
Medium-Chain Length Fatty Acids Enhance Aβ Degradation by Affecting Insulin-Degrading Enzyme

 , , , , and
, , , , and 
{kind=link}
{kind=link}
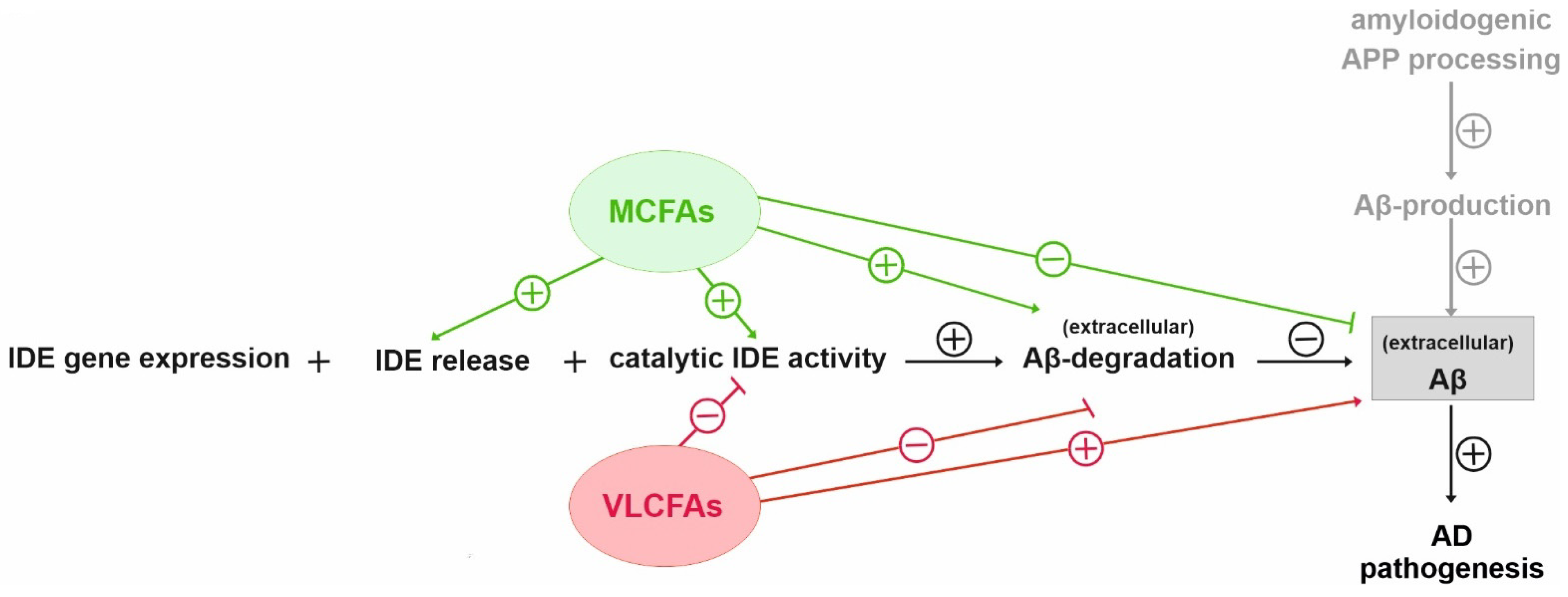{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Culture
2.3. Murine Serum Samples
2.4. Collection of Conditioned Cell Culture Medium and Preparation of Cell Lysates
2.5. Measurement of Total Aβ Degradation
2.6. Determination of Cell Viability
2.7. Quantitative Real-Time PCR
2.8. Measurement of Total Protein Content
2.9. Western Blot Analysis
2.10. Measurement of Constitutive Protein Secretion
2.11. Measurement of IDE Activity in the Presence of Phospholipids
2.12. Measurement of IDE Activity in Murine Serum
2.13. Mass Spectrometry Analysis
2.14. Statistical Analysis
3. Results
3.1. FA Acyl Chain Length Affects IDE-Dependent Aβ Degradation
3.2. FA Acyl Chain Length Affects the Secretion of IDE and Its Catalytic Activity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Masters, C.L.; Selkoe, D.J. Biochemistry of amyloid beta-protein and amyloid deposits in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006262. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef]
- De Strooper, B. Proteases and proteolysis in Alzheimer disease: A multifactorial view on the disease process. Physiol. Rev. 2010, 90, 465–494. [Google Scholar] [CrossRef]
- Grimm, M.O.; Mett, J.; Stahlmann, C.P.; Haupenthal, V.J.; Zimmer, V.C.; Hartmann, T. Neprilysin and Abeta Clearance: Impact of the APP Intracellular Domain in NEP Regulation and Implications in Alzheimer’s Disease. Front. Aging Neurosci. 2013, 5, 98. [Google Scholar] [CrossRef]
- Nalivaeva, N.N.; Turner, A.J. Targeting amyloid clearance in Alzheimer’s disease as a therapeutic strategy. Br. J. Pharmacol. 2019, 176, 3447–3463. [Google Scholar] [CrossRef]
- Saido, T.; Leissring, M.A. Proteolytic degradation of amyloid beta-protein. Cold Spring Harb. Perspect. Med. 2012, 2, a006379. [Google Scholar] [CrossRef] [PubMed]
- Chesneau, V.; Vekrellis, K.; Rosner, M.R.; Selkoe, D.J. Purified recombinant insulin-degrading enzyme degrades amyloid beta-protein but does not promote its oligomerization. Biochem. J. 2000, 351 Pt 2, 509–516. [Google Scholar] [CrossRef]
- Kurochkin, I.V.; Goto, S. Alzheimer’s beta-amyloid peptide specifically interacts with and is degraded by insulin degrading enzyme. FEBS Lett. 1994, 345, 33–37. [Google Scholar] [CrossRef]
- Perez, A.; Morelli, L.; Cresto, J.C.; Castano, E.M. Degradation of soluble amyloid beta-peptides 1-40, 1-42, and the Dutch variant 1-40Q by insulin degrading enzyme from Alzheimer disease and control brains. Neurochem. Res. 2000, 25, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.Q.; Walsh, D.M.; Ye, Z.; Vekrellis, K.; Zhang, J.; Podlisny, M.B.; Rosner, M.R.; Safavi, A.; Hersh, L.B.; Selkoe, D.J. Insulin-degrading enzyme regulates extracellular levels of amyloid beta-protein by degradation. J. Biol. Chem. 1998, 273, 32730–32738. [Google Scholar] [CrossRef] [PubMed]
