et al. Using functional and molecular MRI techniques to detect neuroinflammation and neuroprotection after traumatic brain injury. Brain Behav. Immun. 2017, 64, 344–353. [Google Scholar] [CrossRef]
- Huber, B.R.; Meabon, J.S.; Hoffer, Z.S.; Zhang, J.; Hoekstra, J.G.; Pagulayan, K.F.; McMillan, P.J.; Mayer, C.L.; Banks, W.A.; Kraemer, B.C.; et al. Blast exposure causes dynamic microglial/macrophage responses and microdomains of brain microvessel dysfunction. Neuroscience 2016, 319, 206–220. [Google Scholar] [CrossRef]
- Younger, D.; Murugan, M.; Rama Rao, K.V.; Wu, L.J.; Chandra, N. Microglia Receptors in Animal Models of Traumatic Brain Injury. Mol. Neurobiol. 2019, 56, 5202–5228. [Google Scholar] [CrossRef]
- Han, X.; Zhao, X.; Lan, X.; Li, Q.; Gao, Y.; Liu, X.; Wan, J.; Yang, Z.; Chen, X.; Zang, W.; et al. 20-HETE synthesis inhibition promotes cerebral protection after intracerebral hemorrhage without inhibiting angiogenesis. J. Cereb. Blood Flow Metab. 2019, 39, 1531–1543. [Google Scholar] [CrossRef]
- Wang, W.; Li, M.; Wang, Y.; Wang, Z.; Zhang, W.; Guan, F.; Chen, Q.; Wang, J. GSK-3beta as a target for protection against transient cerebral ischemia. Int. J. Med. Sci. 2017, 14, 333–339. [Google Scholar] [CrossRef]
- Zhao, D.; Xu, X.; Pan, L.; Zhu, W.; Fu, X.; Guo, L.; Lu, Q.; Wang, J. Pharmacologic activation of cholinergic alpha7 nicotinic receptors mitigates depressive-like behavior in a mouse model of chronic stress. J. Neuroinflamm. 2017, 14, 234. [Google Scholar] [CrossRef] [PubMed]
- Ballabh, P.; Braun, A.; Nedergaard, M. The blood-brain barrier: An overview: Structure, regulation, and clinical implications. Neurobiol. Dis. 2004, 16, 1–13. [Google Scholar] [CrossRef]
