
Positive Regulation of Estrogen Receptor Alpha in Breast Tumorigenesis



Abstract
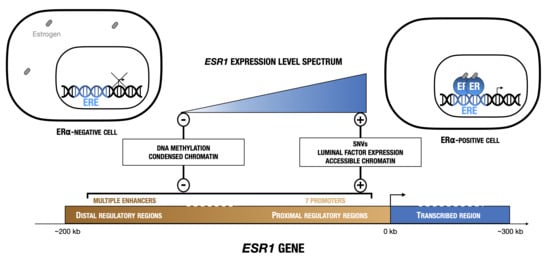
{kind=link}
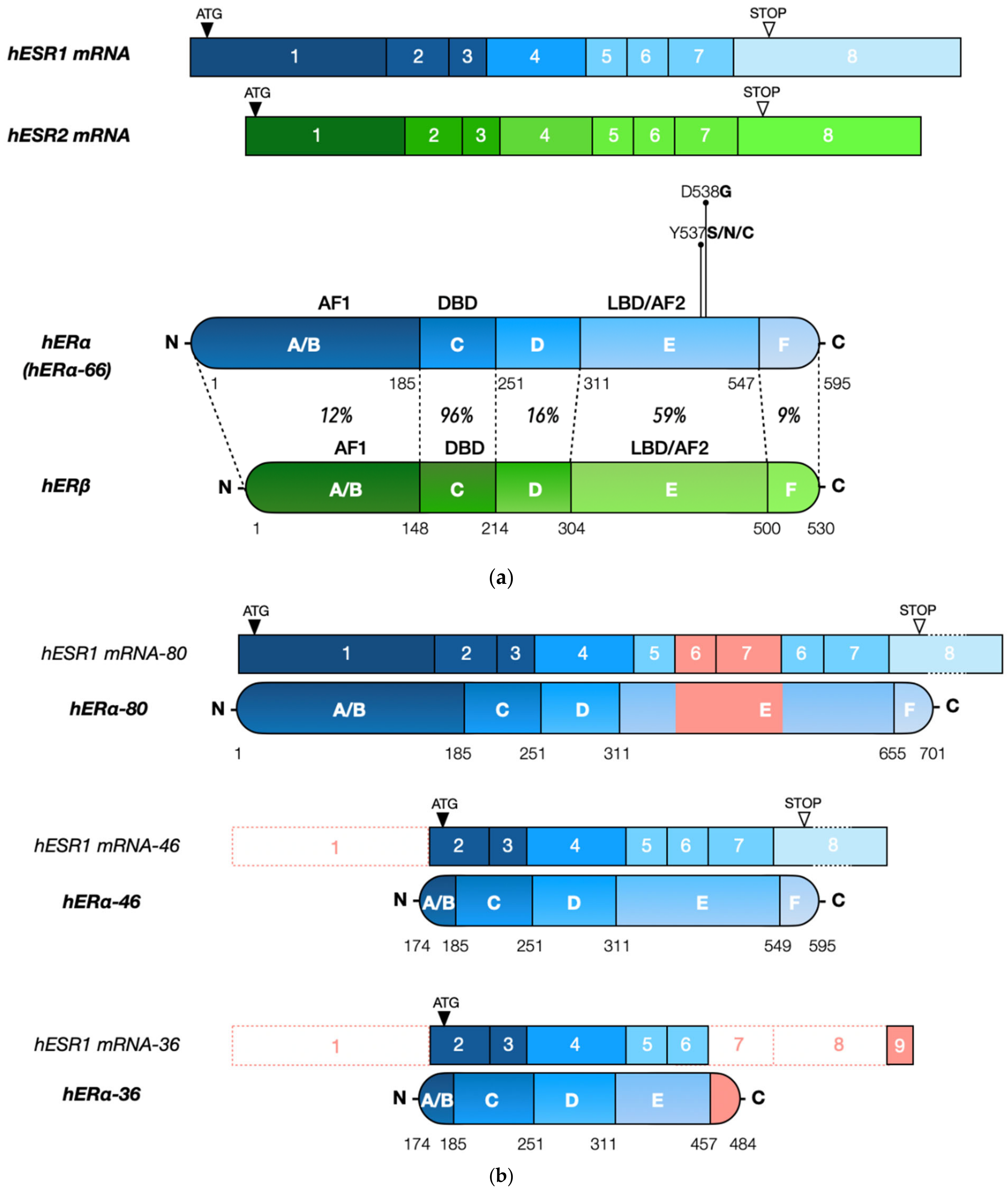{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Expression of ESR1 Is Transcriptionally Regulated in Normal Mammary Tissue and in Breast Tumorigenesis
2.1. Expression in Normal Tissue
2.2. Expression in Tumoral Tissue
3. ESR1 Gene Organization and Regulatory Sequences
3.1. ESR1 Gene Structure and Alternative Transcripts
3.2. ESR1 Alternative Promoters Have Tissue-Specific Activity
3.3. Upstream Enhancers
4. Luminal ESR1 Transcriptional Regulators
4.1. GATA3
4.2. FOXA1
4.3. Estrogen Receptor Alpha
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat. Rev. Dis. Prim. 2019, 5, 66. [Google Scholar] [CrossRef] [PubMed]
- Onitilo, A.A.; Engel, J.M.; Greenlee, R.T.; Mukesh, B.N. Breast Cancer Subtypes Based on ER/PR and Her2 Expression: Comparison of Clinicopathologic Features and Survival. Clin. Med. Res. 2009, 7, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Stierer, M.; Rosen, H.; Weber, R.; Hanak, H.; Spona, J.; Tüchler, H. Immunohistochemical and Biochemical Measurement of Estrogen and Progesterone Receptors in Primary Breast Cancer Correlation of Histopathology and Prognostic Factors. Ann. Surg. 1993, 218, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Traboulsi, T.; El Ezzy, M.; Gleason, J.; Mader, S. Antiestrogens: Structure-activity relationships and use in breast cancer treatment. J. Mol. Endocrinol. 2017, 58, R15–R31. [Google Scholar] [CrossRef] [PubMed]
- Caciolla, J.; Bisi, A.; Belluti, F.; Rampa, A.; Gobbi, S. Reconsidering Aromatase for Breast Cancer Treatment: New Roles for an Old Target. Molecules 2020, 25, 5351. [Google Scholar] [CrossRef]
- Johnston, S.R.; Saccani-Jotti, G.; Smith, I.E.; Salter, J.; Newby, J.; Coppen, M.; Ebbs, S.R.; Dowsett, M. Changes in estrogen receptor, progesterone receptor, and pS2 expression in tamoxifen-resistant human breast cancer. Cancer Res. 1995, 55, 3331–3338. [Google Scholar]
- Lykkesfeldt, A.E. Mechanisms of Tamoxifen Resistance in the Treatment of Advanced Breast Cancer. Acta Oncol. 1996, 35 (Suppl. 5), 9–14. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, M.C.; Detre, S.; Johnston, S.; Mohsin, S.K.; Shou, J.; Allred, D.C.; Schiff, R.; Osborne, C.K.; Dowsett, M. Molecular Changes in Tamoxifen-Resistant Breast Cancer: Relationship Between Estrogen Receptor, HER-2, and p38 Mitogen-Activated Protein Kinase. J. Clin. Oncol. 2005, 23, 2469–2476. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Coombes, R.C. Estrogen Receptor Alpha in Human Breast Cancer: Occurrence and Significance. J. Mammary Gland. Biol. Neoplasia 2000, 5, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Sukocheva, O.A.; Lukina, E.; Friedemann, M.; Menschikowski, M.; Hagelgans, A.; Aliev, G. The crucial role of epigenetic regulation in breast cancer anti-estrogen resistance: Current findings and future perspectives. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Jeffreys, S.A.; Powter, B.; Balakrishnar, B.; Mok, K.; Soon, P.; Franken, A.; Neubauer, H.; de Souza, P.; Becker, T.M. Endocrine Resistance in Breast Cancer: The Role of Estrogen Receptor Stability. Cells 2020, 9, 2077. [Google Scholar] [CrossRef] [PubMed]
- Ascenzi, P.; Bocedi, A.; Marino, M. Structure–function relationship of estrogen receptor α and β: Impact on human health. Mol. Asp. Med. 2006, 27, 299–402. [Google Scholar] [CrossRef]
- Hamilton, K.J.; Hewitt, S.C.; Arao, Y.; Korach, K.S. Estrogen Hormone Biology. Curr. Top. Dev. Biol. 2017, 125, 109–146. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.M.; Mangelsdorf, D.J. Nuclear Receptors, RXR, and the Big Bang. Cell 2014, 157, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Mosselman, S.; Polman, J.; Dijkema, R. ERβ: Identification and characterization of a novel human estrogen receptor. FEBS Lett. 1996, 392, 49–53. [Google Scholar] [CrossRef]
- Sanchez, R.; Nguyen, D.; Rocha, W.; White, J.H.; Mader, S. Diversity in the mechanisms of gene regulation by estrogen receptors. BioEssays 2002, 24, 244–254. [Google Scholar] [CrossRef]
- Klinge, C.M. Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res. 2001, 29, 2905–2919. [Google Scholar] [CrossRef]
- Cotnoir-White, D.; Laperrière, D.; Mader, S. Evolution of the repertoire of nuclear receptor binding sites in genomes. Mol. Cell. Endocrinol. 2011, 334, 76–82. [Google Scholar] [CrossRef]
- Carroll, J.S.; Liu, X.S.; Brodsky, A.S.; Li, W.; Meyer, C.A.; Szary, A.J.; Eeckhoute, J.; Shao, W.; Hestermann, E.V.; Geistlinger, T.R.; et al. Chromosome-Wide Mapping of Estrogen Receptor Binding Reveals Long-Range Regulation Requiring the Forkhead Protein FoxA1. Cell 2005, 122, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Deschênes, J.; Bourdeau, V.; White, J.H.; Mader, S. Regulation of GREB1 Transcription by Estrogen Receptor α through a Multipartite Enhancer Spread Over 20 kb of Upstream Flanking Sequences. J. Biol. Chem. 2007, 282, 17335–17339. [Google Scholar] [CrossRef] [PubMed]
- Barnett, D.H.; Sheng, S.; Charn, T.H.; Waheed, A.; Sly, W.S.; Lin, C.-Y.; Liu, E.T.; Katzenellenbogen, B.S. Estrogen Receptor Regulation of Carbonic Anhydrase XII through a Distal Enhancer in Breast Cancer. Cancer Res. 2008, 68, 3505–3515. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.F.; Wansa, K.D.S.A.; Liu, M.H.; Zhao, B.; Hong, S.Z.; Tan, P.Y.; Lim, K.S.; Bourque, G.; Liu, E.T.; Cheung, E. Regulation of Estrogen Receptor-mediated Long Range Transcription via Evolutionarily Conserved Distal Response Elements. J. Biol. Chem. 2008, 283, 32977–32988. [Google Scholar] [CrossRef]
- Charn, T.H.; Liu, E.T.-B.; Chang, E.C.; Lee, Y.K.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S. Genome-Wide Dynamics of Chromatin Binding of Estrogen Receptors α and β: Mutual Restriction and Competitive Site Selection. Mol. Endocrinol. 2010, 24, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Vega, V.B.; Lin, C.-Y.; Lai, K.S.; Kong, S.L.; Xie, M.; Su, X.; Teh, H.F.; Thomsen, J.S.; Yeo, A.L.; Sung, W.K.; et al. Multiplatform genome-wide identification and modeling of functional human estrogen receptor binding sites. Genome Biol. 2006, 7, R82. [Google Scholar] [CrossRef] [PubMed]
- Joseph, R.; Orlov, Y.; Huss, M.; Sun, W.; Kong, S.L.; Ukil, L.; Pan, Y.F.; Li, G.; Lim, M.; Thomsen, J.S.; et al. Integrative model of genomic factors for determining binding site selection by estrogen receptor-α. Mol. Syst. Biol. 2010, 6, 456. [Google Scholar] [CrossRef]
