
Transcriptome Profiling of Dysregulated GPCRs Reveals Overlapping Patterns across Psychiatric Disorders and Age-Disease Interactions

 ,
,
Abstract
1. Introduction
2. Methods
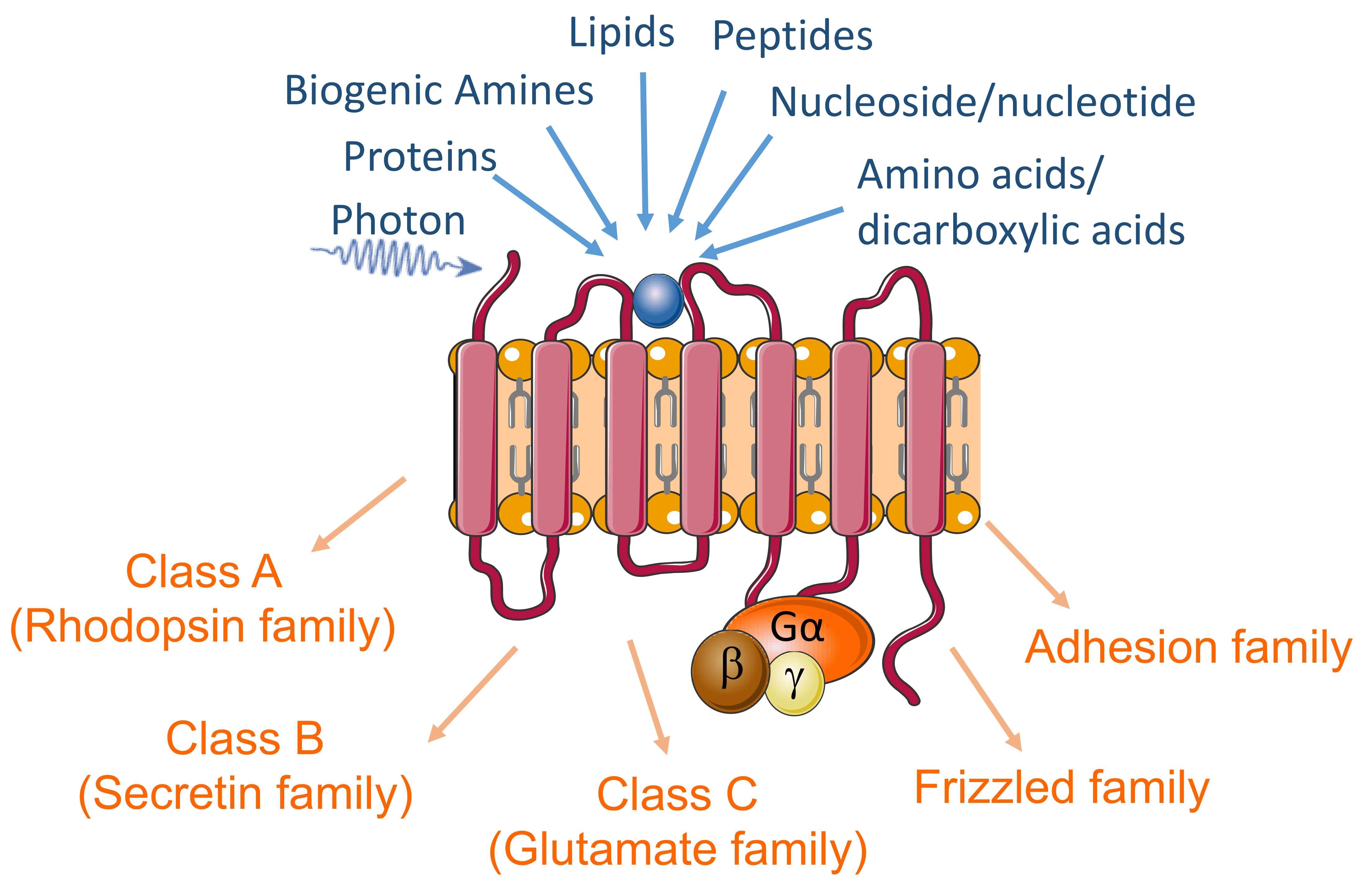
2.1. GPCR Genes
2.2. Transcriptomic Analysis of GPCR Genes in SCZ, ASD, BP, and MDD
2.3. GPCR Differential Expression with Chronological Age
2.4. Analysis of Age-Disease Interaction