- Pivovarova, O.; Hohn, A.; Grune, T.; Pfeiffer, A.F.; Rudovich, N. Insulin-degrading enzyme: New therapeutic target for diabetes and Alzheimer’s disease? Ann. Med. 2016, 48, 614–624. [Google Scholar] [CrossRef]
- Tundo, G.R.; Sbardella, D.; Ciaccio, C.; Grasso, G.; Gioia, M.; Coletta, A.; Polticelli, F.; Di Pierro, D.; Milardi, D.; Van Endert, P.; et al. Multiple functions of insulin-degrading enzyme: A metabolic crosslight? Crit Rev. Biochem. Mol. Biol. 2017, 52, 554–582. [Google Scholar] [CrossRef]
- Farris, W.; Mansourian, S.; Chang, Y.; Lindsley, L.; Eckman, E.A.; Frosch, M.P.; Eckman, C.B.; Tanzi, R.E.; Selkoe, D.J.; Guenette, S. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 4162–4167. [Google Scholar] [CrossRef]
- Leissring, M.A.; Farris, W.; Chang, A.Y.; Walsh, D.M.; Wu, X.; Sun, X.; Frosch, M.P.; Selkoe, D.J. Enhanced proteolysis of beta-amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron 2003, 40, 1087–1093. [Google Scholar] [CrossRef]
- Miller, B.C.; Eckman, E.A.; Sambamurti, K.; Dobbs, N.; Chow, K.M.; Eckman, C.B.; Hersh, L.B.; Thiele, D.L. Amyloid-beta peptide levels in brain are inversely correlated with insulysin activity levels in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 6221–6226. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.G.; Leverenz, J.B.; McMillan, P.J.; Kulstad, J.J.; Ericksen, S.; Roth, R.A.; Schellenberg, G.D.; Jin, L.W.; Kovacina, K.S.; Craft, S. Reduced hippocampal insulin-degrading enzyme in late-onset Alzheimer’s disease is associated with the apolipoprotein E-epsilon4 allele. Am. J. Pathol. 2003, 162, 313–319. [Google Scholar] [CrossRef]
- Stargardt, A.; Gillis, J.; Kamphuis, W.; Wiemhoefer, A.; Kooijman, L.; Raspe, M.; Benckhuijsen, W.; Drijfhout, J.W.; Hol, E.M.; Reits, E. Reduced amyloid-beta degradation in early Alzheimer’s disease but not in the APPswePS1dE9 and 3xTg-AD mouse models. Aging Cell 2013, 12, 499–507. [Google Scholar] [CrossRef]
- Zhao, Z.; Xiang, Z.; Haroutunian, V.; Buxbaum, J.D.; Stetka, B.; Pasinetti, G.M. Insulin degrading enzyme activity selectively decreases in the hippocampal formation of cases at high risk to develop Alzheimer’s disease. Neurobiol. Aging 2007, 28, 824–830. [Google Scholar] [CrossRef]
- Ertekin-Taner, N.; Allen, M.; Fadale, D.; Scanlin, L.; Younkin, L.; Petersen, R.C.; Graff-Radford, N.; Younkin, S.G. Genetic variants in a haplotype block spanning IDE are significantly associated with plasma Abeta42 levels and risk for Alzheimer disease. Hum. Mutat. 2004, 23, 334–342. [Google Scholar] [CrossRef]