- Li, Q.; Han, X.; Lan, X.; Hong, X.; Li, Q.; Gao, Y.; Luo, T.; Yang, Q.; Koehler, R.C.; Zhai, Y.; et al. Inhibition of tPA-induced hemorrhagic transformation involves adenosine A2b receptor activation after cerebral ischemia. Neurobiol. Dis. 2017, 108, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Saint-Pol, J.; Gosselet, F.; Duban-Deweer, S.; Pottiez, G.; Karamanos, Y. Targeting and Crossing the Blood-Brain Barrier with Extracellular Vesicles. Cells 2020, 9, 851. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Rosa, E.; Shin, M.K.; Dhar, M.; Chaubey, K.; Cintron-Perez, C.J.; Tang, X.; Liao, X.; Miller, E.; Koh, Y.; Barker, S.; et al. P7C3-A20 treatment one year after TBI in mice repairs the blood-brain barrier, arrests chronic neurodegeneration, and restores cognition. Proc. Natl. Acad. Sci. USA 2020, 117, 27667–27675. [Google Scholar] [CrossRef] [PubMed]
- Keaney, J.; Campbell, M. The dynamic blood-brain barrier. FEBS J 2015, 282, 4067–4079. [Google Scholar] [CrossRef]
- Begley, D.J.; Brightman, M.W. Structural and functional aspects of the blood-brain barrier. Prog. Drug Res. 2003, 61, 39–78. [Google Scholar] [CrossRef]
- Zlokovic, B.V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.D.; Winkler, E.A.; Sagare, A.P.; Singh, I.; LaRue, B.; Deane, R.; Zlokovic, B.V. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 2010, 68, 409–427. [Google Scholar] [CrossRef] [PubMed]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Sifat, A.E.; Vaidya, B.; Abbruscato, T.J. Blood-Brain Barrier Protection as a Therapeutic Strategy for Acute Ischemic Stroke. AAPS J. 2017, 19, 957–972. [Google Scholar] [CrossRef]
- Cash, A.; Theus, M.H. Mechanisms of Blood-Brain Barrier Dysfunction in Traumatic Brain Injury. Int. J. Mol. Sci. 2020, 21, 3344. [Google Scholar] [CrossRef]
- Marmarou, A. A review of progress in understanding the pathophysiology and treatment of brain edema. Neurosurg. Focus 2007, 22, E1. [Google Scholar] [CrossRef]
- Barros, L.F.; Castro, J.; Bittner, C.X. Ion movements in cell death: From protection to execution. Biol. Res. 2002, 35, 209–214. [Google Scholar] [CrossRef]