3. Results
3.1. GPCRs List
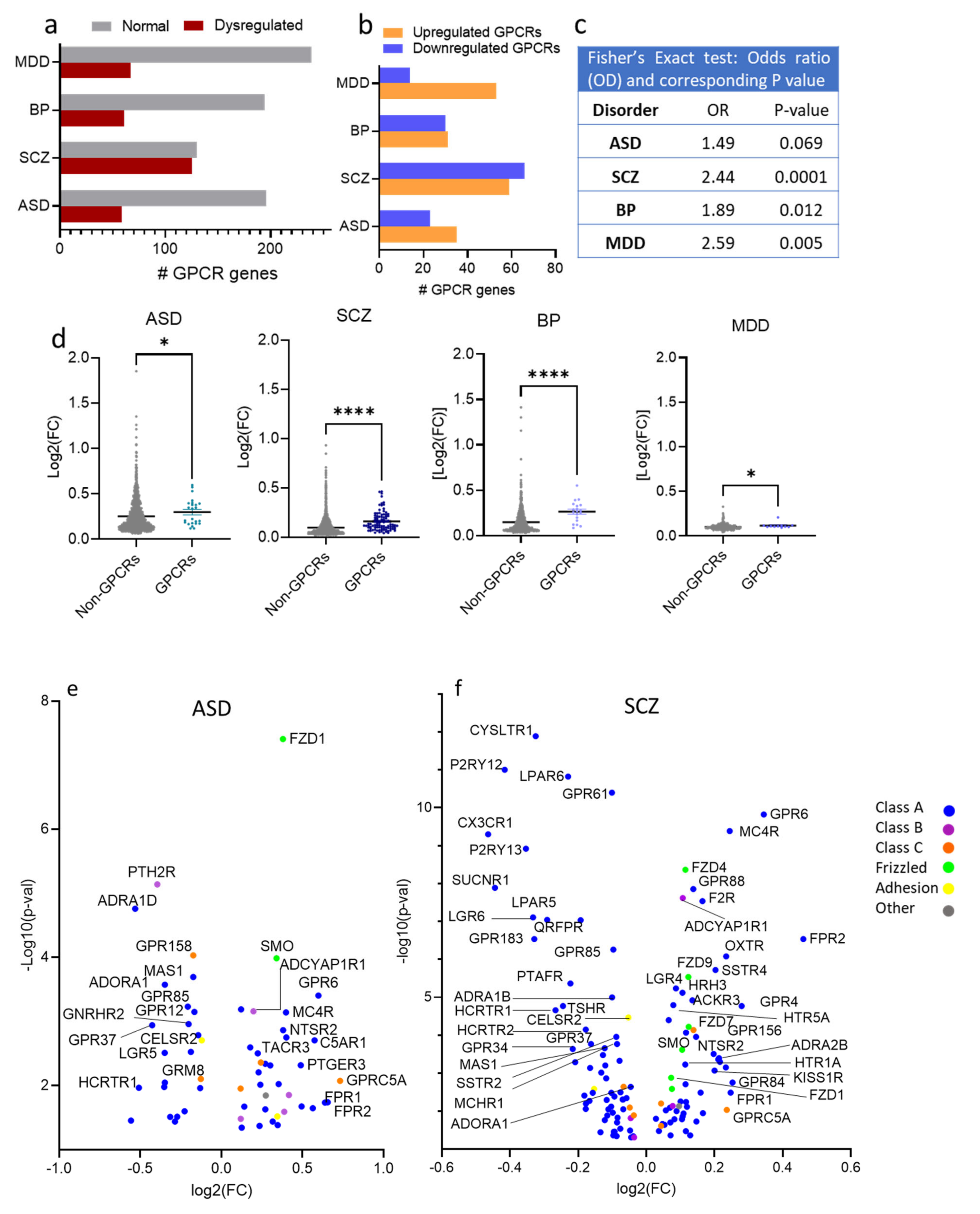
3.2. GPCRs’ Gene Family Is Overrepresented in Psychiatric Disorders’ Transcriptomes
3.3. Dysregulated GPCRs in Psychiatric Disorders Belong to Specific Receptor Subfamilies
3.4. Dysregulated GPCRs in the Four Psychiatric Disorders Couple to Specific G-Proteins
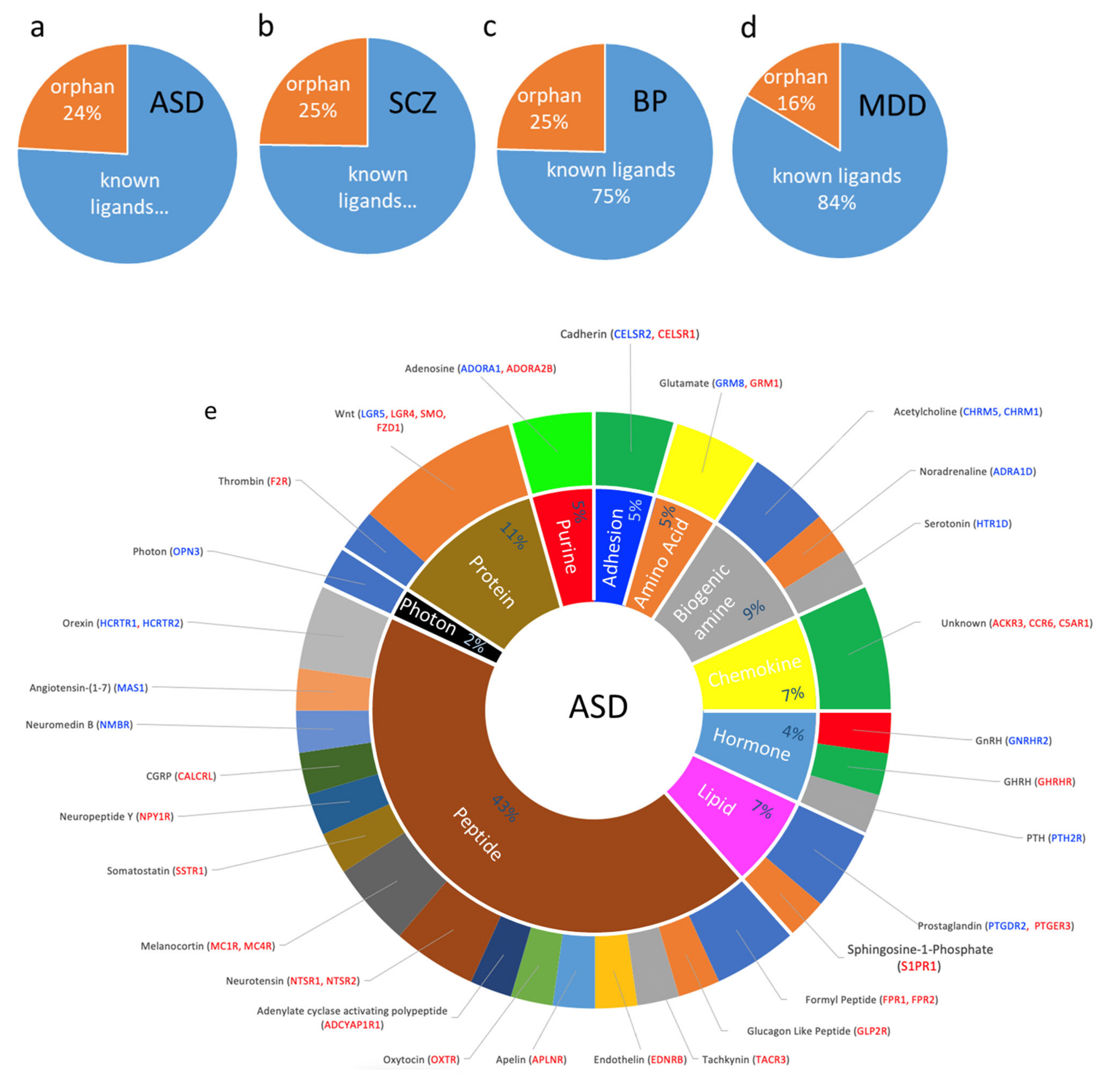
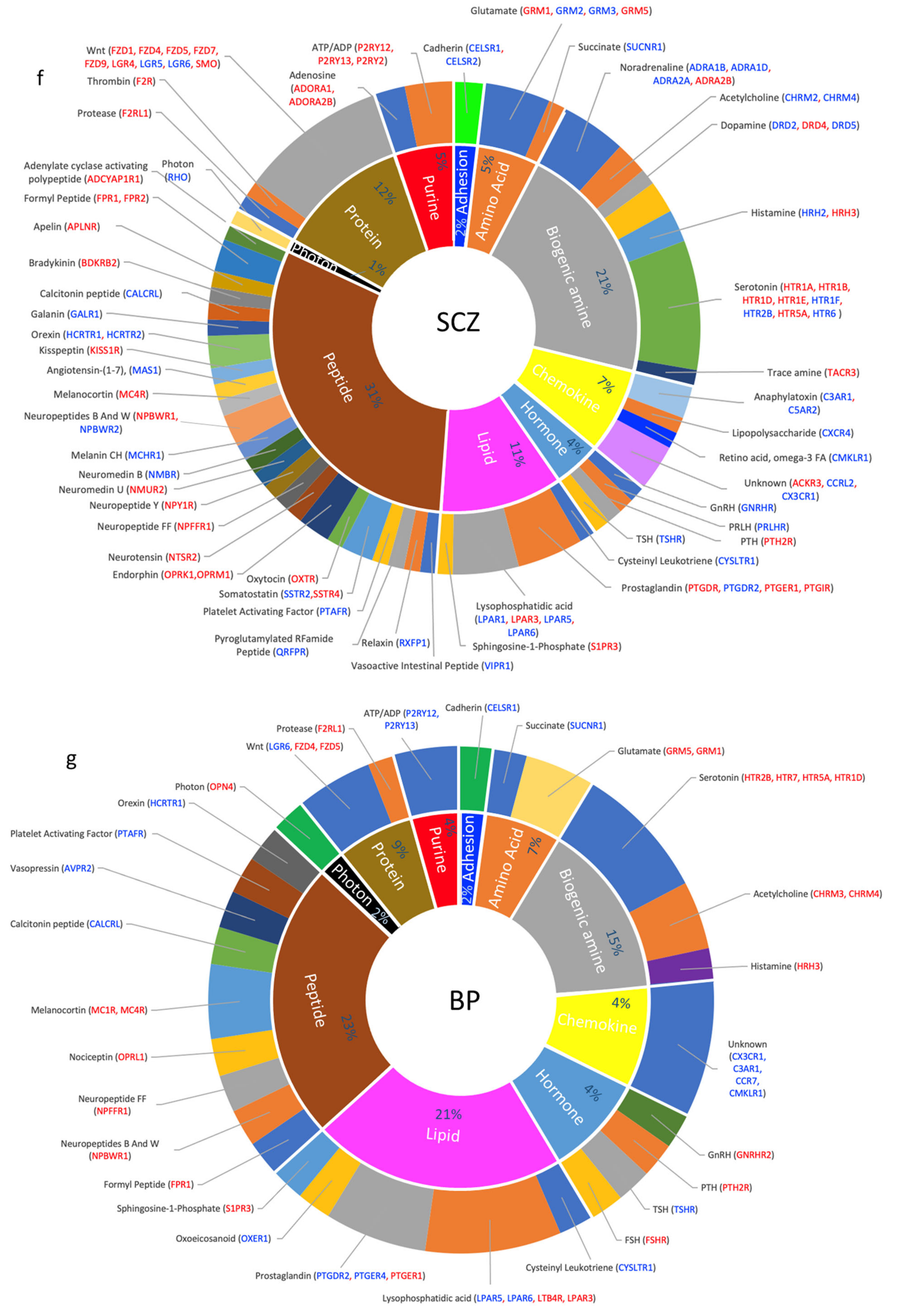
