
The LEDGF/p75 Integrase Binding Domain Interactome Contributes to the Survival, Clonogenicity, and Tumorsphere Formation of Docetaxel-Resistant Prostate Cancer Cells

 ,
, 
 and
and
Abstract
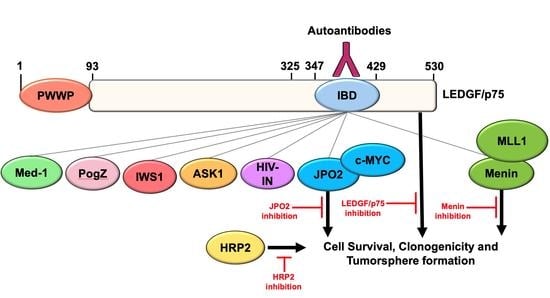
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Antibodies
2.3. Immunoblotting
2.4. Quantitative Real-Time PCR
2.5. MTT Viability Assay and Determination of IC50 Values
2.6. Ingenuity Pathway Analysis
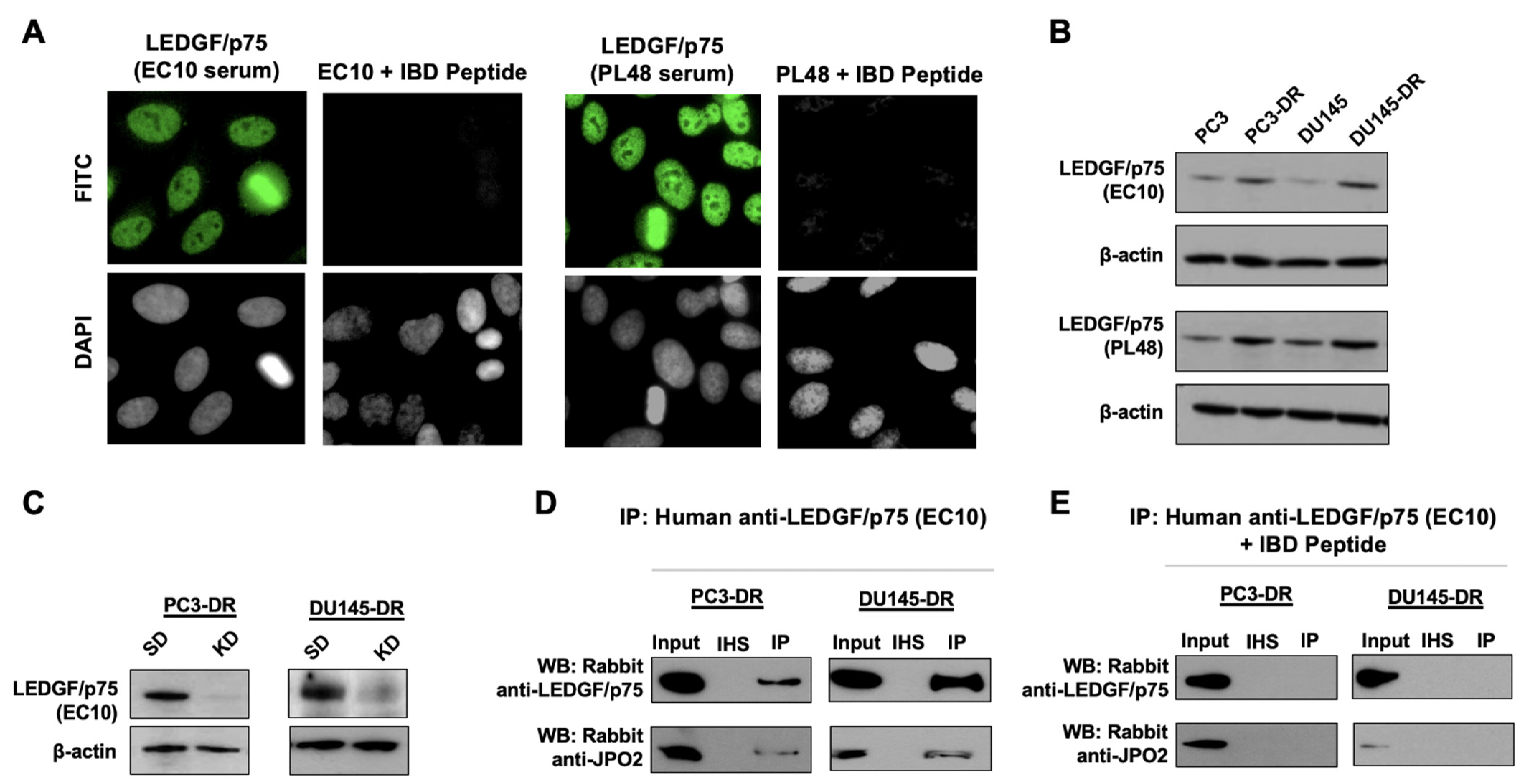
2.7. Validation of Human Anti-LEDGF/p75 Autoantibodies
2.8. Co-Immunoprecipitation
2.9. Confocal Microscopy
2.10. Nuclear Detection of JPO2
2.11. RNA Interference
2.12. Apoptosis Assays
2.13. Clonogenic Assays
2.14. Tumorsphere Formation Assays