- Lupien, M.; Brown, M. Cistromics of hormone-dependent cancer. Endocr.-Relat. Cancer 2009, 16, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Bojcsuk, D.; Nagy, G.; Balint, B.L. Inducible super-enhancers are organized based on canonical signal-specific transcription factor binding elements. Nucleic Acids Res. 2016, 45, 3693–3706. [Google Scholar] [CrossRef] [PubMed]
- Siersbaek, R.; Nielsen, R.; John, S.; Sung, M.-H.; Baek, S.; Loft, A.; Hager, G.L.; Mandrup, S. Extensive chromatin remodelling and establishment of transcription factor ‘hotspots’ during early adipogenesis. EMBO J. 2011, 30, 1459–1472. [Google Scholar] [CrossRef]
- Siersbæk, R.D.; Baek, S.; Rabiee, A.; Nielsen, R.; Traynor, S.; Clark, N.; Sandelin, A.; Jensen, O.N.; Sung, M.-H.; Hager, G.L.; et al. Molecular Architecture of Transcription Factor Hotspots in Early Adipogenesis. Cell Rep. 2014, 7, 1434–1442. [Google Scholar] [CrossRef]
- Liu, Z.; Merkurjev, D.; Yang, F.; Li, W.; Oh, S.; Friedman, M.J.; Song, X.; Zhang, F.; Ma, Q.; Ohgi, K.A.; et al. Enhancer Activation Requires trans-Recruitment of a Mega Transcription Factor Complex. Cell 2014, 159, 358–373. [Google Scholar] [CrossRef]
- Rao, S.S.; Huang, S.-C.; Hilaire, B.G.S.; Engreitz, J.M.; Perez, E.; Kieffer-Kwon, K.-R.; Sanborn, A.L.; Johnstone, S.E.; Bascom, G.D.; Bochkov, I.; et al. Cohesin Loss Eliminates All Loop Domains. Cell 2017, 171, 305–320. [Google Scholar] [CrossRef]
- Huang, J.; Li, K.; Cai, W.; Liu, X.; Zhang, Y.; Orkin, S.H.; Xu, J.; Yuan, G.-C. Dissecting super-enhancer hierarchy based on chromatin interactions. Nat. Commun. 2018, 9, 943. [Google Scholar] [CrossRef] [PubMed]
- Sepehr, E.; Lebl-Rinnova, M.; Mann, M.K.; Pisani, S.L.; Churchwell, M.I.; Korol, D.L.; Katzenellenbogen, J.A.; Doerge, D.R. Pharmacokinetics of the estrogen receptor subtype-selective ligands, PPT and DPN: Quantification using UPLC-ES/MS/MS. J. Pharm. Biomed. Anal. 2012, 71, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Arnal, J.F.; Lenfant, F.; Metivier, R.; Flouriot, G.; Henrion, D.; Adlanmerini, M.; Fontaine, C.; Gourdy, P.; Chambon, P.; Katzenellenbogen, B.; et al. Membrane and Nuclear Estrogen Receptor Alpha Actions: From Tissue Specificity to Medical Implications. Physiol. Rev. 2017, 97, 1045–1087. [Google Scholar] [CrossRef] [PubMed]
- White, J.H.; Fernandes, I.; Mader, S.; Yang, X.-J. Corepressor Recruitment by Agonist-Bound Nuclear Receptors. Vitam. Horm. 2004, 68, 123–143. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.M.; McDonnell, D.P. Coregulators in Nuclear Estrogen Receptor Action: From Concept to Therapeutic Targeting. Mol. Interv. 2005, 5, 343–357. [Google Scholar] [CrossRef] [PubMed]
- Manavathi, B.; Samanthapudi, V.S.K.; Gajulapalli, V.N.R. Estrogen receptor coregulators and pioneer factors: The orchestrators of mammary gland cell fate and development. Front. Cell Dev. Biol. 2014, 2, 34. [Google Scholar] [CrossRef] [PubMed]
- Yi, P.; Wang, Z.; Feng, Q.; Pintilie, G.D.; Foulds, C.; Lanz, R.B.; Ludtke, S.; Schmid, M.F.; Chiu, W.; O’Malley, B.W. Structure of a Biologically Active Estrogen Receptor-Coactivator Complex on DNA. Mol. Cell 2015, 57, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, S.; Korach, K.S. Estrogen Receptors: New Directions in the New Millennium. Endocr. Rev. 2018, 39, 664–675. [Google Scholar] [CrossRef] [PubMed]
- Lannigan, D.A. Estrogen receptor phosphorylation. Steroids 2003, 68, 1–9. [Google Scholar] [CrossRef]
- Le Romancer, M.; Poulard, C.; Cohen, P.; Sentis, S.; Renoir, J.-M.; Corbo, L. Cracking the Estrogen Receptor’s Posttranslational Code in Breast Tumors. Endocr. Rev. 2011, 32, 597–622. [Google Scholar] [CrossRef]
- Wijayaratne, A.L.; McDonnell, D.P. The Human Estrogen Receptor-α Is a Ubiquitinated Protein Whose Stability Is Affected Differentially by Agonists, Antagonists, and Selective Estrogen Receptor Modulators. J. Biol. Chem. 2001, 276, 35684–35692. [Google Scholar] [CrossRef]
- Levin, E.R. Integration of the Extranuclear and Nuclear Actions of Estrogen. Mol. Endocrinol. 2005, 19, 1951–1959. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.; Lu, B.; Leygue, E.; Murphy, L.C. Putative functional characteristics of human estrogen receptor-beta isoforms. J. Mol. Endocrinol. 2003, 30, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Green, S.; Greene, G.; Krust, A.; Bornert, J.M.; Jeltsch, J.M.; Staub, A.; Jensen, E.; Scrace, G.; Waterfield, M. Cloning of the human estrogen receptor cDNA. Proc. Natl. Acad. Sci. USA 1985, 82, 7889–7893. [Google Scholar] [CrossRef]
- Green, S.; Walter, P.; Greene, G.; Krust, A.; Goffin, C.; Jensen, E.; Scrace, G.; Waterfield, M.; Chambon, P. Cloning of the human oestrogen receptor cDNA. J. Steroid Biochem. 1986, 24, 77–83. [Google Scholar] [CrossRef]
- Greene, G.L.; Gilna, P.; Waterfield, M.; Baker, A.; Hort, Y.; Shine, J. Sequence and expression of human estrogen receptor complementary DNA. Science 1986, 231, 1150–1154. [Google Scholar] [CrossRef] [PubMed]
- Ponglikitmongkol, M.; Green, S.; Chambon, P. Genomic organization of the human oestrogen receptor gene. EMBO J. 1988, 7, 3385–3388. [Google Scholar] [CrossRef]
- Koš, M.; Reid, G.; Denger, S.; Gannon, F. Minireview: Genomic Organization of the Human ERα Gene Promoter Region. Mol. Endocrinol. 2001, 15, 2057–2063. [Google Scholar] [CrossRef] [PubMed]
- Hennighausen, L.; Robinson, G.W. Information networks in the mammary gland. Nat. Rev. Mol. Cell Biol. 2005, 6, 715–725. [Google Scholar] [CrossRef]
- Paine, I.S.; Lewis, M.T. The Terminal End Bud: The Little Engine that Could. J. Mammary Gland. Biol. Neoplasia 2017, 22, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.; Clarke, R.B.; Howell, A. Estrogen responsiveness and control of normal human breast proliferation. J. Mammary Gland. Biol. Neoplasia 1998, 3, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Petersen, O.W.; E Høyer, P.; van Deurs, B. Frequency and distribution of estrogen receptor-positive cells in normal, nonlactating human breast tissue. Cancer Res. 1987, 47, 5748–5751. [Google Scholar] [PubMed]
- Macias, H.; Hinck, L. Mammary gland development. Wiley Interdiscip. Rev. Dev. Biol. 2012, 1, 533–557. [Google Scholar] [CrossRef] [PubMed]
- Bondesson, M.; Hao, R.; Lin, C.-Y.; Williams, C.; Gustafsson, J.-. Åke Estrogen receptor signaling during vertebrate development. Biochim. Biophys. Acta (BBA)-Bioenerg. 2015, 1849, 142–151. [Google Scholar] [CrossRef]
- Petersen, O.W.; Polyak, K. Stem Cells in the Human Breast. Cold Spring Harb. Perspect. Biol. 2010, 2, a003160. [Google Scholar] [CrossRef]
- Silberstein, G.B.; van Horn, K.; Shyamala, G.; Daniel, C.W. Essential role of endogenous estrogen in directly stimulating mammary growth demonstrated by implants containing pure antiestrogens. Endocrinology 1994, 134, 84–90. [Google Scholar] [CrossRef]