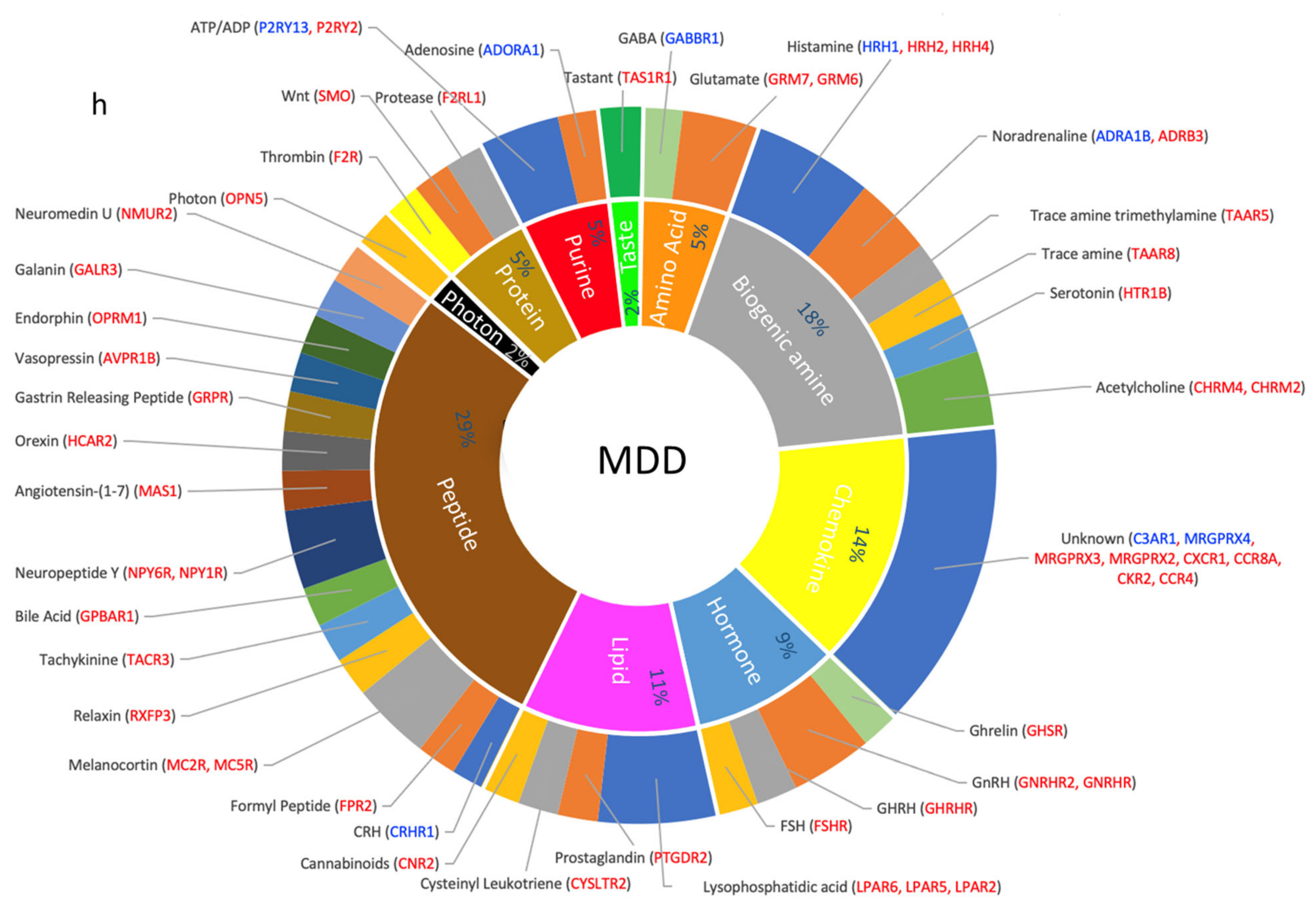
3.5. Dysregulated GPCRs Mediate Signal Transductions of Specific Neurotransmitter and Neuropeptide Systems
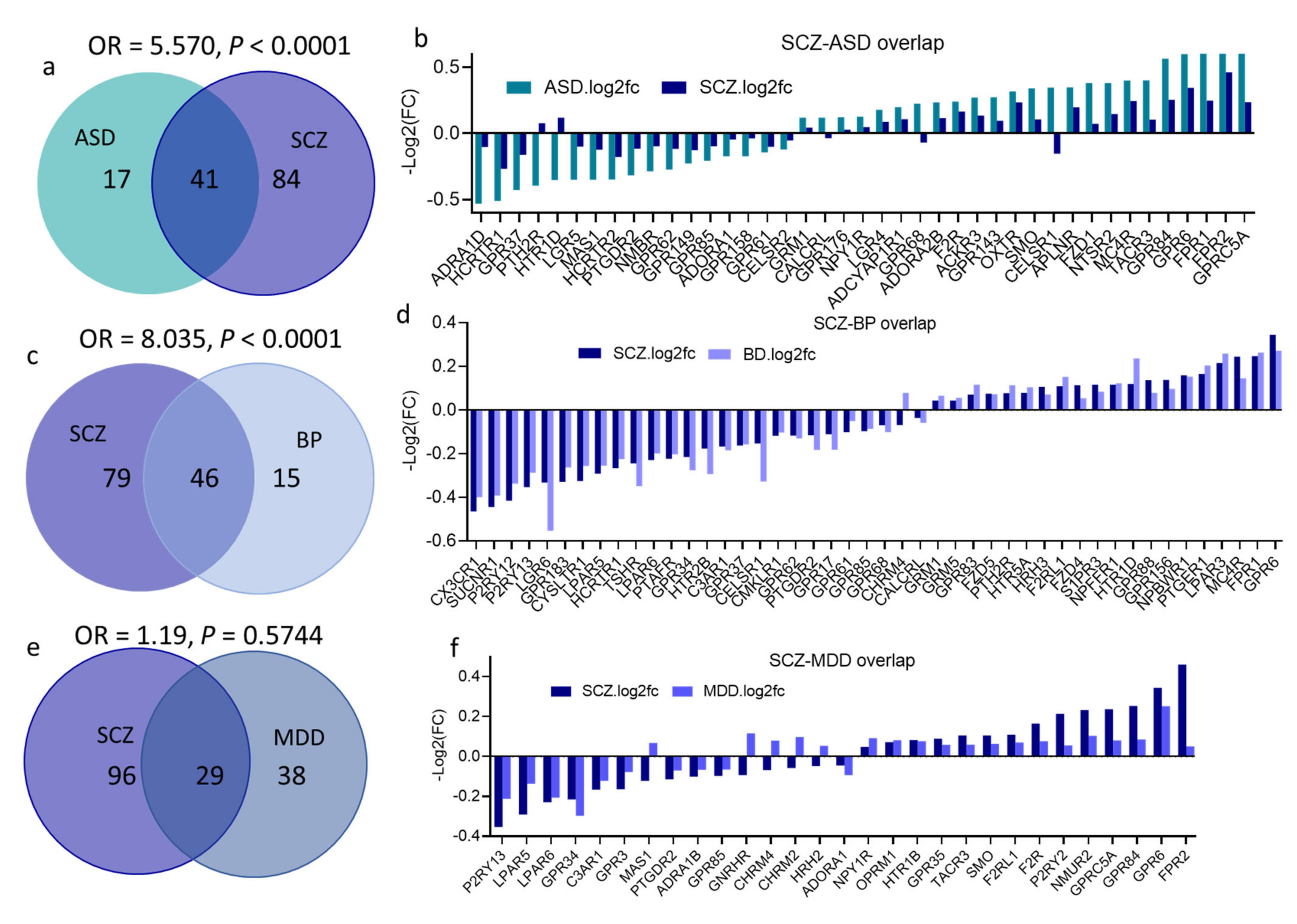
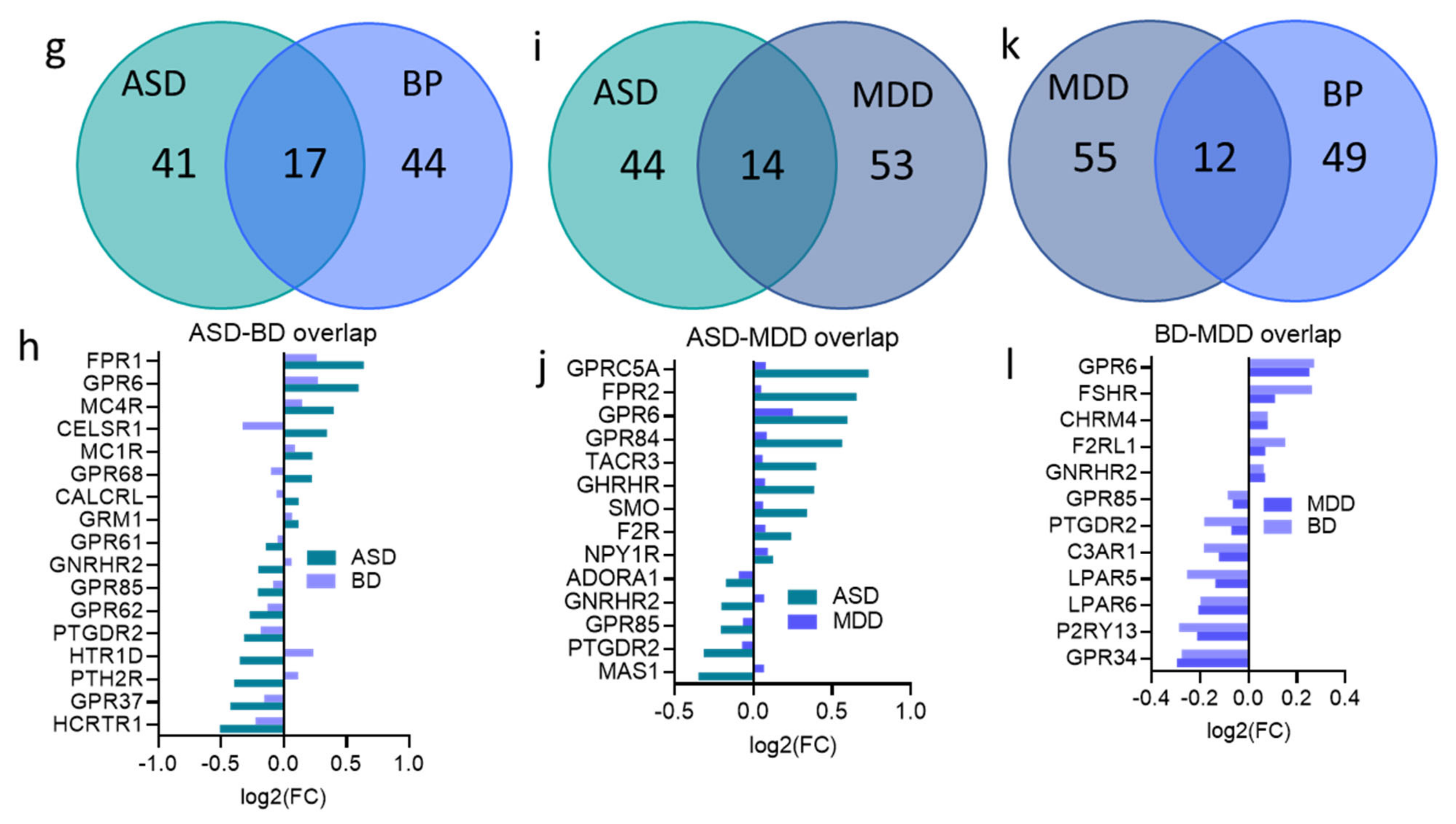
3.6. Overlap across Psychiatric Disorders of GPCR DEs and Their Signaling Systems
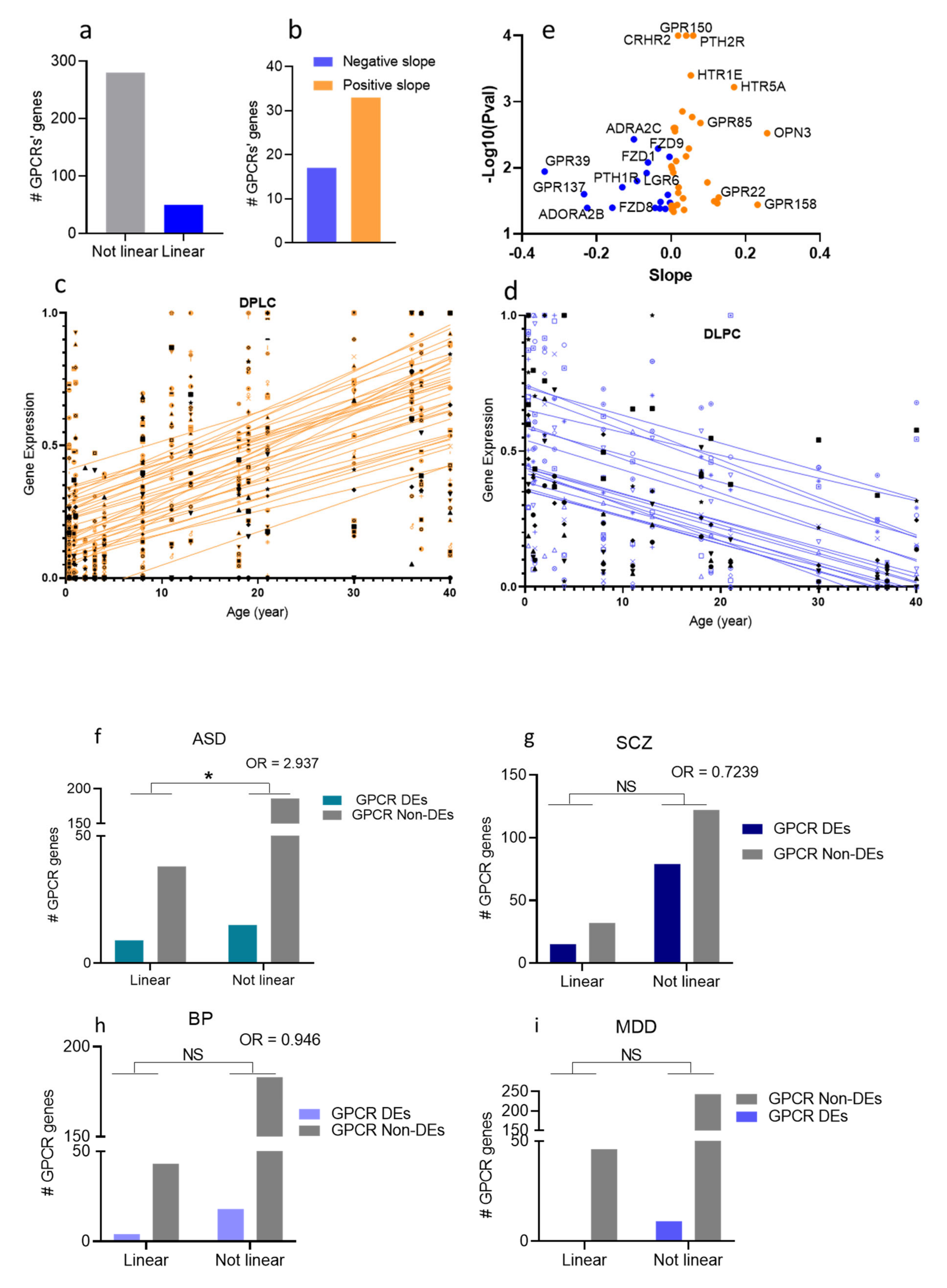
3.7. Age-Association of the GPCR DEs in the Four Psychiatric Disorders
4. Discussion
4.1. GPCR Expressions Are Perturbed in Four Major Psychiatric Disorders
4.2. Disease-Specific GPCR Transcriptomic Signatures
4.2.1. ASD Is Associated with Perturbed Wnt-SMO Pathways and Neuropeptide Transmission
4.2.2. GPCRs Differentially Expressed with Chronological Age Are at a Higher Risk of Dysregulation in ASD than Random GPCRs