- Zuo, X.; Jia, J. Promoter polymorphisms which modulate insulin degrading enzyme expression may increase susceptibility to Alzheimer’s disease. Brain Res. 2009, 1249, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Reitz, C.; Cheng, R.; Schupf, N.; Lee, J.H.; Mehta, P.D.; Rogaeva, E.; St George-Hyslop, P.; Mayeux, R. Association between variants in IDE-KIF11-HHEX and plasma amyloid beta levels. Neurobiol. Aging 2012, 33, 199.e13–199.e17. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, S.; He, F.; Wang, Y. Association between polymorphisms of the insulin-degrading enzyme gene and late-onset Alzheimer disease. J. Geriatr. Psychiatry Neurol. 2015, 28, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.Q.; Ye, Z.; Kholodenko, D.; Seubert, P.; Selkoe, D.J. Degradation of amyloid beta-protein by a metalloprotease secreted by microglia and other neural and non-neural cells. J. Biol. Chem. 1997, 272, 6641–6646. [Google Scholar] [CrossRef] [PubMed]
- Vekrellis, K.; Ye, Z.; Qiu, W.Q.; Walsh, D.; Hartley, D.; Chesneau, V.; Rosner, M.R.; Selkoe, D.J. Neurons regulate extracellular levels of amyloid beta-protein via proteolysis by insulin-degrading enzyme. J. Neurosci. 2000, 20, 1657–1665. [Google Scholar] [CrossRef]
- Bulloj, A.; Leal, M.C.; Xu, H.; Castano, E.M.; Morelli, L. Insulin-degrading enzyme sorting in exosomes: A secretory pathway for a key brain amyloid-beta degrading protease. J. Alzheimers. Dis. 2010, 19, 79–95. [Google Scholar] [CrossRef]
- Tamboli, I.Y.; Barth, E.; Christian, L.; Siepmann, M.; Kumar, S.; Singh, S.; Tolksdorf, K.; Heneka, M.T.; Lutjohann, D.; Wunderlich, P.; et al. Statins promote the degradation of extracellular amyloid {beta}-peptide by microglia via stimulation of exosome-associated insulin-degrading enzyme (IDE) secretion. J. Biol. Chem. 2010, 285, 37405–37414. [Google Scholar] [CrossRef]
- Zhao, J.; Li, L.; Leissring, M.A. Insulin-degrading enzyme is exported via an unconventional protein secretion pathway. Mol. Neurodegener. 2009, 4, 4. [Google Scholar] [CrossRef]
- Liu, Z.; Zhu, H.; Fang, G.G.; Walsh, K.; Mwamburi, M.; Wolozin, B.; Abdul-Hay, S.O.; Ikezu, T.; Leissring, M.A.; Qiu, W.Q. Characterization of insulin degrading enzyme and other amyloid-beta degrading proteases in human serum: A role in Alzheimer’s disease? J. Alzheimers Dis. 2012, 29, 329–340. [Google Scholar] [CrossRef]
- Lauer, A.A.; Mett, J.; Janitschke, D.; Thiel, A.; Stahlmann, C.P.; Bachmann, C.M.; Ritzmann, F.; Schrul, B.; Muller, U.C.; Stein, R.; et al. Regulatory feedback cycle of the insulin-degrading enzyme and the amyloid precursor protein intracellular domain: Implications for Alzheimer’s disease. Aging Cell 2020, 19, e13264. [Google Scholar] [CrossRef]
- Hamel, F.G.; Upward, J.L.; Bennett, R.G. In vitro inhibition of insulin-degrading enzyme by long-chain fatty acids and their coenzyme A thioesters. Endocrinology 2003, 144, 2404–2408. [Google Scholar] [CrossRef]