- Daniel, C.W.; Silberstein, G.B.; Strickland, P. Direct action of 17 beta-estradiol on mouse mammary ducts analyzed by sustained release implants and steroid autoradiography. Cancer Res. 1987, 47, 6052–6057. [Google Scholar] [PubMed]
- Korach, K. Insights from the study of animals lacking functional estrogen receptor. Science 1994, 266, 1524–1527. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, S.; Winuthayanon, W.; Korach, K.S. What’s new in estrogen receptor action in the female reproductive tract. J. Mol. Endocrinol. 2016, 56, R55–R71. [Google Scholar] [CrossRef]
- Atalay, C.; Kanliöz, M.; Altinok, M. Menstrual cycle and hormone receptor status in breast cancer patients. Neoplasma 2002, 49, 278–284. [Google Scholar] [PubMed]
- Bartow, S.A. Use of the autopsy to study ontogeny and expression of the estrogen receptor gene in human breast. J. Mammary Gland. Biol. Neoplasia 1998, 3, 37–48. [Google Scholar] [CrossRef]
- Söderqvist, G.; von Schoultz, B.; Tani, E.; Skoog, L. Estrogen and progesterone receptor content in breast epithelial cells from healthy women during the menstrual cycle. Am. J. Obstet. Gynecol. 1993, 168, 874–879. [Google Scholar] [CrossRef]
- Clarke, R.; Howell, A.; Potten, C.S.; Anderson, E. Dissociation between steroid receptor expression and cell proliferation in the human breast. Cancer Res. 1997, 57, 4987–4991. [Google Scholar] [PubMed]
- Mallepell, S.; Krust, A.; Chambon, P.; Brisken, C. Paracrine signaling through the epithelial estrogen receptor α is required for proliferation and morphogenesis in the mammary gland. Proc. Natl. Acad. Sci. USA 2006, 103, 2196–2201. [Google Scholar] [CrossRef]
- Cheng, G.; Weihua, Z.; Warner, M.; Gustafsson, J.-A. Estrogen receptors ER and ER in proliferation in the rodent mammary gland. Proc. Natl. Acad. Sci. USA 2004, 101, 3739–3746. [Google Scholar] [CrossRef]
- Cheng, G.; Li, Y.; Omoto, Y.; Wang, Y.; Berg, T.; Nord, M.; Vihko, P.; Warner, M.; Piao, Y.-S.; Gustafsson, J.-Å. Differential Regulation of Estrogen Receptor (ER)α and ERβ in Primate Mammary Gland. J. Clin. Endocrinol. Metab. 2005, 90, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.; Zhong, Y.; Pan, Z. Autocrine regulation of cell proliferation by estrogen receptor-alpha in estrogen receptor-alpha-positive breast cancer cell lines. BMC Cancer 2009, 9, 31. [Google Scholar] [CrossRef] [PubMed]
- Stender, J.D.; Frasor, J.; Komm, B.; Chang, K.C.N.; Kraus, W.L.; Katzenellenbogen, B.S. Estrogen-Regulated Gene Networks in Human Breast Cancer Cells: Involvement of E2F1 in the Regulation of Cell Proliferation. Mol. Endocrinol. 2007, 21, 2112–2123. [Google Scholar] [CrossRef]
- Bourdeau, V.; Deschênes, J.; Laperrière, D.; Aid, M.; White, J.; Mader, S. Mechanisms of primary and secondary estrogen target gene regulation in breast cancer cells. Nucleic Acids Res. 2007, 36, 76–93. [Google Scholar] [CrossRef] [PubMed]
- Millour, J.; Constantinidou, D.; Stavropoulou, A.V.; Wilson, M.; Myatt, S.S.; Kwok, J.M.-M.; Sivanandan, K.; Coombes, R.C.; Medema, R.; Hartman, J.; et al. FOXM1 is a transcriptional target of ERα and has a critical role in breast cancer endocrine sensitivity and resistance. Oncogene 2010, 29, 2983–2995. [Google Scholar] [CrossRef] [PubMed]
- JavanMoghadam, S.; Weihua, Z.; Hunt, K.K.; Keyomarsi, K. Estrogen receptor alpha is cell cycle-regulated and regulates the cell cycle in a ligand-dependent fashion. Cell Cycle 2016, 15, 1579–1590. [Google Scholar] [CrossRef] [PubMed]
- Fabian, C.J.; Kimler, B.F.; Zalles, C.M.; Khan, Q.J.; Mayo, M.S.; Phillips, T.A.; Simonsen, M.; Metheny, T.; Petroff, B.K. Reduction in proliferation with six months of letrozole in women on hormone replacement therapy. Breast Cancer Res. Treat. 2007, 106, 75–84. [Google Scholar] [CrossRef]
- Allison, K.H.; Hammond, M.E.H.; Dowsett, M.; McKernin, S.E.; Carey, L.A.; Fitzgibbons, P.L.; Hayes, D.F.; Lakhani, S.R.; Chavez-MacGregor, M.; Perlmutter, J.; et al. Estrogen and Progesterone Receptor Testing in Breast Cancer: ASCO/CAP Guideline Update. J. Clin. Oncol. 2020, 38, 1346–1366. [Google Scholar] [CrossRef] [PubMed]
- Giri, D.D.; Dundas, S.A.C.; Nottingham, J.F.; Underwood, J.C.E. Oestrogen receptors in benign epithelial lesions and intraduct carcinomas of the breast: An immunohistological study. Histopathology 1989, 15, 575–584. [Google Scholar] [CrossRef]
- Frech, M.S.; Halama, E.D.; Tilli, M.T.; Singh, B.; Gunther, E.J.; Chodosh, L.A.; Flaws, J.A.; Furth, P.A. Deregulated estrogen receptor alpha expression in mammary epithelial cells of transgenic mice results in the development of ductal carcinoma in situ. Cancer Res. 2005, 65, 681–685. [Google Scholar]
- Dabydeen, S.A.; Furth, P.A. Genetically engineered ERα-positive breast cancer mouse models. Endocr.-Relat. Cancer 2014, 21, R195–R208. [Google Scholar] [CrossRef] [PubMed]
- Holst, F.; Stahl, P.R.; Ruiz, C.; Hellwinkel, O.; Jehan, Z.; Wendland, M.; Lebeau, A.; Terracciano, L.; Al-Kuraya, K.; Jänicke, F.; et al. Estrogen receptor alpha (ESR1) gene amplification is frequent in breast cancer. Nat. Genet. 2007, 39, 655–660. [Google Scholar] [CrossRef]
- Brown, L.; Hoog, J.; Chin, S.-F.; Tao, Y.; Zayed, A.A.; Chin, K.; E Teschendorff, A.; Quackenbush, J.F.; Marioni, J.C.; Leung, S.; et al. ESR1 gene amplification in breast cancer: A common phenomenon? Nat. Genet. 2008, 40, 806–807. [Google Scholar] [CrossRef] [PubMed]
- Jeselsohn, R.; Buchwalter, G.; de Angelis, C.; Brown, M.; Schiff, R. ESR1 mutations—A mechanism for acquired endocrine resistance in breast cancer. Nat. Rev. Clin. Oncol. 2015, 12, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Toy, W.; Weir, H.; Razavi, P.; Lawson, M.; Goeppert, A.U.; Mazzola, A.M.; Smith, A.; Wilson, J.; Morrow, C.; Wong, W.L.; et al. Activating ESR1 Mutations Differentially Affect the Efficacy of ER Antagonists. Cancer Discov. 2017, 7, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Fanning, S.W.; Jeselsohn, R.; Dharmarajan, V.; Mayne, C.G.; Karimi, M.; Buchwalter, G.; Houtman, R.; Toy, W.; E Fowler, C.; Han, R.; et al. The SERM/SERD bazedoxifene disrupts ESR1 helix 12 to overcome acquired hormone resistance in breast cancer cells. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Zhou, W.; Hafner, M.; Blake, R.A.; Chalouni, C.; Chen, I.; de Bruyn, T.; Giltnane, J.M.; Hartman, S.; Heidersbach, A.; et al. Therapeutic Ligands Antagonize Estrogen Receptor Function by Impairing Its Mobility. Cell 2019, 178, 949–963. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.T.; Gou, X.; Seker, S.; Ellis, M.J. ESR1 alterations and metastasis in estrogen receptor positive breast cancer. J. Cancer Metastasis Treat. 2019, 5, 38. [Google Scholar] [CrossRef] [PubMed]