4.2.3. SCZ: A Dopamine GPCR Disorder?
4.2.4. BP as a Lipid Transmission Disorder
4.2.5. MDD Is Associated with Upregulated GPCRs and Disrupted Biogenic Amine Transmissions
4.3. Dysregulated GPCRs Overlap across Psychiatric Disorders
4.4. Dysregulated GPCRs Are Involved in the Regulation of Sleep–Wake and Feeding
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Maguire, J.J.; Parker, W.A.; Foord, S.M.; Bonner, T.I.; Neubig, R.R.; Davenport, A.P. International Union of Pharmacology. LXXII. Recommendations for trace amine receptor nomenclature. LXXII. Recommendations for trace amine receptor nomenclature. Pharmacol. Rev. 2009, 61, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, D.M.; Rasmussen, S.G.; Kobilka, B.K. The structure and function of G-protein-coupled receptors. Nature 2009, 459, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Gad, A.A.; Balenga, N. The Emerging Role of Adhesion GPCRs in Cancer. ACS Pharmacol. Transl. Sci. 2020, 3, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Kubler, E.; Albrecht, H. Large set data mining reveals overexpressed GPCRs in prostate and breast cancer: Potential for active targeting with engineered anti-cancer nanomedicines. Oncotarget 2018, 9, 24882–24897. [Google Scholar] [CrossRef] [PubMed]
- Nieto Gutierrez, A.; McDonald, P.H. GPCRs: Emerging anti-cancer drug targets. Cell. Signal. 2018, 41, 65–74. [Google Scholar] [CrossRef]
- Lappano, R.; Maggiolini, M. GPCRs and cancer. Acta Pharmacol. Sin. 2012, 33, 351–362. [Google Scholar] [CrossRef]
- Sedvall, G.; Farde, L. Dopamine receptors in schizophrenia. Lancet 1996, 347, 264. [Google Scholar] [CrossRef]
- Seeman, P.; Guan, H.C.; Van Tol, H.H. Dopamine D4 receptors elevated in schizophrenia. Nature 1993, 365, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Crawley, J.C.; Owens, D.G.; Crow, T.J.; Poulter, M.; Johnstone, E.C.; Smith, T.; Oldland, S.R.; Veall, N.; Owen, F.; Zanelli, G.D. Dopamine D2 receptors in schizophrenia studied in vivo. Lancet 1986, 2, 224–225. [Google Scholar] [CrossRef]
- Atwood, B.K.; Lovinger, D.M.; Mathur, B.N. Presynaptic long-term depression mediated by Gi/o-coupled receptors. Trends Neurosci. 2014, 37, 663–673. [Google Scholar] [CrossRef]
- Fabbri, C.; Marsano, A.; Serretti, A. Genetics of serotonin receptors and depression: State of the art. Curr. Drug Targets 2013, 14, 531–548. [Google Scholar] [CrossRef] [PubMed]
- Rask-Andersen, M.; Almen, M.S.; Schioth, H.B. Trends in the exploitation of novel drug targets. Nat. Rev. Drug Discov. 2011, 10, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schioth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef]
- Disease, G.B.D.; Injury, I.; Prevalence, C. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990-2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1211–1259. [Google Scholar] [CrossRef]
- Khan, S.M.; Sung, J.Y.; Hebert, T.E. Gbetagamma subunits-Different spaces, different faces. Pharmacol. Res. 2016, 111, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Bologna, Z.; Teoh, J.P.; Bayoumi, A.S.; Tang, Y.; Kim, I.M. Biased G Protein-Coupled Receptor Signaling: New Player in Modulating Physiology and Pathology. Biomol. Ther. 2017, 25, 12–25. [Google Scholar] [CrossRef]
- Greengard, P. The neurobiology of slow synaptic transmission. Science 2001, 294, 1024–1030. [Google Scholar] [CrossRef]
- Insel, P.A.; Sriram, K.; Gorr, M.W.; Wiley, S.Z.; Michkov, A.; Salmeron, C.; Chinn, A.M. GPCRomics: An Approach to Discover GPCR Drug Targets. Trends Pharmacol. Sci. 2019, 40, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Wise, A.; Gearing, K.; Rees, S. Target validation of G-protein coupled receptors. Drug Discov. Today 2002, 7, 235–246. [Google Scholar] [CrossRef]
- Catapano, L.A.; Manji, H.K. G protein-coupled receptors in major psychiatric disorders. Biochim. Et Biophys. Acta 2007, 1768, 976–993. [Google Scholar] [CrossRef]
- Komatsu, H. Novel Therapeutic GPCRs for Psychiatric Disorders. Int. J. Mol. Sci. 2015, 16, 14109–14121. [Google Scholar] [CrossRef]
- Tomita, H.; Ziegler, M.E.; Kim, H.B.; Evans, S.J.; Choudary, P.V.; Li, J.Z.; Meng, F.; Dai, M.; Myers, R.M.; Neal, C.R.; et al. G protein-linked signaling pathways in bipolar and major depressive disorders. Front. Genet. 2013, 4, 297. [Google Scholar] [CrossRef] [PubMed]
- Senese, N.B.; Rasenick, M.M.; Traynor, J.R. The Role of G-proteins and G-protein Regulating Proteins in Depressive Disorders. Front. Pharmacol. 2018, 9, 1289. [Google Scholar] [CrossRef] [PubMed]
- Boczek, T.; Mackiewicz, J.; Sobolczyk, M.; Wawrzyniak, J.; Lisek, M.; Ferenc, B.; Guo, F.; Zylinska, L. The Role of G Protein-Coupled Receptors (GPCRs) and Calcium Signaling in Schizophrenia. Focus on GPCRs Activated by Neurotransmitters and Chemokines. Focus on GPCRs Activated by Neurotransmitters and Chemokines. Cells 2021, 10, 1228. [Google Scholar] [CrossRef]
- Tiwari, P.; Fanibunda, S.E.; Kapri, D.; Vasaya, S.; Pati, S.; Vaidya, V.A. GPCR signaling: Role in mediating the effects of early adversity in psychiatric disorders. FEBS J. 2021, 288, 2602–2621. [Google Scholar] [CrossRef]
- Seeman, M.V. History of the dopamine hypothesis of antipsychotic action. World J. Psychiatry 2021, 11, 355–364. [Google Scholar] [CrossRef]
- Alexander, S.P.H.; Christopoulos, A.; Davenport, A.P.; Kelly, E.; Mathie, A.; Peters, J.A.; Veale, E.L.; Armstrong, J.F.; Faccenda, E.; Harding, S.D.; et al. The concise guide to pharmacology 2019/20: G protein-coupled receptors. Br. J. Pharmacol. 2019, 176 (Suppl. S1), S21–S141. [Google Scholar] [CrossRef] [PubMed]
- Mombaerts, P. Genes and ligands for odorant, vomeronasal and taste receptors. Nat. Rev. Neurosci. 2004, 5, 263–278. [Google Scholar] [CrossRef]
- Overington, J.P.; Al-Lazikani, B.; Hopkins, A.L. How many drug targets are there? Nat. Rev. Drug Discov. 2006, 5, 993–996. [Google Scholar] [CrossRef]
- Schioth, H.B.; Fredriksson, R. The GRAFS classification system of G-protein coupled receptors in comparative perspective. Gen. Comp. Endocrinol. 2005, 142, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Rask-Andersen, M.; Masuram, S.; Schioth, H.B. The druggable genome: Evaluation of drug targets in clinical trials suggests major shifts in molecular class and indication. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Gardella, T.J.; Vilardaga, J.P. International Union of Basic and Clinical Pharmacology. XCIII. The parathyroid hormone receptors--family B G protein-coupled receptors. Pharmacol. Rev. 2015, 67, 310–337. [Google Scholar] [CrossRef] [PubMed]
- Harmar, A.J. Family-B G-protein-coupled receptors. Genome Biol. 2001, 2, REVIEWS3013. [Google Scholar] [CrossRef]
- Promel, S.; Langenhan, T.; Arac, D. Matching structure with function: The GAIN domain of adhesion-GPCR and PKD1-like proteins. Trends Pharmacol. Sci. 2013, 34, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Gandal, M.J.; Zhang, P.; Hadjimichael, E.; Walker, R.L.; Chen, C.; Liu, S.; Won, H.; van Bakel, H.; Varghese, M.; Wang, Y.; et al. Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 2018, 362. [Google Scholar] [CrossRef] [PubMed]
- Gandal, M.J.; Haney, J.R.; Parikshak, N.N.; Leppa, V.; Ramaswami, G.; Hartl, C.; Schork, A.J.; Appadurai, V.; Buil, A.; Werge, T.M.; et al. Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science 2018, 359, 693–697. [Google Scholar] [CrossRef] [PubMed]