- Grimm, M.O.; Mett, J.; Stahlmann, C.P.; Haupenthal, V.J.; Blumel, T.; Stotzel, H.; Grimm, H.S.; Hartmann, T. Eicosapentaenoic acid and docosahexaenoic acid increase the degradation of amyloid-beta by affecting insulin-degrading enzyme. Biochem. Cell Biol. 2016, 94, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Zhang, L.; Liu, S.; Wang, Z. Palmitic acid and docosahexaenoic acid opposingly regulate the expression of insulin-degrading enzyme in neurons. Pharmazie 2010, 65, 231–232. [Google Scholar]
- Grimm, M.O.; Mett, J.; Grimm, H.S.; Hartmann, T. APP Function and Lipids: A Bidirectional Link. Front. Mol. Neurosci. 2017, 10, 63. [Google Scholar] [CrossRef]
- Mett, J.; Hartmann, T.; Grimm, M.O.W. The Effects of Glycerophospholipids and Fatty Acids on APP Processing: Implications for Alzheimer’s Disease. In Handbook of Lipids in Human Function; Elsevier: Urbana, IL, USA, 2016; pp. 377–421. [Google Scholar]
- Fernando, W.M.; Martins, I.J.; Goozee, K.G.; Brennan, C.S.; Jayasena, V.; Martins, R.N. The role of dietary coconut for the prevention and treatment of Alzheimer’s disease: Potential mechanisms of action. Br. J. Nutr. 2015, 114, 1–14. [Google Scholar] [CrossRef]
- Chatterjee, P.; Fernando, M.; Fernando, B.; Dias, C.B.; Shah, T.; Silva, R.; Williams, S.; Pedrini, S.; Hillebrandt, H.; Goozee, K.; et al. Potential of coconut oil and medium chain triglycerides in the prevention and treatment of Alzheimer’s disease. Mech. Ageing Dev. 2020, 186, 111209. [Google Scholar] [CrossRef] [PubMed]
- Henderson, S.T.; Vogel, J.L.; Barr, L.J.; Garvin, F.; Jones, J.J.; Costantini, L.C. Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer’s disease: A randomized, double-blind, placebo-controlled, multicenter trial. Nutr. Metab. 2009, 6, 31. [Google Scholar] [CrossRef] [PubMed]
- Rebello, C.J.; Keller, J.N.; Liu, A.G.; Johnson, W.D.; Greenway, F.L. Pilot feasibility and safety study examining the effect of medium chain triglyceride supplementation in subjects with mild cognitive impairment: A randomized controlled trial. BBA Clin. 2015, 3, 123–125. [Google Scholar] [CrossRef]
- Reger, M.A.; Henderson, S.T.; Hale, C.; Cholerton, B.; Baker, L.D.; Watson, G.S.; Hyde, K.; Chapman, D.; Craft, S. Effects of beta-hydroxybutyrate on cognition in memory-impaired adults. Neurobiol. Aging 2004, 25, 311–314. [Google Scholar] [CrossRef]
- Taylor, M.K.; Sullivan, D.K.; Mahnken, J.D.; Burns, J.M.; Swerdlow, R.H. Feasibility and efficacy data from a ketogenic diet intervention in Alzheimer’s disease. Alzheimers Dement. Transl. Res. Clin. Interv. 2018, 4, 28–36. [Google Scholar] [CrossRef]
- Ota, M.; Matsuo, J.; Ishida, I.; Takano, H.; Yokoi, Y.; Hori, H.; Yoshida, S.; Ashida, K.; Nakamura, K.; Takahashi, T.; et al. Effects of a medium-chain triglyceride-based ketogenic formula on cognitive function in patients with mild-to-moderate Alzheimer’s disease. Neurosci. Lett. 2019, 690, 232–236. [Google Scholar] [CrossRef]