- De Bock, M.; Van Haver, V.; Vandenbroucke, R.E.; Decrock, E.; Wang, N.; Leybaert, L. Into rather unexplored terrain-transcellular transport across the blood-brain barrier. Glia 2016, 64, 1097–1123. [Google Scholar] [CrossRef]
- Tait, M.J.; Saadoun, S.; Bell, B.A.; Papadopoulos, M.C. Water movements in the brain: Role of aquaporins. Trends Neurosci. 2008, 31, 37–43. [Google Scholar] [CrossRef]
- Bhowmick, S.; D’Mello, V.; Caruso, D.; Wallerstein, A.; Abdul-Muneer, P.M. Impairment of pericyte-endothelium crosstalk leads to blood-brain barrier dysfunction following traumatic brain injury. Exp. Neurol. 2019, 317, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef]
- Obermeier, B.; Daneman, R.; Ransohoff, R.M. Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 2013, 19, 1584–1596. [Google Scholar] [CrossRef]
- Hellstrom, M.; Kalen, M.; Lindahl, P.; Abramsson, A.; Betsholtz, C. Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development 1999, 126, 3047–3055. [Google Scholar] [PubMed]
- Winkler, E.A.; Sengillo, J.D.; Bell, R.D.; Wang, J.; Zlokovic, B.V. Blood-spinal cord barrier pericyte reductions contribute to increased capillary permeability. J. Cereb. Blood Flow Metab. 2012, 32, 1841–1852. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.A.; Birk, H.; Burkhardt, J.K.; Chen, X.; Yue, J.K.; Guo, D.; Rutledge, W.C.; Lasker, G.F.; Partow, C.; Tihan, T.; et al. Reductions in brain pericytes are associated with arteriovenous malformation vascular instability. J. Neurosurg. 2018, 129, 1464–1474. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef]
- Wan, J.; Ren, H.; Wang, J. Iron toxicity, lipid peroxidation and ferroptosis after intracerebral haemorrhage. Stroke Vasc. Neurol. 2019, 4, 93–95. [Google Scholar] [CrossRef]
- Weiland, A.; Wang, Y.; Wu, W.; Lan, X.; Han, X.; Li, Q.; Wang, J. Ferroptosis and Its Role in Diverse Brain Diseases. Mol. Neurobiol. 2019, 56, 4880–4893. [Google Scholar] [CrossRef]