- Fromer, M.; Roussos, P.; Sieberts, S.K.; Johnson, J.S.; Kavanagh, D.H.; Perumal, T.M.; Ruderfer, D.M.; Oh, E.C.; Topol, A.; Shah, H.R.; et al. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat. Neurosci. 2016, 19, 1442–1453. [Google Scholar] [CrossRef] [PubMed]
- Parikshak, N.N.; Swarup, V.; Belgard, T.G.; Irimia, M.; Ramaswami, G.; Gandal, M.J.; Hartl, C.; Leppa, V.; Ubieta, L.T.; Huang, J.; et al. Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature 2016, 540, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.J.; Kawasawa, Y.I.; Cheng, F.; Zhu, Y.; Xu, X.; Li, M.; Sousa, A.M.; Pletikos, M.; Meyer, K.A.; Sedmak, G.; et al. Spatio-temporal transcriptome of the human brain. Nature 2011, 478, 483–489. [Google Scholar] [CrossRef]
- Chang, L.C.; Jamain, S.; Lin, C.W.; Rujescu, D.; Tseng, G.C.; Sibille, E. A conserved BDNF, glutamate- and GABA-enriched gene module related to human depression identified by coexpression meta-analysis and DNA variant genome-wide association studies. PLoS ONE 2014, 9, e90980. [Google Scholar] [CrossRef] [PubMed]
- Bishop, J.R.; Ellingrod, V.L.; Moline, J.; Miller, D. Pilot study of the G-protein beta3 subunit gene (C825T) polymorphism and clinical response to olanzapine or olanzapine-related weight gain in persons with schizophrenia. Med. Sci. Monit. 2006, 12, BR47–BR50. [Google Scholar]
- Vuoristo, J.T.; Berrettini, W.H.; Overhauser, J.; Prockop, D.J.; Ferraro, T.N.; Ala-Kokko, L. Sequence and genomic organization of the human G-protein Golfalpha gene (GNAL) on chromosome 18p11, a susceptibility region for bipolar disorder and schizophrenia. Mol. Psychiatry 2000, 5, 495–501. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Huang, L.; Max, M.; Margolskee, R.F.; Su, H.; Masland, R.H.; Euler, T. G protein subunit G gamma 13 is coexpressed with G alpha o, G beta 3, and G beta 4 in retinal ON bipolar cells. J. Comp. Neurol. 2003, 455, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Busnelli, M.; Sauliere, A.; Manning, M.; Bouvier, M.; Gales, C.; Chini, B. Functional selective oxytocin-derived agonists discriminate between individual G protein family subtypes. J. Biol. Chem. 2012, 287, 3617–3629. [Google Scholar] [CrossRef]
- Fang, L.; Zhou, C.; Bai, S.; Huang, C.; Pan, J.; Wang, L.; Wang, X.; Mao, Q.; Sun, L.; Xie, P. The C825T Polymorphism of the G-Protein beta3 Gene as a Risk Factor for Depression: A Meta-Analysis. PLoS ONE 2015, 10, e0132274. [Google Scholar] [CrossRef]
- Miller, J.A.; Ding, S.L.; Sunkin, S.M.; Smith, K.A.; Ng, L.; Szafer, A.; Ebbert, A.; Riley, Z.L.; Royall, J.J.; Aiona, K.; et al. Transcriptional landscape of the prenatal human brain. Nature 2014, 508, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Sriram, K.; Moyung, K.; Corriden, R.; Carter, H.; Insel, P.A. GPCRs show widespread differential mRNA expression and frequent mutation and copy number variation in solid tumors. PLoS Biol. 2019, 17, e3000434. [Google Scholar] [CrossRef] [PubMed]
- Dijksterhuis, J.P.; Petersen, J.; Schulte, G. WNT/Frizzled signalling: Receptor-ligand selectivity with focus on FZD-G protein signalling and its physiological relevance: IUPHAR Review 3. Br. J. Pharmacol. 2014, 171, 1195–1209. [Google Scholar] [CrossRef]
- Liu, X.; Low, S.K.; Atkins, J.R.; Wu, J.Q.; Reay, W.R.; Cairns, H.M.; Green, M.J.; Schall, U.; Jablensky, A.; Mowry, B.; et al. Wnt receptor gene FZD1 was associated with schizophrenia in genome-wide SNP analysis of the Australian Schizophrenia Research Bank cohort. Aust. N. Z. J. Psychiatry 2020, 54, 902–908. [Google Scholar] [CrossRef] [PubMed]
- Modahl, C.; Fein, D.; Waterhouse, L.; Newton, N. Does oxytocin deficiency mediate social deficits in autism? J. Autism Dev. Disord. 1992, 22, 449–451. [Google Scholar] [CrossRef]
- Gregory, S.G.; Connelly, J.J.; Towers, A.J.; Johnson, J.; Biscocho, D.; Markunas, C.A.; Lintas, C.; Abramson, R.K.; Wright, H.H.; Ellis, P.; et al. Genomic and epigenetic evidence for oxytocin receptor deficiency in autism. BMC Med. 2009, 7, 62. [Google Scholar] [CrossRef]
- Davis, M.C.; Horan, W.P.; Nurmi, E.L.; Rizzo, S.; Li, W.; Sugar, C.A.; Green, M.F. Associations between oxytocin receptor genotypes and social cognitive performance in individuals with schizophrenia. Schizophr. Res. 2014, 159, 353–357. [Google Scholar] [CrossRef]
- Panda, D.; Goltzman, D.; Juppner, H.; Karaplis, A.C. TIP39/parathyroid hormone type 2 receptor signaling is a potent inhibitor of chondrocyte proliferation and differentiation. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E1125–E1136. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, M.C.; Yeung, H.M.; Kuo, J.; Usdin, T.B. Incubation of Fear Is Regulated by TIP39 Peptide Signaling in the Medial Nucleus of the Amygdala. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 12152–12161. [Google Scholar] [CrossRef]
- Dobolyi, A.; Dimitrov, E.; Palkovits, M.; Usdin, T.B. The neuroendocrine functions of the parathyroid hormone 2 receptor. Front. Endocrinol. 2012, 3, 121. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Chen, C.; Martinez, R.M.; Fan, Y.T.; Liu, C.C.; Chen, C.Y.; Cheng, Y. An amygdala-centered hyper-connectivity signature of threatening face processing predicts anxiety in youths with autism spectrum conditions. Autism Res. 2021. [Google Scholar] [CrossRef]
- Herrington, J.D.; Maddox, B.B.; Kerns, C.M.; Rump, K.; Worley, J.A.; Bush, J.C.; McVey, A.J.; Schultz, R.T.; Miller, J.S. Amygdala Volume Differences in Autism Spectrum Disorder Are Related to Anxiety. J. Autism Dev. Disord. 2017, 47, 3682–3691. [Google Scholar] [CrossRef] [PubMed]
- Rajagopal, S.; Shenoy, S.K. GPCR desensitization: Acute and prolonged phases. Cell. Signal. 2018, 41, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Martins, M.C.; Spadari, R.C. Stress-induced desensitization of the cardiovascular response to noradrenaline in unanesthetized rats. Braz. J. Med. Biol. Res. 1990, 23, 1041–1044. [Google Scholar]
- Kiss, A.; Aguilera, G. Role of alpha-1-adrenergic receptors in the regulation of corticotropin-releasing hormone mRNA in the paraventricular nucleus of the hypothalamus during stress. Cell. Mol. Neurobiol. 2000, 20, 683–694. [Google Scholar] [CrossRef]
- Meerson, F.Z.; Kopylov Iu, N.; Baldenkov, G.N. [Alpha1 desensitization of the heart during adaptation to stress]. Fiziolohichnyi Zhurnal 1991, 37, 3–6. [Google Scholar]
- Campeau, S.; Nyhuis, T.J.; Kryskow, E.M.; Masini, C.V.; Babb, J.A.; Sasse, S.K.; Greenwood, B.N.; Fleshner, M.; Day, H.E. Stress rapidly increases alpha 1d adrenergic receptor mRNA in the rat dentate gyrus. Brain Res. 2010, 1323, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Arredondo, S.B.; Valenzuela-Bezanilla, D.; Mardones, M.D.; Varela-Nallar, L. Role of Wnt Signaling in Adult Hippocampal Neurogenesis in Health and Disease. Front. Cell Dev. Biol. 2020, 8, 860. [Google Scholar] [CrossRef] [PubMed]
- Zwamborn, R.A.J.; Snijders, C.; An, N.; Thomson, A.; Rutten, B.P.F.; de Nijs, L. Wnt Signaling in the Hippocampus in Relation to Neurogenesis, Neuroplasticity, Stress and Epigenetics. Prog. Mol. Biol. Transl. Sci. 2018, 158, 129–157. [Google Scholar] [CrossRef]
- Inestrosa, N.C.; Varela-Nallar, L. Wnt signalling in neuronal differentiation and development. Cell Tissue Res. 2015, 359, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Hoseth, E.Z.; Krull, F.; Dieset, I.; Morch, R.H.; Hope, S.; Gardsjord, E.S.; Steen, N.E.; Melle, I.; Brattbakk, H.R.; Steen, V.M.; et al. Exploring the Wnt signaling pathway in schizophrenia and bipolar disorder. Transl. Psychiatry 2018, 8, 55. [Google Scholar] [CrossRef] [PubMed]