- Hu Yang, I.; De la Rubia Orti, J.E.; Selvi Sabater, P.; Sancho Castillo, S.; Rochina, M.J.; Manresa Ramon, N.; Montoya-Castilla, I. Coconut Oil: Non-Alternative Drug Treatment against Alzheimer S Disease. Nutr. Hosp. 2015, 32, 2822–2827. [Google Scholar] [CrossRef]
- de la Rubia Orti, J.E.; Garcia-Pardo, M.P.; Drehmer, E.; Sancho Cantus, D.; Julian Rochina, M.; Aguilar, M.A.; Hu Yang, I. Improvement of Main Cognitive Functions in Patients with Alzheimer’s Disease after Treatment with Coconut Oil Enriched Mediterranean Diet: A Pilot Study. J. Alzheimers Dis. 2018, 65, 577–587. [Google Scholar] [CrossRef]
- Mett, J.; Muller, U. The medium-chain fatty acid decanoic acid reduces oxidative stress levels in neuroblastoma cells. Sci. Rep. 2021, 11, 6135. [Google Scholar] [CrossRef]
- Grimm, M.O.; Stahlmann, C.P.; Mett, J.; Haupenthal, V.J.; Zimmer, V.C.; Lehmann, J.; Hundsdorfer, B.; Endres, K.; Grimm, H.S.; Hartmann, T. Vitamin E: Curse or Benefit in Alzheimer’s Disease? A Systematic Investigation of the Impact of alpha-, gamma- and delta-Tocopherol on Ass Generation and Degradation in Neuroblastoma Cells. J. Nutr. Health Aging 2015, 19, 646–656. [Google Scholar] [CrossRef]
- Grimm, M.O.; Haupenthal, V.J.; Rothhaar, T.L.; Zimmer, V.C.; Grosgen, S.; Hundsdorfer, B.; Lehmann, J.; Grimm, H.S.; Hartmann, T. Effect of Different Phospholipids on alpha-Secretase Activity in the Non-Amyloidogenic Pathway of Alzheimer’s Disease. Int. J. Mol. Sci. 2013, 14, 5879–5898. [Google Scholar] [CrossRef]
- Jankowsky, J.L.; Fadale, D.J.; Anderson, J.; Xu, G.M.; Gonzales, V.; Jenkins, N.A.; Copeland, N.G.; Lee, M.K.; Younkin, L.H.; Wagner, S.L.; et al. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: Evidence for augmentation of a 42-specific gamma secretase. Hum. Mol. Genet. 2004, 13, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Nagarajan, A.; Uchil, P.D. Analysis of Cell Viability by the Lactate Dehydrogenase Assay. Cold Spring Harb. Protoc. 2018, 2018, pdb.prot095497. [Google Scholar] [CrossRef]
- Crowley, L.C.; Scott, A.P.; Marfell, B.J.; Boughaba, J.A.; Chojnowski, G.; Waterhouse, N.J. Measuring Cell Death by Propidium Iodide Uptake and Flow Cytometry. Cold Spring Harb. Protoc. 2016, 2016, pdb-prot087163. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]
- Janitschke, D.; Nelke, C.; Lauer, A.A.; Regner, L.; Winkler, J.; Thiel, A.; Grimm, H.S.; Hartmann, T.; Grimm, M.O.W. Effect of Caffeine and Other Methylxanthines on Abeta-Homeostasis in SH-SY5Y Cells. Biomolecules 2019, 9, 689. [Google Scholar] [CrossRef] [PubMed]
- Gorr, S.U.; Moore, Y.R. Sorting of a constitutive secretory protein to the regulated secretory pathway of exocrine cells. Biochem. Biophys. Res. Commun. 1999, 257, 545–548. [Google Scholar] [CrossRef]
- Grimm, M.O.; Grosgen, S.; Riemenschneider, M.; Tanila, H.; Grimm, H.S.; Hartmann, T. From brain to food: Analysis of phosphatidylcholins, lyso-phosphatidylcholins and phosphatidylcholin-plasmalogens derivates in Alzheimer’s disease human post mortem brains and mice model via mass spectrometry. J. Chromatogr. A 2011, 1218, 7713–7722. [Google Scholar] [CrossRef]