- Sørlie, T.; Tibshirani, R.; Parker, J.; Hastie, T.; Marron, J.S.; Nobel, A.; Deng, S.; Johnsen, H.; Pesich, R.; Geisler, S.; et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc. Natl. Acad. Sci. USA 2003, 100, 8418–8423. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, A.; Liu, M.C.; Zwart, A.; Lee, R.Y.; Gallagher, A.; Wang, Y.; Miller, W.R.; Dixon, J.M.; Clarke, R. Estrogen receptor alpha positive breast tumors and breast cancer cell lines share similarities in their transcriptome data structures. Int. J. Oncol. 2006, 29, 1581–1589. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lapidus, R.G.; Nass, S.J.; A Butash, K.; Parl, F.F.; A Weitzman, S.; Graff, J.G.; Herman, J.G.; Davidson, N.E. Mapping of ER gene CpG island methylation-specific polymerase chain reaction. Cancer Res. 1998, 58, 2515–2519. [Google Scholar] [PubMed]
- Ferguson, A.T.; Lapidus, R.G.; Baylin, S.B.; E Davidson, N. Demethylation of the estrogen receptor gene in estrogen receptor-negative breast cancer cells can reactivate estrogen receptor gene expression. Cancer Res. 1995, 55, 2279–2283. [Google Scholar] [PubMed]
- Yang, X.; Phillips, D.L.; Ferguson, A.T.; Nelson, W.G.; Herman, J.G.; Davidson, N.E. Synergistic activation of functional estrogen receptor (ER)-alpha by DNA methyltransferase and histone deacetylase inhibition in human ER-alpha-negative breast cancer cells. Cancer Res. 2001, 61, 7025–7029. [Google Scholar] [PubMed]
- Fan, J.; Yin, W.-J.; Lu, J.-S.; Wang, L.; Wu, J.; Wu, F.-Y.; Di, G.-H.; Shen, Z.-Z.; Shao, Z.-M. ERα negative breast cancer cells restore response to endocrine therapy by combination treatment with both HDAC inhibitor and DNMT inhibitor. J. Cancer Res. Clin. Oncol. 2008, 134, 883–890. [Google Scholar] [CrossRef]
- Billam, M.; Witt, A.E.; Davidson, N.E. The silent estrogen receptor—Can we make it speak? Cancer Biol. Ther. 2009, 8, 485–496. [Google Scholar] [CrossRef]
- De Cremoux, P.; Dalvai, M.; N’Doye, O.; Moutahir, F.; Rolland, G.; Chouchane-Mlik, O.; Assayag, F.; Lehmann-Che, J.; Kraus-Berthie, L.; Nicolas, A.; et al. HDAC inhibition does not induce estrogen receptor in human triple-negative breast cancer cell lines and patient-derived xenografts. Breast Cancer Res. Treat. 2015, 149, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Alao, J.P.; Lam, E.; Ali, S.; Buluwela, L.; Bordogna, W.; Lockey, P.; Varshochi, R.; Stavropoulou, A.V.; Coombes, R.C.; Vigushin, D.M. Histone Deacetylase Inhibitor Trichostatin A Represses Estrogen Receptor α-Dependent Transcription and Promotes Proteasomal Degradation of Cyclin D1 in Human Breast Carcinoma Cell Lines. Clin. Cancer Res. 2004, 10, 8094–8104. [Google Scholar] [CrossRef] [PubMed]
- Reid, G.; Métivier, R.; Lin, C.-Y.; Denger, S.; Ibberson, D.; Ivacevic, T.; Brand, H.; Benes, V.; Liu, E.T.; Gannon, F. Multiple mechanisms induce transcriptional silencing of a subset of genes, including oestrogen receptor α, in response to deacetylase inhibition by valproic acid and trichostatin A. Oncogene 2005, 24, 4894–4907. [Google Scholar] [CrossRef] [PubMed]
- Margueron, R.; Duong, V.; Bonnet, S.; Escande, A.; Vignon, F.; Balaguer, P.; Cavailles, V. Histone deacetylase inhibition and estrogen receptor alpha levels modulate the transcriptional activity of partial antiestrogens. J. Mol. Endocrinol. 2004, 32, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Rocha, W.; Sanchez, R.; Deschênes, J.; Auger, A.; Hébert, E.; White, J.H.; Mader, S. Opposite Effects of Histone Deacetylase Inhibitors on Glucocorticoid and Estrogen Signaling in Human Endometrial Ishikawa Cells. Mol. Pharmacol. 2005, 68, 1852–1862. [Google Scholar] [CrossRef]
- Yomtoubian, S.; Lee, S.B.; Verma, A.; Izzo, F.; Markowitz, G.; Choi, H.; Cerchietti, L.; Vahdat, L.; Brown, K.A.; Andreopoulou, E.; et al. Inhibition of EZH2 Catalytic Activity Selectively Targets a Metastatic Subpopulation in Triple-Negative Breast Cancer. Cell Rep. 2020, 30, 755–770. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.D.; Desai, K.; Kron, K.J.; Mazrooei, P.; Sinnott-Armstrong, N.A.; Treloar, A.E.; Dowar, M.; Thu, K.L.; Cescon, D.W.; Silvester, J.; et al. Noncoding somatic and inherited single-nucleotide variants converge to promote ESR1 expression in breast cancer. Nat. Genet. 2016, 48, 1260–1266. [Google Scholar] [CrossRef]
- Lemieux, S.; Sargeant, T.; Laperrière, D.; Ismail, H.; Boucher, G.; Rozendaal, M.; Lavallée, V.-P.; Ashton-Beaucage, D.; Wilhelm, B.; Hébert, J.; et al. MiSTIC, an integrated platform for the analysis of heterogeneity in large tumour transcriptome datasets. Nucleic Acids Res. 2017, 45, e122. [Google Scholar] [CrossRef] [PubMed]
- Keaveney, M.; Klug, J.; Dawson, M.; Nestor, P.; Neilan, J.; Forde, R.; Gannon, F. EVIDENCE FOR A PREVIOUSLY UNIDENTIFIED UPSTREAM EXON IN THE HUMAN OESTROGEN RECEPTOR GENE. J. Mol. Endocrinol. 1991, 6, 111–115. [Google Scholar] [CrossRef]
- Green, S.; Walter, P.; Kumar, V.; Krust, A.; Bornert, J.-M.; Argos, P.; Chambon, P. Human oestrogen receptor cDNA: Sequence, expression and homology to v-erb-A. Nat. Cell Biol. 1986, 320, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Flouriot, G.; Brand, H.; Denger, S.; Métivier, R.; Koš, M.; Reid, G.; Sonntag-Buck, V.; Gannon, F. Identification of a new isoform of the human estrogen receptor-alpha (hER-α) that is encoded by distinct transcripts and that is able to repress hER-α activation function 1. EMBO J. 2000, 19, 4688–4700. [Google Scholar] [CrossRef] [PubMed]
- Hattori, Y.; Ishii, H.; Morita, A.; Sakuma, Y.; Ozawa, H. Characterization of the fundamental properties of the N-terminal truncation (Δ exon 1) variant of estrogen receptor α in the rat. Gene 2015, 571, 117–125. [Google Scholar] [CrossRef] [PubMed]
- A Stevens, T.; Meech, R. BARX2 and estrogen receptor-α (ESR1) coordinately regulate the production of alternatively spliced ESR1 isoforms and control breast cancer cell growth and invasion. Oncogene 2006, 25, 5426–5435. [Google Scholar] [CrossRef] [PubMed]
- Ohe, K.; Miyajima, S.; Abe, I.; Tanaka, T.; Hamaguchi, Y.; Harada, Y.; Horita, Y.; Beppu, Y.; Ito, F.; Yamasaki, T.; et al. HMGA1a induces alternative splicing of estrogen receptor alpha in MCF-7 human breast cancer cells. J. Steroid Biochem. Mol. Biol. 2018, 182, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Penot, G.; Le Péron, C.; Mérot, Y.; Grimaud-Fanouillére, E.; Ferriere, F.; Boujrad, N.; Kah, O.; Saligaut, C.; Ducouret, B.; Métivier, R.; et al. The Human Estrogen Receptor-α Isoform hERα46 Antagonizes the Proliferative Influence of hERα66 in MCF7 Breast Cancer Cells. Endocrinology 2005, 146, 5474–5484. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, X.; Shen, P.; Loggie, B.W.; Chang, Y.; Deuel, T.F. Identification, cloning, and expression of human estrogen receptor-α36, a novel variant of human estrogen receptor-α66. Biochem. Biophys. Res. Commun. 2005, 336, 1023–1027. [Google Scholar] [CrossRef]