- Katsu, T.; Ujike, H.; Nakano, T.; Tanaka, Y.; Nomura, A.; Nakata, K.; Takaki, M.; Sakai, A.; Uchida, N.; Imamura, T.; et al. The human frizzled-3 (FZD3) gene on chromosome 8p21, a receptor gene for Wnt ligands, is associated with the susceptibility to schizophrenia. Neurosci. Lett. 2003, 353, 53–56. [Google Scholar] [CrossRef]
- Luchins, D. The dopamine hypothesis of schizophrenia. A critical analysis. Neuropsychobiology 1975, 1, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Howes, O.D.; Kapur, S. The dopamine hypothesis of schizophrenia: Version III--the final common pathway. Schizophr. Bull. 2009, 35, 549–562. [Google Scholar] [CrossRef]
- Snyder, S.H. Amphetamine psychosis: A “model” schizophrenia mediated by catecholamines. Am. J. Psychiatry 1973, 130, 61–67. [Google Scholar] [CrossRef]
- Arrang, J.M. Histamine and schizophrenia. Int. Rev. Neurobiol. 2007, 78, 247–287. [Google Scholar] [CrossRef] [PubMed]
- Montastruc, F.; Palmaro, A.; Bagheri, H.; Schmitt, L.; Montastruc, J.L.; Lapeyre-Mestre, M. Role of serotonin 5-HT2C and histamine H1 receptors in antipsychotic-induced diabetes: A pharmacoepidemiological-pharmacodynamic study in VigiBase. Eur. Neuropsychopharmacol. 2015, 25, 1556–1565. [Google Scholar] [CrossRef] [PubMed]
- Tokuda, K. [Pharmacological action of antipsychotic drugs]. Nihon Yakurigaku Zasshi Folia Pharmacol. Jpn. 2006, 128, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Tallima, H.; El Ridi, R. Arachidonic acid: Physiological roles and potential health benefits - A review. J. Adv. Res. 2018, 11, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, S.I. Arachidonic acid and the brain. J. Nutr. 2008, 138, 2515–2520. [Google Scholar] [CrossRef] [PubMed]
- Bosetti, F. Arachidonic acid metabolism in brain physiology and pathology: Lessons from genetically altered mouse models. J. Neurochem. 2007, 102, 577–586. [Google Scholar] [CrossRef]
- Duncan, R.E.; Bazinet, R.P. Brain arachidonic acid uptake and turnover: Implications for signaling and bipolar disorder. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 130–138. [Google Scholar] [CrossRef]
- Bavaresco, D.V.; Uggioni, M.L.R.; Simon, C.S.; Colonetti, T.; Ferraz, S.D.; Cruz, M.V.B.; Valvassori, S.S.; Quevedo, J.; da Rosa, M.I. Evaluation of the arachidonic acid pathway in bipolar disorder: A systematic review. Mol. Biol. Rep. 2020, 47, 8209–8217. [Google Scholar] [CrossRef]
- Rapoport, S.I. Lithium and the other mood stabilizers effective in bipolar disorder target the rat brain arachidonic acid cascade. ACS Chem. Neurosci. 2014, 5, 459–467. [Google Scholar] [CrossRef]
- Basselin, M.; Chang, L.; Bell, J.M.; Rapoport, S.I. Chronic lithium chloride administration attenuates brain NMDA receptor-initiated signaling via arachidonic acid in unanesthetized rats. Neuropsychopharmacology 2006, 31, 1659–1674. [Google Scholar] [CrossRef]
- Wang, L.N.; Liu, L.F.; Zhang, J.X.; Zhao, G.F. [Plasma levels of nociceptin/orphanin FQ in patients with bipolar disorders and health adults]. Zhonghua Yi Xue Za Zhi 2009, 89, 916–918. [Google Scholar]
- Asth, L.; Tiago, P.R.F.; Costa, L.R.F.; Holanda, V.A.D.; Pacifico, S.; Zaveri, N.T.; Calo, G.; Ruzza, C.; Gavioli, E.C. Effects of non-peptide nociceptin/orphanin FQ receptor ligands on methylphenidate-induced hyperactivity in mice: Implications for bipolar disorders. Neuropeptides 2020, 82, 102059. [Google Scholar] [CrossRef]
- de Montigny, C.; Blier, P. Desensitization of terminal 5-HT autoreceptors by 5-HT reuptake blockers. Arch. Gen. Psychiatry 1991, 48, 483–484. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, J.P.; Medvedev, I.O.; Caron, M.G. The 5-HT deficiency theory of depression: Perspectives from a naturalistic 5-HT deficiency model, the tryptophan hydroxylase 2Arg439His knockin mouse. Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 2444–2459. [Google Scholar] [CrossRef] [PubMed]
- Ribas, C.; Miralles, A.; Busquets, X.; Garcia-Sevilla, J.A. Brain alpha(2)-adrenoceptors in monoamine-depleted rats: Increased receptor density, G coupling proteins, receptor turnover and receptor mRNA. Br. J. Pharmacol. 2001, 132, 1467–1476. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.S.; Szczypka, M.S.; Palmiter, R.D. Dopamine-deficient mice are hypersensitive to dopamine receptor agonists. J. Neurosci. Off. J. Soc. Neurosci. 2000, 20, 4405–4413. [Google Scholar] [CrossRef]
- Mulinari, S. Monoamine theories of depression: Historical impact on biomedical research. J. Hist. Neurosci. 2012, 21, 366–392. [Google Scholar] [CrossRef] [PubMed]
- Waters, R.P.; Rivalan, M.; Bangasser, D.A.; Deussing, J.M.; Ising, M.; Wood, S.K.; Holsboer, F.; Summers, C.H. Evidence for the role of corticotropin-releasing factor in major depressive disorder. Neurosci. Biobehav. Rev. 2015, 58, 63–78. [Google Scholar] [CrossRef] [PubMed]
- Davis, E.G.; Keller, J.; Hallmayer, J.; Pankow, H.R.; Murphy, G.M., Jr.; Gotlib, I.H.; Schatzberg, A.F. Corticotropin-releasing factor 1 receptor haplotype and cognitive features of major depression. Transl. Psychiatry 2018, 8, 5. [Google Scholar] [CrossRef]
- Heim, C.; Mletzko, T.; Purselle, D.; Musselman, D.L.; Nemeroff, C.B. The dexamethasone/corticotropin-releasing factor test in men with major depression: Role of childhood trauma. Biol. Psychiatry 2008, 63, 398–405. [Google Scholar] [CrossRef]
- Heim, C.; Owens, M.J.; Plotsky, P.M.; Nemeroff, C.B. The role of early adverse life events in the etiology of depression and posttraumatic stress disorder. Focus on corticotropin-releasing factor. Ann. N. Y. Acad. Sci. 1997, 821, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Lobo, M.K.; Cui, Y.; Ostlund, S.B.; Balleine, B.W.; Yang, X.W. Genetic control of instrumental conditioning by striatopallidal neuron-specific S1P receptor Gpr6. Nat. Neurosci. 2007, 10, 1395–1397. [Google Scholar] [CrossRef]
- Shrader, S.H.; Song, Z.H. Discovery of endogenous inverse agonists for G protein-coupled receptor 6. Biochem. Biophys. Res. Commun. 2020, 522, 1041–1045. [Google Scholar] [CrossRef] [PubMed]
- Preissler, J.; Grosche, A.; Lede, V.; Le Duc, D.; Krugel, K.; Matyash, V.; Szulzewsky, F.; Kallendrusch, S.; Immig, K.; Kettenmann, H.; et al. Altered microglial phagocytosis in GPR34-deficient mice. Glia 2015, 63, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Milior, G.; Morin-Brureau, M.; Chali, F.; Le Duigou, C.; Savary, E.; Huberfeld, G.; Rouach, N.; Pallud, J.; Capelle, L.; Navarro, V.; et al. Distinct P2Y Receptors Mediate Extension and Retraction of Microglial Processes in Epileptic and Peritumoral Human Tissue. J. Neurosci. Off. J. Soc. Neurosci. 2020, 40, 1373–1388. [Google Scholar] [CrossRef] [PubMed]
- Stefani, J.; Tschesnokowa, O.; Parrilla, M.; Robaye, B.; Boeynaems, J.M.; Acker-Palmer, A.; Zimmermann, H.; Gampe, K. Disruption of the Microglial ADP Receptor P2Y13 Enhances Adult Hippocampal Neurogenesis. Front. Cell. Neurosci. 2018, 12, 134. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Yamanaka, H.; Yanamoto, F.; Okubo, M.; Noguchi, K. Multiple P2Y subtypes in spinal microglia are involved in neuropathic pain after peripheral nerve injury. Glia 2012, 60, 1529–1539. [Google Scholar] [CrossRef]