- Li, Q.; Han, X.; Lan, X.; Gao, Y.; Wan, J.; Durham, F.; Cheng, T.; Yang, J.; Wang, Z.; Jiang, C.; et al. Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI Insight 2017, 2, e90777. [Google Scholar] [CrossRef]
- Li, Q.; Weiland, A.; Chen, X.; Lan, X.; Han, X.; Durham, F.; Liu, X.; Wan, J.; Ziai, W.C.; Hanley, D.F.; et al. Ultrastructural Characteristics of Neuronal Death and White Matter Injury in Mouse Brain Tissues After Intracerebral Hemorrhage: Coexistence of Ferroptosis, Autophagy, and Necrosis. Front. Neurol. 2018, 9, 581. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.S.; Wang, Y.Q.; Lin, Y.; Mao, Q.; Feng, J.F.; Gao, G.Y.; Jiang, J.Y. Inhibition of ferroptosis attenuates tissue damage and improves long-term outcomes after traumatic brain injury in mice. CNS Neurosci. Ther. 2019, 25, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Yang, X.; Wang, J. Ferroptosis in Nervous System Diseases. In Ferroptosis in Health and Disease; Springer Nature: Cham, Switzerland, 2019; pp. 173–195. [Google Scholar] [CrossRef]
- Hua, W.; Chen, X.; Wang, J.; Zang, W.; Jiang, C.; Ren, H.; Hong, M.; Wang, J.; Wu, H.; Wang, J. Mechanisms and potential therapeutic targets for spontaneous intracerebral hemorrhage. Brain Hemorrhages 2020, 1, 99–104. [Google Scholar] [CrossRef]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef]
- Yagoda, N.; von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 864–868. [Google Scholar] [CrossRef]
- Magtanong, L.; Dixon, S.J. Ferroptosis and Brain Injury. Dev. Neurosci. 2018, 40, 382–395. [Google Scholar] [CrossRef]
- Cao, J.Y.; Dixon, S.J. Mechanisms of ferroptosis. Cell Mol. Life Sci. 2016, 73, 2195–2209. [Google Scholar] [CrossRef]