- Omarjee, S.; Jacquemetton, J.; Poulard, C.; Rochel, N.; Dejaegere, A.; Chebaro, Y.; Treilleux, I.; Marangoni, E.; Corbo, L.; Le Romancer, M. The molecular mechanisms underlying the ERα-36-mediated signaling in breast cancer. Oncogene 2016, 36, 2503–2514. [Google Scholar] [CrossRef]
- Wang, Q.; Jiang, J.; Ying, G.; Xie, X.-Q.; Zhang, X.; Xu, W.; Zhang, X.; Song, E.; Bu, H.; Ping, Y.-F.; et al. Tamoxifen enhances stemness and promotes metastasis of ERα36+ breast cancer by upregulating ALDH1A1 in cancer cells. Cell Res. 2018, 28, 336–358. [Google Scholar] [CrossRef]
- Pink, J.J.; Wu, S.-Q.; Wolf, D.M.; Bilimoria, M.M.; Jordan, V.C. A Novel 80 kDa Human Estrogen Receptor Containing a Duplication of Exons 6 and 7. Nucleic Acids Res. 1996, 24, 962–969. [Google Scholar] [CrossRef]
- Hartmaier, R.; Trabucco, S.; Priedigkeit, N.; Chung, J.; Parachoniak, C.; Borre, P.V.; Morley, S.; Rosenzweig, M.; Gay, L.; Goldberg, M.; et al. Recurrent hyperactive ESR1 fusion proteins in endocrine therapy-resistant breast cancer. Ann. Oncol. 2018, 29, 872–880. [Google Scholar] [CrossRef] [PubMed]
- Grandien, K.; Berkenstam, A.; Gustafsson, J.-Å. The estrogen receptor gene: Promoter organization and expression. Int. J. Biochem. Cell Biol. 1997, 29, 1343–1369. [Google Scholar] [CrossRef]
- Becherini, L.; Gennari, L.; Masi, L.; Mansani, R.; Massart, F.; Morelli, A.; Falchetti, A.; Gonnelli, S.; Fiorelli, G.; Tanini, A.; et al. Evidence of a linkage disequilibrium between polymorphisms in the human estrogen receptor alpha gene and their relationship to bone mass variation in postmenopausal Italian women. Hum. Mol. Genet. 2000, 9, 2043–2050. [Google Scholar] [CrossRef] [PubMed]
- Flouriot, G.; Griffin, C.; Kenealy, M.; Sonntag-Buck, V.; Gannon, F. Differentially Expressed Messenger RNA Isoforms of the Human Estrogen Receptor-α Gene Are Generated by Alternative Splicing and Promoter Usage. Mol. Endocrinol. 1998, 12, 1939–1954. [Google Scholar] [CrossRef] [PubMed]
- Tsuboi, K.; Nagatomo, T.; Gohno, T.; Higuchi, T.; Sasaki, S.; Fujiki, N.; Kurosumi, M.; Takei, H.; Yamaguchi, Y.; Niwa, T.; et al. Single CpG site methylation controls estrogen receptor gene transcription and correlates with hormone therapy resistance. J. Steroid Biochem. Mol. Biol. 2017, 171, 209–217. [Google Scholar] [CrossRef]
- Castles, C.; Oesterreich, S.; Hansen, R.; Fuqua, S. Auto-regulation of the estrogen receptor promoter. J. Steroid Biochem. Mol. Biol. 1997, 62, 155–163. [Google Scholar] [CrossRef]
- Hosey, A.M.; Gorski, J.J.; Murray, M.M.; Quinn, J.E.; Chung, W.Y.; Stewart, G.E.; James, C.R.; Farragher, S.M.; Mulligan, J.M.; Scott, A.N.; et al. Molecular Basis for Estrogen Receptor Deficiency in BRCA1-Linked Breast Cancer. J. Natl. Cancer Inst. 2007, 99, 1683–1694. [Google Scholar] [CrossRef] [PubMed]
- Shirley, S.H.; Rundhaug, J.E.; Tian, J.; Cullinan-Ammann, N.; Lambertz, I.; Conti, C.J.; Fuchs-Young, R. Transcriptional Regulation of Estrogen Receptor-α by p53 in Human Breast Cancer Cells. Cancer Res. 2009, 69, 3405–3414. [Google Scholar] [CrossRef]
- Degraffenried, L.A.; Hilsenbeck, S.G.; Fuqua, S.A. Sp1 is essential for estrogen receptor α gene transcription. J. Steroid Biochem. Mol. Biol. 2002, 82, 7–18. [Google Scholar] [CrossRef]
- Yu-Rice, Y.; Jin, Y.; Han, B.; Qu, Y.; Johnson, J.; Watanabe, T.; Cheng, L.; Deng, N.; Tanaka, H.; Gao, B.; et al. FOXC1 is involved in ERα silencing by counteracting GATA3 binding and is implicated in endocrine resistance. Oncogene 2016, 35, 5400–5411. [Google Scholar] [CrossRef] [PubMed]
- Tanimoto, K.; Eguchi, H.; Yoshida, T.; Hajiro-Nakanishi, K.; Hayashi, S.-I. Regulation of estrogen receptor gene mediated by promoter B responsible for its enhanced expression in human breast cancer. Nucleic Acids Res. 1999, 27, 903–909. [Google Scholar] [CrossRef] [PubMed]
- McPherson, L.A.; Baichwal, V.R.; Weigel, R. Identification of ERF-1 as a member of the AP2 transcription factor family. Proc. Natl. Acad. Sci. USA 1997, 94, 4342–4347. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Eguchi, H.; Nakachi, K.; Tanimoto, K.; Higashi, Y.; Suemasu, K.; Iino, Y.; Morishita, Y.; Hayashi, S.-I. Distinct mechanisms of loss of estrogen receptor alpha gene expression in human breast cancer: Methylation of the gene and alteration of trans-acting factors. Carcinogenesis 2000, 21, 2193–2201. [Google Scholar] [CrossRef] [PubMed]
- Deconinck, E.C.; A McPherson, L.; Weigel, R.J. Transcriptional regulation of estrogen receptor in breast carcinomas. Mol. Cell. Biol. 1995, 15, 2191–2196. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Treilleux, I.; Brown, M. A transcriptional enhancer required for the differential expression of the human estrogen receptor in breast cancers. Mol. Cell. Biol. 1997, 17, 1274–1280. [Google Scholar] [CrossRef] [PubMed]
- Friedrichs, N.; Steiner, S.; Buettner, R.; Knoepfle, G. Immunohistochemical expression patterns of AP2α and AP2γ in the developing fetal human breast. Histopathology 2007, 51, 814–823. [Google Scholar] [CrossRef]
- Cyr, A.R.; Kulak, M.V.; Park, J.M.; Bogachek, M.V.; Spanheimer, P.M.; Woodfield, G.W.; White-Baer, L.S.; O’Malley, Y.Q.; Sugg, S.L.; Olivier, A.K.; et al. TFAP2C governs the luminal epithelial phenotype in mammary development and carcinogenesis. Oncogene 2014, 34, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Woodfield, G.W.; Horan, A.D.; Chen, Y.; Weigel, R. TFAP2C Controls Hormone Response in Breast Cancer Cells through Multiple Pathways of Estrogen Signaling. Cancer Res. 2007, 67, 8439–8443. [Google Scholar] [CrossRef] [PubMed]
- Bogachek, M.V.; Chen, Y.; Kulak, M.; Woodfield, G.W.; Cyr, A.R.; Park, J.M.; Spanheimer, P.M.; Li, Y.; Li, T.; Weigel, R.J. Sumoylation Pathway Is Required to Maintain the Basal Breast Cancer Subtype. Cancer Cell 2014, 25, 748–761. [Google Scholar] [CrossRef] [PubMed]
- Angeloni, S.V.; Martin, M.B.; Garcia-Morales, P.; Castro-Galache, M.D.; A Ferragut, J.; Saceda, M. Regulation of estrogen receptor-alpha expression by the tumor suppressor gene p53 in MCF-7 cells. J. Endocrinol. 2004, 180, 497–504. [Google Scholar] [CrossRef]
- Denger, S.; Reid, G.; Brand, H.; Kos, M.; Gannon, F. Tissue-specific expression of human ERα and ERβ in the male. Mol. Cell. Endocrinol. 2001, 178, 155–160. [Google Scholar] [CrossRef]