- Sastry, P.S. Lipids of nervous tissue: Composition and metabolism. Prog. Lipid Res. 1985, 24, 69–176. [Google Scholar] [CrossRef]
- Liberato, M.V.; Nascimento, A.S.; Ayers, S.D.; Lin, J.Z.; Cvoro, A.; Silveira, R.L.; Martinez, L.; Souza, P.C.; Saidemberg, D.; Deng, T.; et al. Medium chain fatty acids are selective peroxisome proliferator activated receptor (PPAR) gamma activators and pan-PPAR partial agonists. PLoS ONE 2012, 7, e36297. [Google Scholar] [CrossRef]
- Du, J.; Zhang, L.; Liu, S.; Zhang, C.; Huang, X.; Li, J.; Zhao, N.; Wang, Z. PPARgamma transcriptionally regulates the expression of insulin-degrading enzyme in primary neurons. Biochem. Biophys. Res. Commun. 2009, 383, 485–490. [Google Scholar] [CrossRef]
- Kadowaki, H.; Grant, M.A. Relationship of membrane phospholipid composition, lactosylceramide molecular species, and the specificity of CMP-N-acetylneuraminate:lactosylceramide alpha 2,3-sialyltransferase to the molecular species composition of GM3 ganglioside. J. Lipid Res. 1995, 36, 1274–1282. [Google Scholar] [CrossRef]
- Werlein, A.; Peters, A.; Ngoune, R.; Winkler, K.; Putz, G. Interference of phosphatidylcholines with in-vitro cell proliferation—No flock without black sheep. Biochim. Biophys. Acta 2015, 1848, 1599–1608. [Google Scholar] [CrossRef]
- Llorente, A.; van Deurs, B.; Sandvig, K. Cholesterol regulates prostasome release from secretory lysosomes in PC-3 human prostate cancer cells. Eur. J. Cell Biol. 2007, 86, 405–415. [Google Scholar] [CrossRef]
- Phuyal, S.; Hessvik, N.P.; Skotland, T.; Sandvig, K.; Llorente, A. Regulation of exosome release by glycosphingolipids and flotillins. FEBS J. 2014, 281, 2214–2227. [Google Scholar] [CrossRef] [PubMed]
- Record, M.; Carayon, K.; Poirot, M.; Silvente-Poirot, S. Exosomes as new vesicular lipid transporters involved in cell-cell communication and various pathophysiologies. Biochim. Biophys. Acta 2014, 1841, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brugger, B.; Simons, M. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 2008, 319, 1244–1247. [Google Scholar] [CrossRef]
- Song, E.S.; Rodgers, D.W.; Hersh, L.B. Insulin-degrading enzyme is not secreted from cultured cells. Sci. Rep. 2018, 8, 2335. [Google Scholar] [CrossRef]
- Leissring, M.A. Insulin-Degrading Enzyme: Paradoxes and Possibilities. Cells 2021, 10, 2445. [Google Scholar] [CrossRef]
- DeMattos, R.B.; Bales, K.R.; Cummins, D.J.; Dodart, J.C.; Paul, S.M.; Holtzman, D.M. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2001, 98, 8850–8855. [Google Scholar] [CrossRef]
- Zhang, Y.; Lee, D.H. Sink hypothesis and therapeutic strategies for attenuating Abeta levels. Neuroscientist 2011, 17, 163–173. [Google Scholar] [CrossRef]