- Long, X.; Deng, S.; Mattner, J.; Zang, Z.; Zhou, D.; McNary, N.; Goff, R.D.; Teyton, L.; Bendelac, A.; Savage, P.B. Synthesis and evaluation of stimulatory properties of Sphingomonadaceae glycolipids. Nat. Chem. Biol. 2007, 3, 559–564. [Google Scholar] [CrossRef]
- Li, Q.; Wan, J.; Lan, X.; Han, X.; Wang, Z.; Wang, J. Neuroprotection of brain-permeable iron chelator VK-28 against intracerebral hemorrhage in mice. J. Cereb. Blood Flow Metab. 2017, 37, 3110–3123. [Google Scholar] [CrossRef]
- Kim, S.E.; Zhang, L.; Ma, K.; Riegman, M.; Chen, F.; Ingold, I.; Conrad, M.; Turker, M.Z.; Gao, M.; Jiang, X.; et al. Ultrasmall nanoparticles induce ferroptosis in nutrient-deprived cancer cells and suppress tumour growth. Nat. Nanotechnol. 2016, 11, 977–985. [Google Scholar] [CrossRef]
- Boltze, J.; Aronowski, J.A.; Badaut, J.; Buckwalter, M.S.; Caleo, M.; Chopp, M.; Dave, K.R.; Didwischus, N.; Dijkhuizen, R.M.; Doeppner, T.R.; et al. New Mechanistic Insights, Novel Treatment Paradigms, and Clinical Progress in Cerebrovascular Diseases. Front. Aging Neurosci. 2021, 13, 623751. [Google Scholar] [CrossRef] [PubMed]
- Amyot, F.; Arciniegas, D.B.; Brazaitis, M.P.; Curley, K.C.; Diaz-Arrastia, R.; Gandjbakhche, A.; Herscovitch, P.; Hinds, S.R., 2nd; Manley, G.T.; Pacifico, A.; et al. A Review of the Effectiveness of Neuroimaging Modalities for the Detection of Traumatic Brain Injury. J. Neurotrauma 2015, 32, 1693–1721. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, W.; Jiang, S.; Zhang, Y.; Heo, H.Y.; Wang, X.; Peng, Y.; Wang, J.; Zhou, J. Amide proton transfer-weighted MRI detection of traumatic brain injury in rats. J. Cereb. Blood Flow Metab. 2017, 37, 3422–3432. [Google Scholar] [CrossRef]
- Wang, M.; Hong, X.; Chang, C.F.; Li, Q.; Ma, B.; Zhang, H.; Xiang, S.; Heo, H.Y.; Zhang, Y.; Lee, D.H.; et al. Simultaneous detection and separation of hyperacute intracerebral hemorrhage and cerebral ischemia using amide proton transfer MRI. Magn. Reson Med. 2015, 74, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Bai, Y.; Lin, Y.; Hong, X.; Liu, T.; Ma, L.; Haacke, E.M.; Zhou, J.; Wang, J.; Wang, M. Amide proton transfer magnetic resonance imaging in detecting intracranial hemorrhage at different stages: A comparative study with susceptibility weighted imaging. Sci. Rep. 2017, 7, 45696. [Google Scholar] [CrossRef]
- Wang, J.; Jiang, C.; Zhang, K.; Lan, X.; Chen, X.; Zang, W.; Wang, Z.; Guan, F.; Zhu, C.; Yang, X.; et al. Melatonin receptor activation provides cerebral protection after traumatic brain injury by mitigating oxidative stress and inflammation via the Nrf2 signaling pathway. Free Radic. Biol. Med. 2019, 131, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.; Yang, B.; Li, D.; Ma, S.; Tian, Y.; Qu, R.; Zhang, W.; Zhang, Y.; Hu, K.; Guan, F.; et al. Wharton’s Jelly Transplantation Improves Neurologic Function in a Rat Model of Traumatic Brain Injury. Cell Mol. Neurobiol. 2015, 35, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, H.; Yu, J.; Hong, M.; Zhou, J.; Zhu, L.; Wang, Y.; Luo, M.; Xia, Z.; Yang, Z.J.; et al. Protective Effects of Chinese Herbal Medicine Rhizoma drynariae in Rats After Traumatic Brain Injury and Identification of Active Compound. Mol. Neurobiol. 2016, 53, 4809–4820. [Google Scholar] [CrossRef] [PubMed]








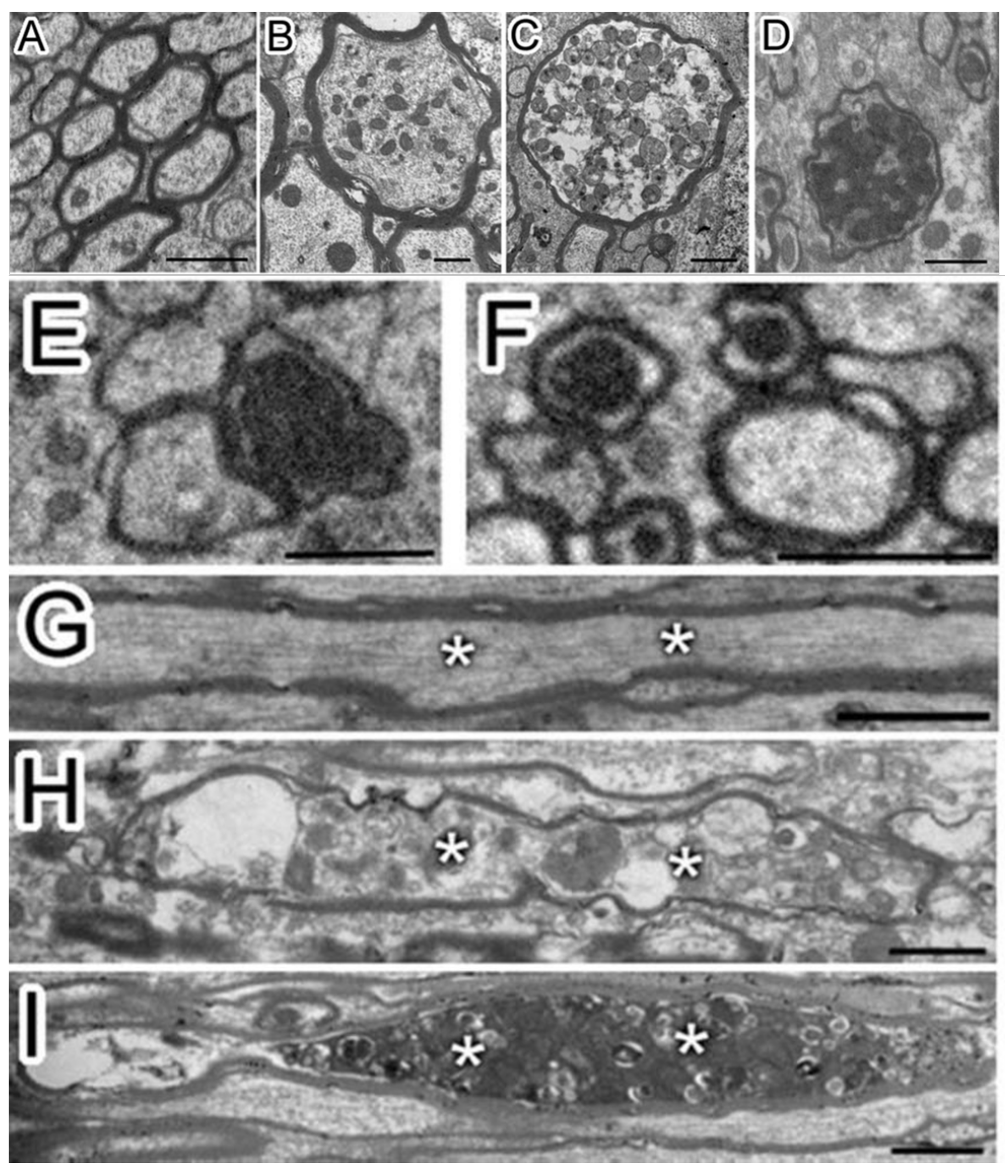{kind=link}
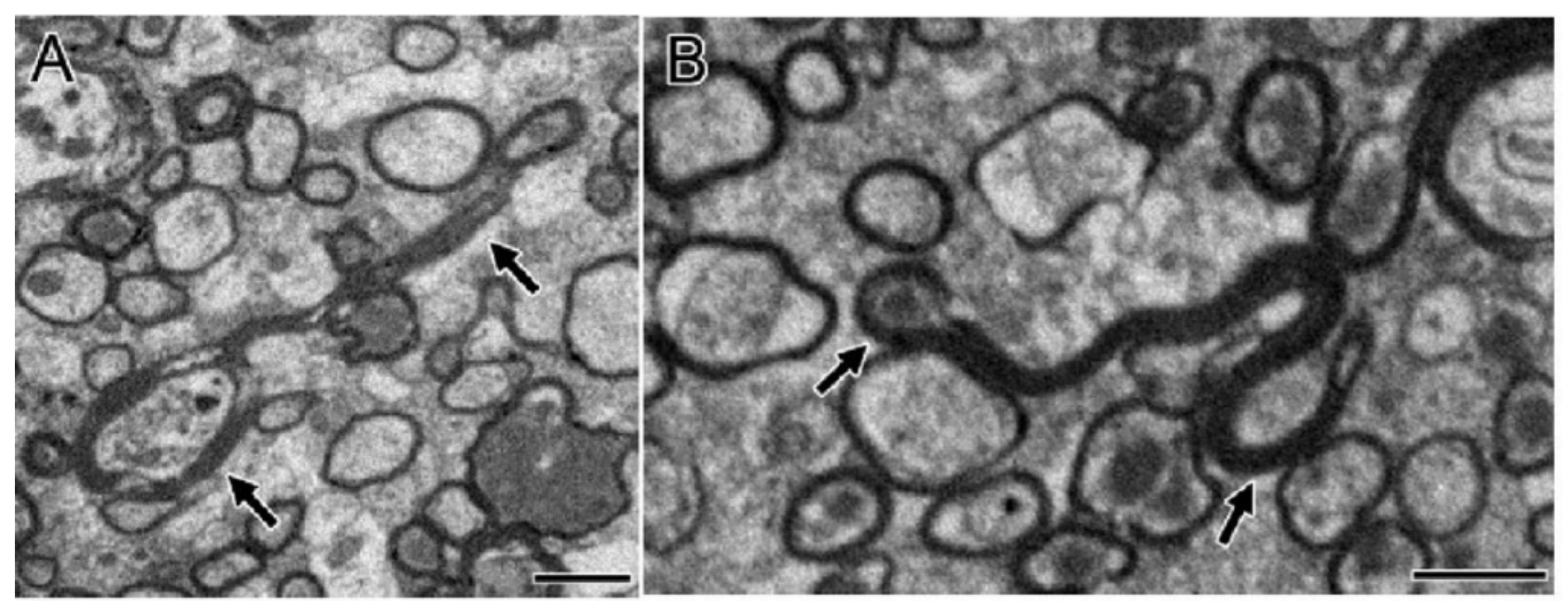{kind=link}
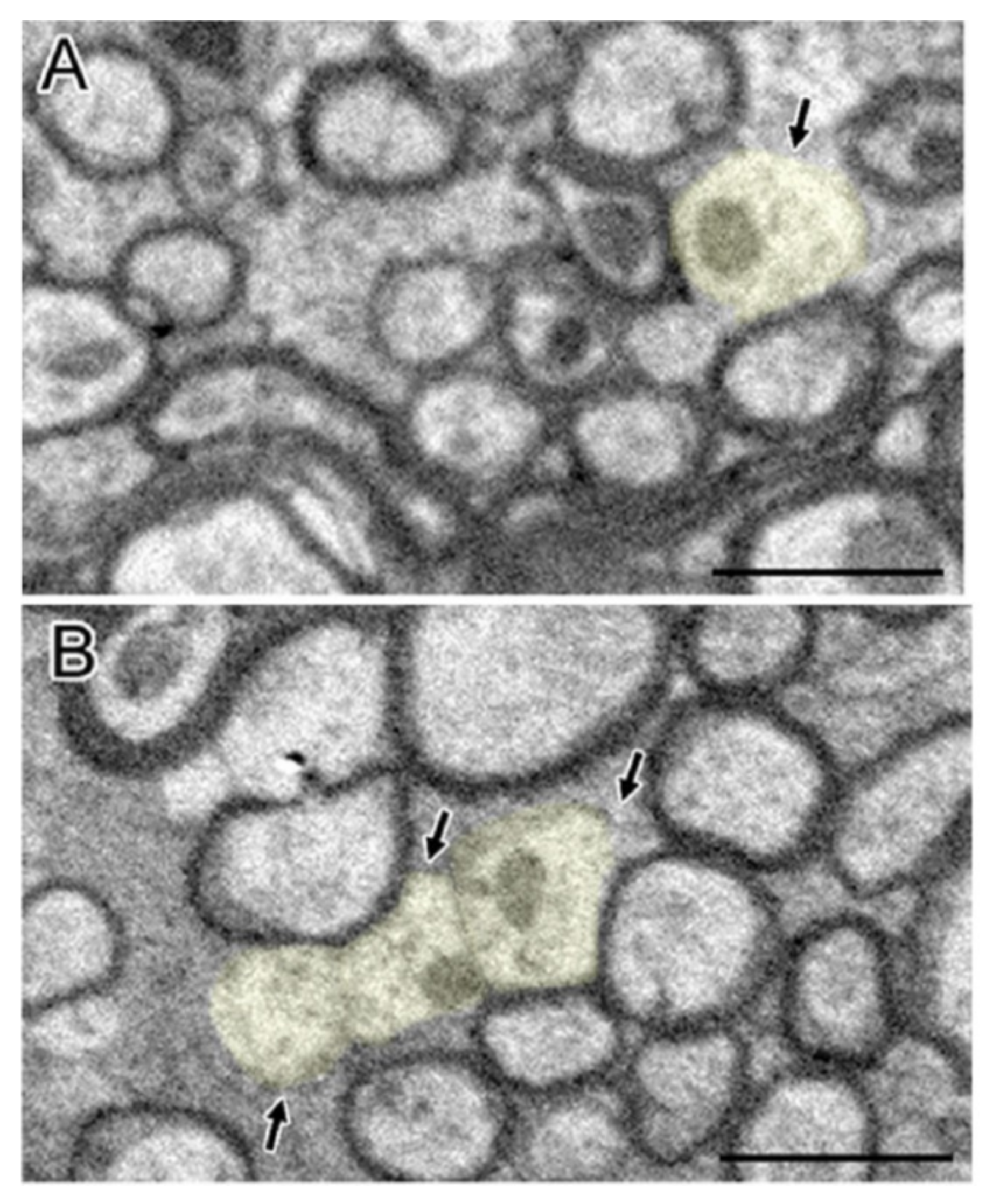{kind=link}
{kind=link}
{kind=link}
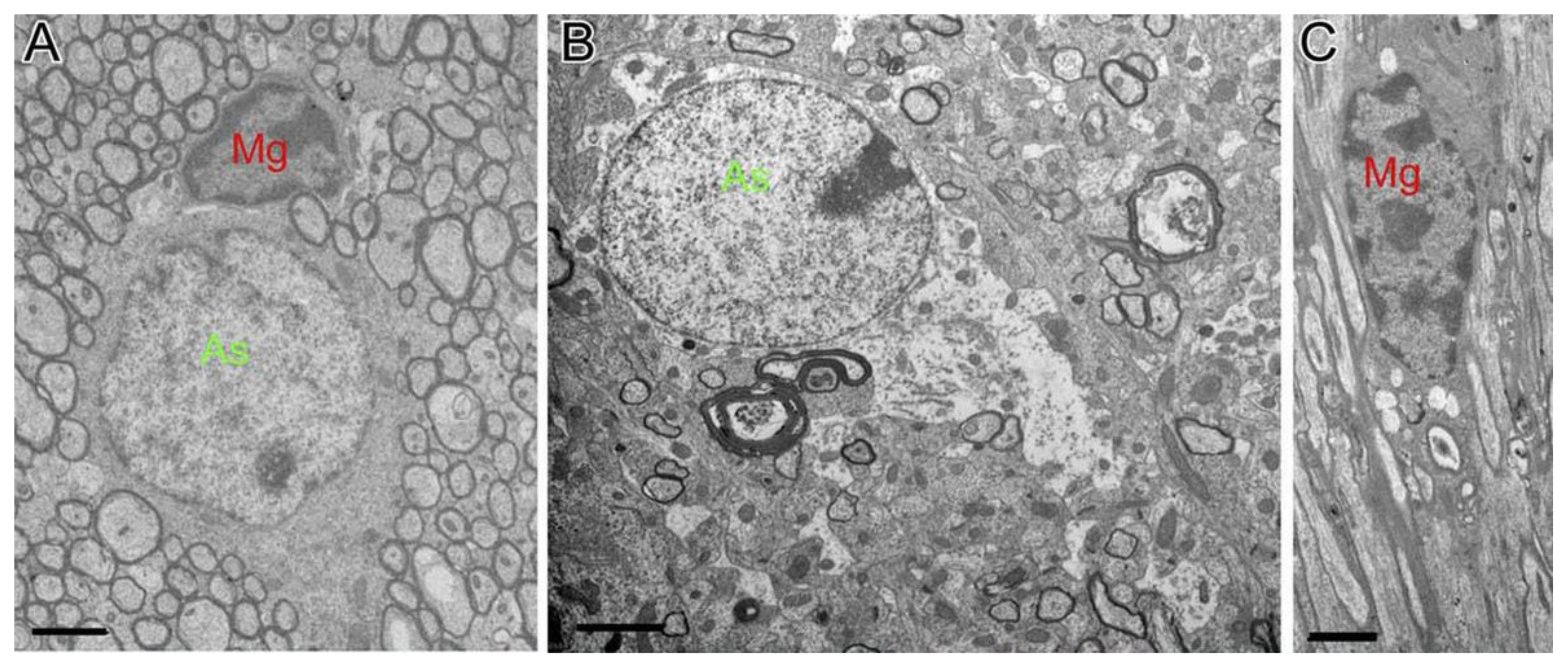{kind=link}
{kind=link}
| Cell | Ultrastructural | After TBI | Reference |
|---|---|---|---|
| Neuron | Cell body | Swelling (or hydropic disintegration) | [1] |
| Cell membrane | Fracture | [1] | |
| Nuclear | Heterochromatin loss | [1] | |
| Organelle | Abnormality; Loss | [1,19] | |
| Mitochondrion | Fracture; Swelling; Membrane rupture; Atrophy (Ferroptosis); Crista collapse and disorder; Mitochondrial density decreased | [1,19,21,22,34,35,164] | |