- Penolazzi, L.; Lambertini, E.; Giordano, S.; Sollazzo, V.; Traina, G.; del Senno, L.; Piva, R. Methylation analysis of the promoter F of estrogen receptor α gene: Effects on the level of transcription on human osteoblastic cells. J. Steroid Biochem. Mol. Biol. 2004, 91, 1–9. [Google Scholar] [CrossRef]
- Brand, H.; Koš, M.; Denger, S.; Flouriot, G.; Gromoll, J.; Gannon, F.; Reid, G. A Novel Promoter Is Involved in the Expression of Estrogen Receptor α in Human Testis and Epididymis. Endocrinology 2002, 143, 3397–3404. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bulger, M.; Groudine, M. Looping versus linking: Toward a model for long-distance gene activation. Genes Dev. 1999, 13, 2465–2477. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.S.; A Meyer, C.; Song, J.; Li, W.; Geistlinger, T.R.; Eeckhoute, J.; Brodsky, A.S.; Keeton, E.K.; Fertuck, K.C.; Hall, G.F.; et al. Genome-wide analysis of estrogen receptor binding sites. Nat. Genet. 2006, 38, 1289–1297. [Google Scholar] [CrossRef]
- Kong, S.L.; Li, G.; Loh, S.L.; Sung, W.-K.; Liu, E.T. Cellular reprogramming by the conjoint action of ERα, FOXA1, and GATA3 to a ligand-inducible growth state. Mol. Syst. Biol. 2011, 7, 526. [Google Scholar] [CrossRef] [PubMed]
- A Serandour, A.; Brown, G.D.; Cohen, J.D.; Carroll, J.S. Development of an Illumina-based ChIP-exonuclease method provides insight into FoxA1-DNA binding properties. Genome Biol. 2013, 14, R147. [Google Scholar] [CrossRef] [PubMed]
- Mader, S.; Leroy, P.; Chen, J.; Chambon, P. Multiple parameters control the selectivity of nuclear receptors for their response elements. Selectivity and promiscuity in response element recognition by retinoic acid receptors and retinoid X receptors. J. Biol. Chem. 1993, 268, 591–600. [Google Scholar] [CrossRef]
- Driscoll, M.D.; Sathya, G.; Muyan, M.; Klinge, C.; Hilf, R.; Bambara, R.A. Sequence Requirements for Estrogen Receptor Binding to Estrogen Response Elements. J. Biol. Chem. 1998, 273, 29321–29330. [Google Scholar] [CrossRef] [PubMed]
- Bourdeau, V.; Deschênes, J.; Métivier, R.; Nagai, Y.; Nguyen, D.; Bretschneider, N.; Gannon, F.; White, J.H.; Mader, S. Genome-Wide Identification of High-Affinity Estrogen Response Elements in Human and Mouse. Mol. Endocrinol. 2004, 18, 1411–1427. [Google Scholar] [CrossRef]
- Overdier, D.G.; Porcella, A.; Costa, R.H. The DNA-binding specificity of the hepatocyte nuclear factor 3/forkhead domain is influenced by amino-acid residues adjacent to the recognition helix. Mol. Cell. Biol. 1994, 14, 2755–2766. [Google Scholar] [CrossRef] [PubMed]
- Lentjes, M.H.F.M.; Niessen, H.E.C.; Akiyama, Y.; de Bruine, A.P.; Melotte, V.; van Engeland, M. The emerging role of GATA transcription factors in development and disease. Expert Rev. Mol. Med. 2016, 18, e3. [Google Scholar] [CrossRef] [PubMed]
- Merika, M.; Orkin, S.H. DNA-binding specificity of GATA family transcription factors. Mol. Cell. Biol. 1993, 13, 3999–4010. [Google Scholar] [CrossRef]
- Bates, D.L.; Chen, Y.; Kim, G.; Guo, L.; Chen, L. Crystal Structures of Multiple GATA Zinc Fingers Bound to DNA Reveal New Insights into DNA Recognition and Self-Association by GATA. J. Mol. Biol. 2008, 381, 1292–1306. [Google Scholar] [CrossRef] [PubMed]
- Hanstein, B.; Eckner, R.; DiRenzo, J.; Halachmi, S.; Liu, H.; Searcy, B.; Kurokawa, R.; Brown, M. p300 is a component of an estrogen receptor coactivator complex. Proc. Natl. Acad. Sci. USA 1996, 93, 11540–11545. [Google Scholar] [CrossRef] [PubMed]
- Zwart, W.; Theodorou, V.; Kok, M.; Canisius, S.; Linn, S.; Carroll, J.S. Oestrogen receptor-co-factor-chromatin specificity in the transcriptional regulation of breast cancer. EMBO J. 2011, 30, 4764–4776. [Google Scholar] [CrossRef]
- Fullwood, M.; Liu, M.H.; Pan, Y.F.; Liu, J.; Xu, H.; Bin Mohamed, Y.; Orlov, Y.; Velkov, S.; Thoreau, H.; Mei, P.H.; et al. An oestrogen-receptor-α-bound human chromatin interactome. Nat. Cell Biol. 2009, 462, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Frietze, S.; Wang, R.; Yao, L.; Tak, Y.G.; Ye, Z.; Gaddis, M.; Witt, H.; Farnham, P.J.; Jin, V.X. Cell type-specific binding patterns reveal that TCF7L2 can be tethered to the genome by association with GATA3. Genome Biol. 2012, 13, 1–18. [Google Scholar] [CrossRef]
- Smith, V.M.; Lee, P.P.; Szychowski, S.; Winoto, A. GATA-3 Dominant Negative Mutant. J. Biol. Chem. 1995, 270, 1515–1520. [Google Scholar] [CrossRef]
- Shinnakasu, R.; Yamashita, M.; Shinoda, K.; Endo, Y.; Hosokawa, H.; Hasegawa, A.; Ikemizu, S.; Nakayama, T. Critical YxKxHxxxRP Motif in the C-Terminal Region of GATA3 for Its DNA Binding and Function. J. Immunol. 2006, 177, 5801–5810. [Google Scholar] [CrossRef] [PubMed]
- Crossley, M.; Merika, M.; Orkin, S.H. Self-association of the erythroid transcription factor GATA-1 mediated by its zinc finger domains. Mol. Cell. Biol. 1995, 15, 2448–2456. [Google Scholar] [CrossRef]
- Li, X.; Jin, J.; Yang, S.; Xu, W.; Meng, X.; Deng, H.; Zhan, J.; Gao, S.; Zhang, H. GATA3 acetylation at K119 by CBP inhibits cell migration and invasion in lung adenocarcinoma. Biochem. Biophys. Res. Commun. 2018, 497, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, M.; Leclercq, G. About GATA3, HNF3A, and XBP1, three genes co-expressed with the oestrogen receptor-α gene (ESR1) in breast cancer. Mol. Cell. Endocrinol. 2004, 219, 1–7. [Google Scholar] [CrossRef]
- Kouros-Mehr, H.; Werb, Z. Candidate regulators of mammary branching morphogenesis identified by genome-wide transcript analysis. Dev. Dyn. 2006, 235, 3404–3412. [Google Scholar] [CrossRef] [PubMed]
- Kouros-Mehr, H.; Slorach, E.M.; Sternlicht, M.D.; Werb, Z. GATA-3 Maintains the Differentiation of the Luminal Cell Fate in the Mammary Gland. Cell 2006, 127, 1041–1055. [Google Scholar] [CrossRef]
- Kouros-Mehr, H.; Kim, J.-W.; Bechis, S.K.; Werb, Z. GATA-3 and the regulation of the mammary luminal cell fate. Curr. Opin. Cell Biol. 2008, 20, 164–170. [Google Scholar] [CrossRef]
- Asselin-Labat, M.-L.; Sutherland, K.D.; Barker, H.; Thomas, R.; Shackleton, M.; Forrest, N.C.; Hartley, L.; Robb, L.; Grosveld, F.G.; van der Wees, J.; et al. Gata-3 is an essential regulator of mammary-gland morphogenesis and luminal-cell differentiation. Nat. Cell Biol. 2007, 9, 201–209. [Google Scholar] [CrossRef]
- Eeckhoute, J.; Keeton, E.K.; Lupien, M.; Krum, S.A.; Carroll, J.; Brown, M. Positive Cross-Regulatory Loop Ties GATA-3 to Estrogen Receptor α Expression in Breast Cancer. Cancer Res. 2007, 67, 6477–6483. [Google Scholar] [CrossRef]