- Spector, R. Fatty acid transport through the blood-brain barrier. J. Neurochem. 1988, 50, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Augustin, K.; Khabbush, A.; Williams, S.; Eaton, S.; Orford, M.; Cross, J.H.; Heales, S.J.R.; Walker, M.C.; Williams, R.S.B. Mechanisms of action for the medium-chain triglyceride ketogenic diet in neurological and metabolic disorders. Lancet Neurol. 2018, 17, 84–93. [Google Scholar] [CrossRef]
- Studzinski, C.M.; MacKay, W.A.; Beckett, T.L.; Henderson, S.T.; Murphy, M.P.; Sullivan, P.G.; Burnham, W.M. Induction of ketosis may improve mitochondrial function and decrease steady-state amyloid-beta precursor protein (APP) levels in the aged dog. Brain Res. 2008, 1226, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Maianti, J.P.; Tan, G.A.; Vetere, A.; Welsh, A.J.; Wagner, B.K.; Seeliger, M.A.; Liu, D.R. Substrate-selective inhibitors that reprogram the activity of insulin-degrading enzyme. Nat. Chem. Biol. 2019, 15, 565–574. [Google Scholar] [CrossRef]
- Tamaki, C.; Ohtsuki, S.; Terasaki, T. Insulin facilitates the hepatic clearance of plasma amyloid beta-peptide (1 40) by intracellular translocation of low-density lipoprotein receptor-related protein 1 (LRP-1) to the plasma membrane in hepatocytes. Mol. Pharmacol. 2007, 72, 850–855. [Google Scholar] [CrossRef] [PubMed]
- Henderson, S.T.; Poirier, J. Pharmacogenetic analysis of the effects of polymorphisms in APOE, IDE and IL1B on a ketone body based therapeutic on cognition in mild to moderate Alzheimer’s disease; a randomized, double-blind, placebo-controlled study. BMC Med. Genet. 2011, 12, 137. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mett, J.; Lauer, A.A.; Janitschke, D.; Griebsch, L.V.; Theiss, E.L.; Grimm, H.S.; Koivisto, H.; Tanila, H.; Hartmann, T.; Grimm, M.O.W. Medium-Chain Length Fatty Acids Enhance Aβ Degradation by Affecting Insulin-Degrading Enzyme. Cells 2021, 10, 2941. https://doi.org/10.3390/cells10112941
Mett J, Lauer AA, Janitschke D, Griebsch LV, Theiss EL, Grimm HS, Koivisto H, Tanila H, Hartmann T, Grimm MOW. Medium-Chain Length Fatty Acids Enhance Aβ Degradation by Affecting Insulin-Degrading Enzyme. Cells. 2021; 10(11):2941. https://doi.org/10.3390/cells10112941
Chicago/Turabian StyleMett, Janine, Anna A. Lauer, Daniel Janitschke, Lea V. Griebsch, Elena L. Theiss, Heike S. Grimm, Hennariikka Koivisto, Heikki Tanila, Tobias Hartmann, and Marcus O. W. Grimm. 2021. "Medium-Chain Length Fatty Acids Enhance Aβ Degradation by Affecting Insulin-Degrading Enzyme" Cells 10, no. 11: 2941. https://doi.org/10.3390/cells10112941
APA StyleMett, J., Lauer, A. A., Janitschke, D., Griebsch, L. V., Theiss, E. L., Grimm, H. S., Koivisto, H., Tanila, H., Hartmann, T., & Grimm, M. O. W. (2021). Medium-Chain Length Fatty Acids Enhance Aβ Degradation by Affecting Insulin-Degrading Enzyme. Cells, 10(11), 2941. https://doi.org/10.3390/cells10112941