- McIlvain, H.B.; Ma, L.; Ludwig, B.; Manners, M.T.; Martone, R.L.; Dunlop, J.; Kaftan, E.J.; Kennedy, J.D.; Whiteside, G.T. Purinergic receptor-mediated morphological changes in microglia are transient and independent from inflammatory cytokine release. Eur. J. Pharmacol. 2010, 643, 202–210. [Google Scholar] [CrossRef]
- Sheng, X.; Yung, Y.C.; Chen, A.; Chun, J. Lysophosphatidic acid signalling in development. Development 2015, 142, 1390–1395. [Google Scholar] [CrossRef] [PubMed]
- Pasciuto, E.; Burton, O.T.; Roca, C.P.; Lagou, V.; Rajan, W.D.; Theys, T.; Mancuso, R.; Tito, R.Y.; Kouser, L.; Callaerts-Vegh, Z.; et al. Microglia Require CD4 T Cells to Complete the Fetal-to-Adult Transition. Cell 2020, 182, 625–640.e24. [Google Scholar] [CrossRef]
- Gisbert Gustemps, L.; Lugo Marin, J.; Setien Ramos, I.; Ibanez Jimenez, P.; Romero Santo-Tomas, O.; Jurado Luque, M.J.; Ballester Navarro, P.; Esteve Cruella, A.; Diez Villoria, E.; Canal Bedia, R.; et al. Sleep disturbances in autism spectrum disorder without intellectual impairment: Relationship with executive function and psychiatric symptoms. Sleep Med. 2021, 83, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Mazzone, L.; Postorino, V.; Siracusano, M.; Riccioni, A.; Curatolo, P. The Relationship between Sleep Problems, Neurobiological Alterations, Core Symptoms of Autism Spectrum Disorder, and Psychiatric Comorbidities. J. Clin. Med. 2018, 7, 102. [Google Scholar] [CrossRef]
- Meyer, N.; Faulkner, S.M.; McCutcheon, R.A.; Pillinger, T.; Dijk, D.J.; MacCabe, J.H. Sleep and Circadian Rhythm Disturbance in Remitted Schizophrenia and Bipolar Disorder: A Systematic Review and Meta-analysis. Schizophr. Bull. 2020, 46, 1126–1143. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.C.; Liao, C.C.; Wu, P.L.; Lee, S.D.; Lane, H.Y. Metabolic Abnormality and Sleep Disturbance are Associated with Clinical Severity of Patients with Schizophrenia. BioMedicine 2014, 4, 6. [Google Scholar] [CrossRef]
- Ng, T.H.; Chung, K.F.; Ho, F.Y.; Yeung, W.F.; Yung, K.P.; Lam, T.H. Sleep-wake disturbance in interepisode bipolar disorder and high-risk individuals: A systematic review and meta-analysis. Sleep Med. Rev. 2015, 20, 46–58. [Google Scholar] [CrossRef]
- Harvey, A.G.; Talbot, L.S.; Gershon, A. Sleep Disturbance in Bipolar Disorder Across the Lifespan. Clin. Psychol. Sci. Pract. 2009, 16, 256–277. [Google Scholar] [CrossRef]
- Cui, F.; Liu, Q.; Lv, X.; Leonhart, R.; Tian, H.; Wei, J.; Zhang, K.; Zhu, G.; Chen, Q.; Wang, G.; et al. Severe sleep disturbance is associated with executive function impairment in patients with first-episode, treatment-naive major depressive disorders. BMC Psychiatry 2021, 21, 198. [Google Scholar] [CrossRef]
- Seow, L.S.; Subramaniam, M.; Abdin, E.; Vaingankar, J.A.; Chong, S.A. Sleep disturbance among people with major depressive disorders (MDD) in Singapore. J. Ment. Health 2016, 25, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Emery, P.C.; Wilson, K.G.; Kowal, J. Major depressive disorder and sleep disturbance in patients with chronic pain. Pain Res. Manag. 2014, 19, 35–41. [Google Scholar] [CrossRef]
- Kudlow, P.A.; Cha, D.S.; Lam, R.W.; McIntyre, R.S. Sleep architecture variation: A mediator of metabolic disturbance in individuals with major depressive disorder. Sleep Med. 2013, 14, 943–949. [Google Scholar] [CrossRef]
- Espana, R.A.; Scammell, T.E. Sleep neurobiology from a clinical perspective. Sleep 2011, 34, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Eban-Rothschild, A.; Appelbaum, L.; de Lecea, L. Neuronal Mechanisms for Sleep/Wake Regulation and Modulatory Drive. Neuropsychopharmacology 2018, 43, 937–952. [Google Scholar] [CrossRef] [PubMed]
- Richter, C.; Woods, I.G.; Schier, A.F. Neuropeptidergic control of sleep and wakefulness. Annu. Rev. Neurosci. 2014, 37, 503–531. [Google Scholar] [CrossRef]
- De Girolamo, P.; Dieguez, C. Neuropeptides and control of food intake. Int. J. Endocrinol. 2014, 2014, 910912. [Google Scholar] [CrossRef] [PubMed]
- Hillebrand, J.J.; de Wied, D.; Adan, R.A. Neuropeptides, food intake and body weight regulation: A hypothalamic focus. Peptides 2002, 23, 2283–2306. [Google Scholar] [CrossRef]
- Mills, J.G.; Thomas, S.J.; Larkin, T.A.; Pai, N.B.; Deng, C. Problematic eating behaviours, changes in appetite, and weight gain in Major Depressive Disorder: The role of leptin. J. Affect. Disord. 2018, 240, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Privitera, G.J.; Misenheimer, M.L.; Doraiswamy, P.M. From weight loss to weight gain: Appetite changes in major depressive disorder as a mirror into brain-environment interactions. Front. Psychol. 2013, 4, 873. [Google Scholar] [CrossRef][Green Version]
- Kouidrat, Y.; Amad, A.; Lalau, J.D.; Loas, G. Eating disorders in schizophrenia: Implications for research and management. Schizophr. Res. Treat. 2014, 2014, 791573. [Google Scholar] [CrossRef] [PubMed]
- Nickel, K.; Maier, S.; Endres, D.; Joos, A.; Maier, V.; Tebartz van Elst, L.; Zeeck, A. Systematic Review: Overlap Between Eating, Autism Spectrum, and Attention-Deficit/Hyperactivity Disorder. Front. Psychiatry 2019, 10, 708. [Google Scholar] [CrossRef]
- Balzafiore, D.R.; Rasgon, N.L.; Yuen, L.D.; Shah, S.; Kim, H.; Goffin, K.C.; Miller, S.; Wang, P.W.; Ketter, T.A. Lifetime eating disorder comorbidity associated with delayed depressive recovery in bipolar disorder. Int. J. Bipolar Disord. 2017, 5, 25. [Google Scholar] [CrossRef]
- Segura-Garcia, C.; Caroleo, M.; Rania, M.; Barbuto, E.; Sinopoli, F.; Aloi, M.; Arturi, F.; De Fazio, P. Binge Eating Disorder and Bipolar Spectrum disorders in obesity: Psychopathological and eating behaviors differences according to comorbidities. J. Affect. Disord. 2017, 208, 424–430. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classes | # from iuphar-db.org (Accessed on 4 May 2021) | # GPCRs in SCZ, ASD, BP Transcriptomics | # GPCRs in MDD Transcriptomics |
|---|---|---|---|
| Class A | 290 | 207 | 253 |
| Class B | 15 | 12 | 14 |
| Class C | 22 | 17 | 20 |
| Frizzled | 11 | 11 | 11 |
| Adhesion | 33 | 3 | 3 |
| Others | 5 | 5 | 5 |
| Total | 376 | 255 | 306 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monfared, R.V.; Alhassen, W.; Truong, T.M.; Gonzales, M.A.M.; Vachirakorntong, V.; Chen, S.; Baldi, P.; Civelli, O.; Alachkar, A. Transcriptome Profiling of Dysregulated GPCRs Reveals Overlapping Patterns across Psychiatric Disorders and Age-Disease Interactions. Cells 2021, 10, 2967. https://doi.org/10.3390/cells10112967
Monfared RV, Alhassen W, Truong TM, Gonzales MAM, Vachirakorntong V, Chen S, Baldi P, Civelli O, Alachkar A. Transcriptome Profiling of Dysregulated GPCRs Reveals Overlapping Patterns across Psychiatric Disorders and Age-Disease Interactions. Cells. 2021; 10(11):2967. https://doi.org/10.3390/cells10112967
Chicago/Turabian StyleMonfared, Roudabeh Vakil, Wedad Alhassen, Tri Minh Truong, Michael Angelo Maglalang Gonzales, Vincent Vachirakorntong, Siwei Chen, Pierre Baldi, Olivier Civelli, and Amal Alachkar. 2021. "Transcriptome Profiling of Dysregulated GPCRs Reveals Overlapping Patterns across Psychiatric Disorders and Age-Disease Interactions" Cells 10, no. 11: 2967. https://doi.org/10.3390/cells10112967
APA StyleMonfared, R. V., Alhassen, W., Truong, T. M., Gonzales, M. A. M., Vachirakorntong, V., Chen, S., Baldi, P., Civelli, O., & Alachkar, A. (2021). Transcriptome Profiling of Dysregulated GPCRs Reveals Overlapping Patterns across Psychiatric Disorders and Age-Disease Interactions. Cells, 10(11), 2967. https://doi.org/10.3390/cells10112967