- Chu, I.M.; Lai, W.-C.; Aprelikova, O.; El Touny, L.H.; Kouros-Mehr, H.; Green, J.E. Expression of GATA3 in MDA-MB-231 Triple-negative Breast Cancer Cells Induces a Growth Inhibitory Response to TGFß. PLoS ONE 2013, 8, e61125. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Cao, Q.J.; Arenas, R.B.; Bentley, B.; Shao, R. GATA3 Inhibits Breast Cancer Metastasis through the Reversal of Epithelial-Mesenchymal Transition. J. Biol. Chem. 2010, 285, 14042–14051. [Google Scholar] [CrossRef] [PubMed]
- Theodorou, V.; Stark, R.; Menon, S.; Carroll, J.S. GATA3 acts upstream of FOXA1 in mediating ESR1 binding by shaping enhancer accessibility. Genome Res. 2013, 23, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Takaku, M.; Grimm, S.A.; Shimbo, T.; Perera, L.; Menafra, R.; Stunnenberg, H.G.; Archer, T.K.; Machida, S.; Kurumizaka, H.; Wade, P.A. GATA3-dependent cellular reprogramming requires activation-domain dependent recruitment of a chromatin remodeler. Genome Biol. 2016, 17, 36. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Takizawa, Y.; Takaku, M.; Kato, D.; Kumagawa, Y.; Grimm, S.A.; Wade, P.A.; Kurumizaka, H. Interaction of the pioneer transcription factor GATA3 with nucleosomes. Nat. Commun. 2020, 11, 4136. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Lambros, M.B.; Horlings, H.M.; Pearson, A.; Sharpe, R.; Natrajan, R.; Geyer, F.C.; van Kouwenhove, M.; Kreike, B.; Mackay, A.; et al. Integrative molecular profiling of triple negative breast cancers identifies amplicon drivers and potential therapeutic targets. Oncogene 2010, 29, 2013–2023. [Google Scholar] [CrossRef] [PubMed]
- Usary, J.; Llaca, V.; Karaca, G.; Presswala, S.; Karaca, M.; He, X.; Langerød, A.; Kåresen, R.; Oh, D.S.; Dressler, L.G.; et al. Mutation of GATA3 in human breast tumors. Oncogene 2004, 23, 7669–7678. [Google Scholar] [CrossRef]
- Ciriello, G.; Gatza, M.; Beck, A.H.; Wilkerson, M.D.; Rhie, S.; Pastore, A.; Zhang, H.; McLellan, M.; Yau, C.; Kandoth, C.; et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 2015, 163, 506–519. [Google Scholar] [CrossRef] [PubMed]
- Emmanuel, N.; Lofgren, K.A.; Peterson, E.A.; Meier, D.R.; Jung, E.H.; Kenny, P.A. Mutant GATA3 Actively Promotes the Growth of Normal and Malignant Mammary Cells. Anticancer Res. 2018, 38, 4435–4441. [Google Scholar] [CrossRef]
- Afzaljavan, F.; Sadr, A.S.; Savas, S.; Pasdar, A. GATA3 somatic mutations are associated with clinicopathological features and expression profile in TCGA breast cancer patients. Sci. Rep. 2021, 11, 1679. [Google Scholar] [CrossRef] [PubMed]
- Adomas, A.B.; A Grimm, S.; Malone, C.; Takaku, M.; Sims, J.K.; A Wade, P. Breast tumor specific mutation in GATA3 affects physiological mechanisms regulating transcription factor turnover. BMC Cancer 2014, 14, 278. [Google Scholar] [CrossRef] [PubMed]
- Takaku, M.; A Grimm, S.; de Kumar, B.; Bennett, B.D.; A Wade, P. Cancer-specific mutation of GATA3 disrupts the transcriptional regulatory network governed by Estrogen Receptor alpha, FOXA1 and GATA3. Nucleic Acids Res. 2020, 48, 4756–4768. [Google Scholar] [CrossRef]
- Hruschka, N.; Kalisz, M.; Subijana, M.; Graña-Castro, O.; Del Cano-Ochoa, F.; Brunet, L.P.; Chernukhin, I.; Sagrera, A.; de Reynies, A.; Kloesch, B.; et al. The GATA3 X308_Splice breast cancer mutation is a hormone context-dependent oncogenic driver. Oncogene 2020, 39, 5455–5467. [Google Scholar] [CrossRef] [PubMed]
- Clark, K.L.; Halay, E.D.; Lai, E.; Burley, S. Co-crystal structure of the HNF-3/fork head DNA-recognition motif resembles histone H5. Nat. Cell Biol. 1993, 364, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Lam, E.W.-F.; Brosens, J.; Gomes, A.R.; Koo, C.Y. Forkhead box proteins: Tuning forks for transcriptional harmony. Nat. Rev. Cancer 2013, 13, 482–495. [Google Scholar] [CrossRef]
- Marsden, I.; Jin, C.; Liao, X. Structural changes in the region directly adjacent to the DNA-binding helix highlight a possible mechanism to explain the observed changes in the sequence-specific binding of winged helix proteins. J. Mol. Biol. 1998, 278, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, G.M.; Lozada, K.L.; Miedler, J.D.; Harburg, G.; Hewitt, S.; Mosley, J.; Godwin, A.K.; Korach, K.; Visvader, J.E.; Kaestner, K.H.; et al. FOXA1 is an essential determinant of ERα expression and mammary ductal morphogenesis. Development 2010, 137, 2045–2054. [Google Scholar] [CrossRef] [PubMed]
- Caizzi, L.; Ferrero, G.; Cutrupi, S.; Cordero, F.; Ballaré, C.; Miano, V.; Reineri, S.; Ricci, L.; Friard, O.; Testori, A.; et al. Genome-wide activity of unliganded estrogen receptor-α in breast cancer cells. Proc. Natl. Acad. Sci. USA 2014, 111, 4892–4897. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, G.M.; Bebek, G.; Ginther, C.L.; Sizemore, S.T.; Lozada, K.L.; Miedler, J.D.; A Anderson, L.; Godwin, A.K.; Abdul-Karim, F.W.; Slamon, D.J.; et al. FOXA1 represses the molecular phenotype of basal breast cancer cells. Oncogene 2013, 32, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, L.A.; McPherson, C.E.; Bossard, P.; Stevens, K.; Cherian, S.; Shim, E.Y.; Clark, K.L.; Burley, S.; Zaret, K.S. Binding of the winged-helix transcription factor HNF3 to a linker histone site on the nucleosome. EMBO J. 1998, 17, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, L.A.; Lin, F.R.; Cuesta, I.; Friedman, D.; Jarnik, M.; Zaret, K.S. Opening of Compacted Chromatin by Early Developmental Transcription Factors HNF3 (FoxA) and GATA-4. Mol. Cell 2002, 9, 279–289. [Google Scholar] [CrossRef]
- Laganiere, J.; Deblois, G.; Lefebvre, C.; Bataille, A.R.; Robert, F.; Giguere, V. Location analysis of estrogen receptor target promoters reveals that FOXA1 defines a domain of the estrogen response. Proc. Natl. Acad. Sci. USA 2005, 102, 11651–11656. [Google Scholar] [CrossRef]
- Lupien, M.; Eeckhoute, J.; Meyer, C.A.; Wang, Q.; Zhang, Y.; Li, W.; Carroll, J.S.; Liu, X.S.; Brown, M. FoxA1 Translates Epigenetic Signatures into Enhancer-Driven Lineage-Specific Transcription. Cell 2008, 132, 958–970. [Google Scholar] [CrossRef] [PubMed]
- Sahu, B.; Laakso, M.; Ovaska, K.; Mirtti, T.; Lundin, J.; Rannikko, A.; Sankila, A.; Turunen, J.-P.; Lundin, M.; Konsti, J.; et al. Dual role of FoxA1 in androgen receptor binding to chromatin, androgen signalling and prostate cancer. EMBO J. 2011, 30, 3962–3976. [Google Scholar] [CrossRef]
- Fu, X.; Jeselsohn, R.; Pereira, R.; Hollingsworth, E.F.; Creighton, C.J.; Li, F.; Shea, M.; Nardone, A.; de Angelis, C.; Heiser, L.M.; et al. FOXA1 overexpression mediates endocrine resistance by altering the ER transcriptome and IL-8 expression in ER-positive breast cancer. Proc. Natl. Acad. Sci. USA 2016, 113, E6600–E6609. [Google Scholar] [CrossRef]