| Endoplasmic reticulum | Swelling; Dissolution | [1,56] | |
| Cytoskeleton | Broken; Local loss | [1,19] | |
| Process | Hydropic disintegration | [1] | |
| Axon | Destruction; Demyelination and myelination | [1,20,21,22] | |
| Astrocyte | Cell body | Swelling; Prolonged protrusion | [20,88,89] |
| Foot process | Swelling; Vacuolization | [90,91] | |
| Microglia | Cytoplasm | Myelin fragment appeared; Cell body hypertrophy; Elongated processes (forming a hexagonal honeycomb structure) | [115,116,117,118] |
| Endothelia | Cytoplasm | Dense granulation and multivesical body appeared | [91] |
| Surface | Longitudinal folds; invagination appeared; end-feet swelling | [90,91] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qin, D.; Wang, J.; Le, A.; Wang, T.J.; Chen, X.; Wang, J. Traumatic Brain Injury: Ultrastructural Features in Neuronal Ferroptosis, Glial Cell Activation and Polarization, and Blood–Brain Barrier Breakdown. Cells 2021, 10, 1009. https://doi.org/10.3390/cells10051009
Qin D, Wang J, Le A, Wang TJ, Chen X, Wang J. Traumatic Brain Injury: Ultrastructural Features in Neuronal Ferroptosis, Glial Cell Activation and Polarization, and Blood–Brain Barrier Breakdown. Cells. 2021; 10(5):1009. https://doi.org/10.3390/cells10051009
Chicago/Turabian StyleQin, Delong, Junmin Wang, Anh Le, Tom J. Wang, Xuemei Chen, and Jian Wang. 2021. "Traumatic Brain Injury: Ultrastructural Features in Neuronal Ferroptosis, Glial Cell Activation and Polarization, and Blood–Brain Barrier Breakdown" Cells 10, no. 5: 1009. https://doi.org/10.3390/cells10051009
APA StyleQin, D., Wang, J., Le, A., Wang, T. J., Chen, X., & Wang, J. (2021). Traumatic Brain Injury: Ultrastructural Features in Neuronal Ferroptosis, Glial Cell Activation and Polarization, and Blood–Brain Barrier Breakdown. Cells, 10(5), 1009. https://doi.org/10.3390/cells10051009