- Arruabarrena-Aristorena, A.; Maag, J.L.; Kittane, S.; Cai, Y.; Karthaus, W.R.; Ladewig, E.; Park, J.; Kannan, S.; Ferrando, L.; Cocco, E.; et al. FOXA1 Mutations Reveal Distinct Chromatin Profiles and Influence Therapeutic Response in Breast Cancer. Cancer Cell 2020, 38, 534–550. [Google Scholar] [CrossRef] [PubMed]
- Rheinbay, E.; Parasuraman, P.; Grimsby, J.; Tiao, G.; Engreitz, J.M.; Kim, J.; Lawrence, M.S.; Taylor-Weiner, A.; Rodriguez-Cuevas, S.; Rosenberg, M.; et al. Recurrent and functional regulatory mutations in breast cancer. Nat. Cell Biol. 2017, 547, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Bullock, M. FOXO factors and breast cancer: Outfoxing endocrine resistance. Endocr.-Relat. Cancer 2015, 23, R113–R130. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Sonenshein, G.E. Forkhead Box Transcription Factor FOXO3a Regulates Estrogen Receptor Alpha Expression and Is Repressed by the Her-2/neu/Phosphatidylinositol 3-Kinase/Akt Signaling Pathway. Mol. Cell. Biol. 2004, 24, 8681–8690. [Google Scholar] [CrossRef]
- Zou, Y.; Tsai, W.-B.; Cheng, C.-J.; Hsu, C.; Chung, Y.M.; Li, P.-C.; Lin, S.-H.; Hu, M.C.-T. Forkhead box transcription factor FOXO3a suppresses estrogen-dependent breast cancer cell proliferation and tumorigenesis. Breast Cancer Res. 2008, 10, R21. [Google Scholar] [CrossRef] [PubMed]
- Alasiri, G.; Jiramongkol, Y.; Zona, S.; Fan, L.Y.-N.; Mahmud, Z.; Gong, G.; Lee, H.J.; Lam, E.W.-F. Regulation of PERK expression by FOXO3: A vulnerability of drug-resistant cancer cells. Oncogene 2019, 38, 6382–6398. [Google Scholar] [CrossRef] [PubMed]
- Madureira, P.; Varshochi, R.; Constantinidou, D.; Francis, R.E.; Coombes, R.C.; Yao, K.-M.; Lam, E. The Forkhead Box M1 Protein Regulates the Transcription of the Estrogen Receptor α in Breast Cancer Cells. J. Biol. Chem. 2006, 281, 25167–25176. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Wang, Q.; Xie, Y.; Qiao, X.; Zhang, S.; Wang, Y.; Yang, Y.; Zhang, B. Identification of FOXM1 as a specific marker for triple-negative breast cancer. Int. J. Oncol. 2018, 54, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Carr-Ascher, J.; Kiefer, M.M.; Park, H.J.; Li, J.; Wang, Z.; Fontanarosa, J.; DeWaal, D.; Kopanja, D.; Benevolenskaya, E.V.; Guzman, G.; et al. FoxM1 Regulates Mammary Luminal Cell Fate. Cell Rep. 2012, 1, 715–729. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ray, P.S.; Sim, M.-S.; Zhou, X.Z.; Lu, K.P.; Lee, A.V.; Lin, X.; Bagaria, S.P.; Giuliano, A.E.; Cui, X. FOXC1 regulates the functions of human basal-like breast cancer cells by activating NF-κB signaling. Oncogene 2012, 31, 4798–4802. [Google Scholar] [CrossRef] [PubMed]
- Taube, J.; Herschkowitz, J.I.; Komurov, K.; Zhou, A.; Gupta, S.; Yang, J.; Hartwell, K.; Onder, T.; Gupta, P.B.; Evans, K.W.; et al. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc. Natl. Acad. Sci. USA 2010, 107, 15449–15454. [Google Scholar] [CrossRef] [PubMed]
- Welboren, W.-J.; A van Driel, M.; Janssen-Megens, E.M.; van Heeringen, S.; Sweep, F.; Span, P.; Stunnenberg, H.G. ChIP-Seq of ERα and RNA polymerase II defines genes differentially responding to ligands. EMBO J. 2009, 28, 1418–1428. [Google Scholar] [CrossRef]
- Read, L.D.; Greene, G.L.; Katzenellenbogen, B.S. Regulation of Estrogen Receptor Messenger Ribonucleic Acid and Protein Levels in Human Breast Cancer Cell Lines by Sex Steroid Hormones, Their Antagonists, and Growth Factors. Mol. Endocrinol. 1989, 3, 295–304. [Google Scholar] [CrossRef]
- Grober, O.M.V.; Mutarelli, M.; Giurato, G.; Ravo, M.; Cicatiello, L.; de Filippo, M.R.; Ferraro, L.; Nassa, G.; Papa, M.F.; Paris, O.; et al. Global analysis of estrogen receptor beta binding to breast cancer cell genome reveals an extensive interplay with estrogen receptor alpha for target gene regulation. BMC Genom. 2011, 12, 36. [Google Scholar] [CrossRef]
- Matthews, J.; Gustafsson, J.A. Estrogen signaling: A subtle balance between ER alpha and ER beta. Mol. Interv. 2003, 3, 281–292. [Google Scholar] [CrossRef]
- Cotnoir-White, D.; El Ezzy, M.; Boulay, P.-L.; Rozendaal, M.; Bouvier, M.; Gagnon, E.; Mader, S. Monitoring ligand-dependent assembly of receptor ternary complexes in live cells by BRETFect. Proc. Natl. Acad. Sci. USA 2018, 115, E2653–E2662. [Google Scholar] [CrossRef]
- Eckert, R.L.; Mullick, A.; Rorke, E.A.; Katzenellenbogen, B.S. Estrogen Receptor Synthesis and Turnover in MCF-7 Breast Cancer Cells Measured by a Density Shift Technique. Endocrinology 1984, 114, 629–637. [Google Scholar] [CrossRef]
- A Khan, S.; Sachdeva, A.; Naim, S.; Meguid, M.M.; Marx, W.; Simon, H.; Halverson, J.D.; Numann, P.J. The normal breast epithelium of women with breast cancer displays an aberrant response to estradiol. Cancer Epidemiol. Biomark. Prev. 1999, 8, 867–872. [Google Scholar]
- Macaluso, M.; Cinti, C.; Russo, G.; Russo, A.; Giordano, A. pRb2/p130-E2F4/5-HDAC1-SUV39H1-p300 and pRb2/p130-E2F4/5-HDAC1-SUV39H1-DNMT1 multimolecular complexes mediate the transcription of estrogen receptor-α in breast cancer. Oncogene 2003, 22, 3511–3517. [Google Scholar] [CrossRef] [PubMed]
- Kalyuga, M.; Gallego-Ortega, D.; Lee, H.; Roden, D.L.; Cowley, M.; Caldon, C.E.; Stone, A.; Allerdice, S.L.; Valdes-Mora, F.; Launchbury, R.; et al. ELF5 Suppresses Estrogen Sensitivity and Underpins the Acquisition of Antiestrogen Resistance in Luminal Breast Cancer. PLoS Biol. 2012, 10, e1001461. [Google Scholar] [CrossRef] [PubMed]
- Dhasarathy, A.; Kajita, M.; Wade, P.A. The Transcription Factor Snail Mediates Epithelial to Mesenchymal Transitions by Repression of Estrogen Receptor-α. Mol. Endocrinol. 2007, 21, 2907–2918. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhou, C.; Jiang, H.; Liang, L.; Shi, W.; Zhang, Q.; Sun, P.; Xiang, R.; Wang, Y.; Yang, S. ZEB1 induces ER-α promoter hypermethylation and confers antiestrogen resistance in breast cancer. Cell Death Dis. 2017, 8, e2732. [Google Scholar] [CrossRef]
- Klinge, C.M. miRNAs and estrogen action. Trends Endocrinol. Metab. 2012, 23, 223–233. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Porras, L.; Ismail, H.; Mader, S. Positive Regulation of Estrogen Receptor Alpha in Breast Tumorigenesis. Cells 2021, 10, 2966. https://doi.org/10.3390/cells10112966
Porras L, Ismail H, Mader S. Positive Regulation of Estrogen Receptor Alpha in Breast Tumorigenesis. Cells. 2021; 10(11):2966. https://doi.org/10.3390/cells10112966
Chicago/Turabian StylePorras, Lucas, Houssam Ismail, and Sylvie Mader. 2021. "Positive Regulation of Estrogen Receptor Alpha in Breast Tumorigenesis" Cells 10, no. 11: 2966. https://doi.org/10.3390/cells10112966
APA StylePorras, L., Ismail, H., & Mader, S. (2021). Positive Regulation of Estrogen Receptor Alpha in Breast Tumorigenesis. Cells, 10(11), 2966. https://doi.org/10.3390/cells10112966