2.15. Measurement of Surface CD44 Antigen
2.16. Statistical Analysis
3. Results
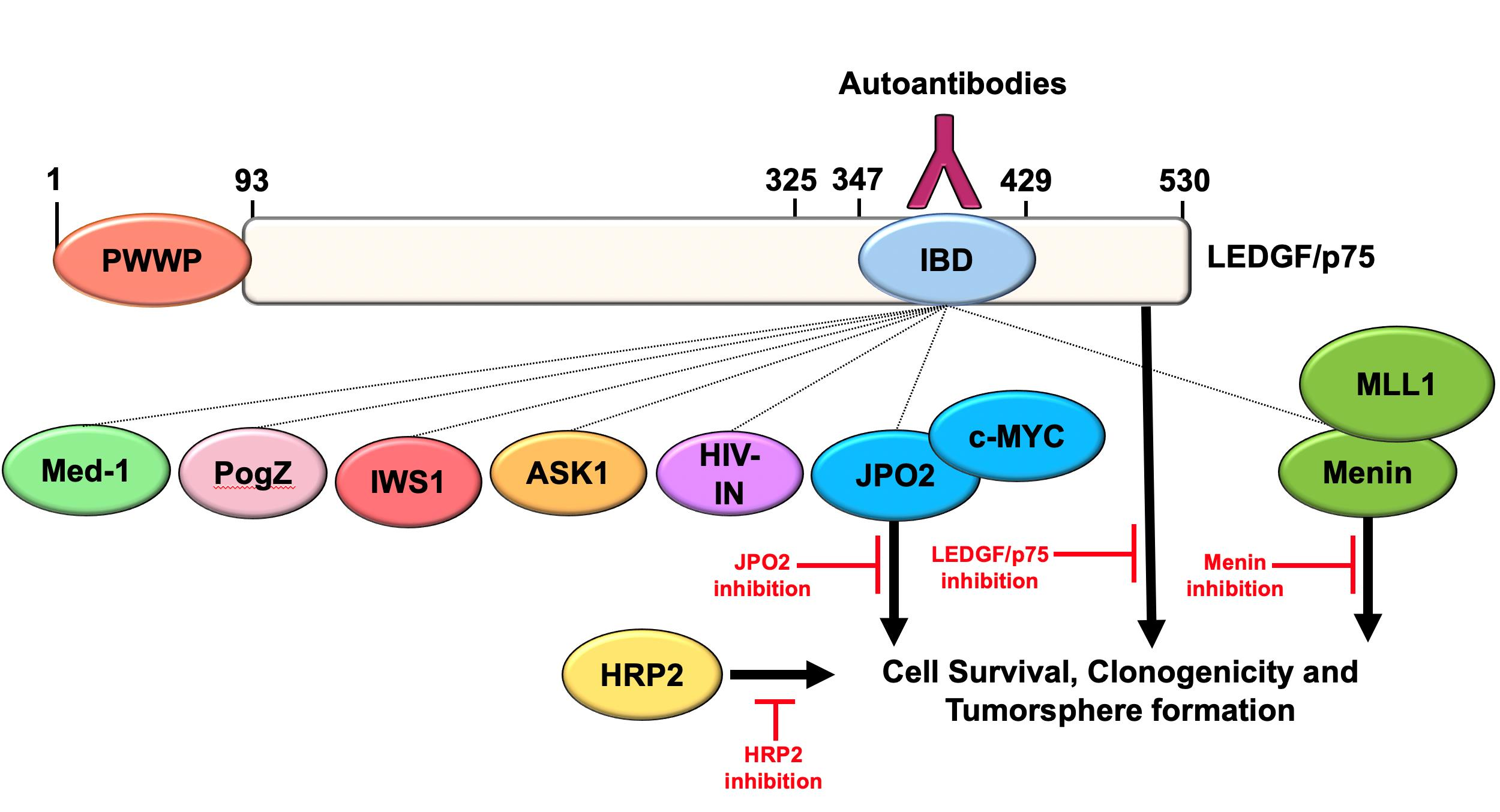
3.1. The LEDGF/p75 IBD Interactome Is Endogenously Overexpressed in DTX-Resistant PCa Cells
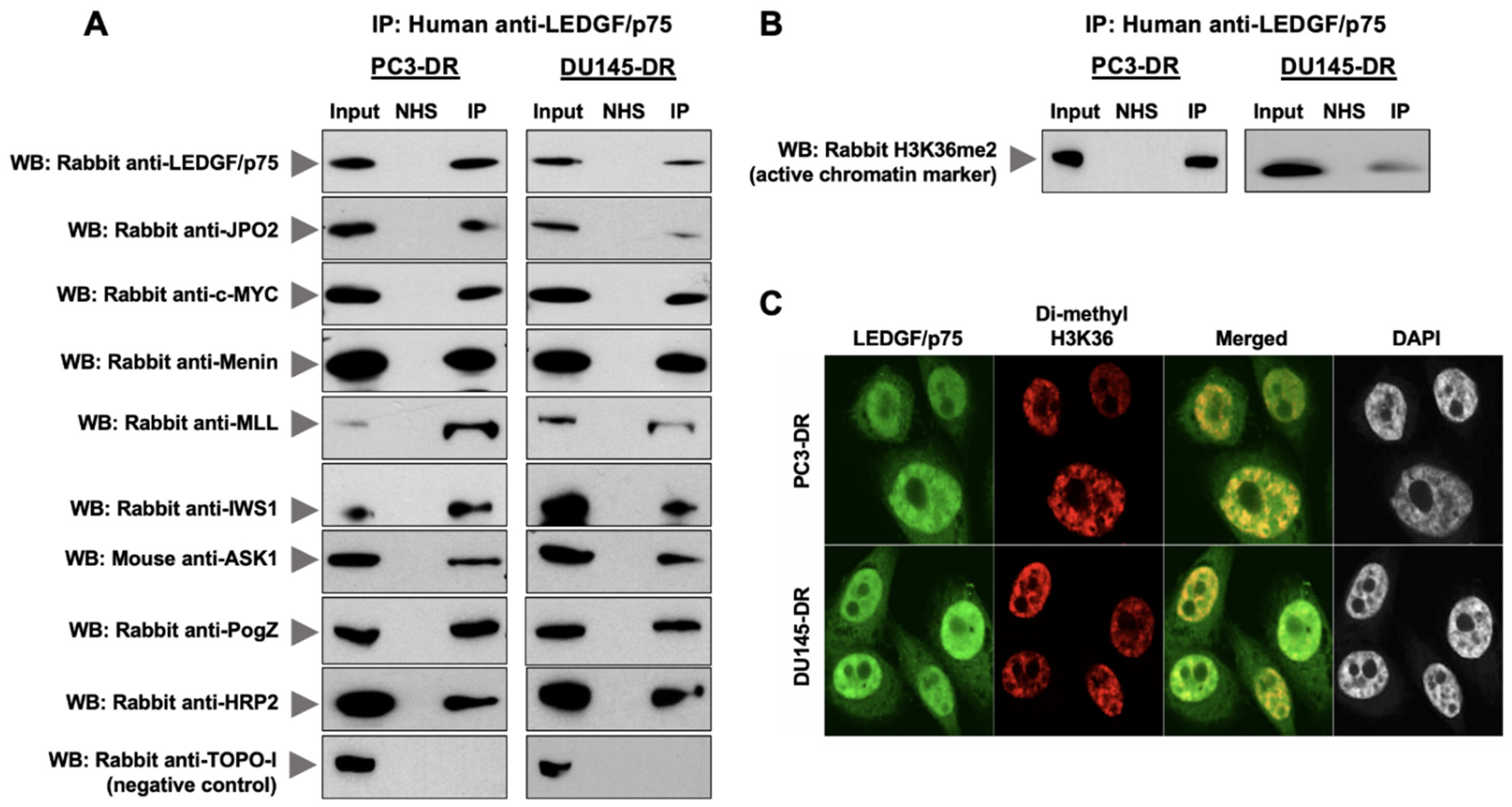
3.2. LEDGF/p75 Interacts Endogenously with IBD-Binding Partners in DTX-Resistant PCa Cells
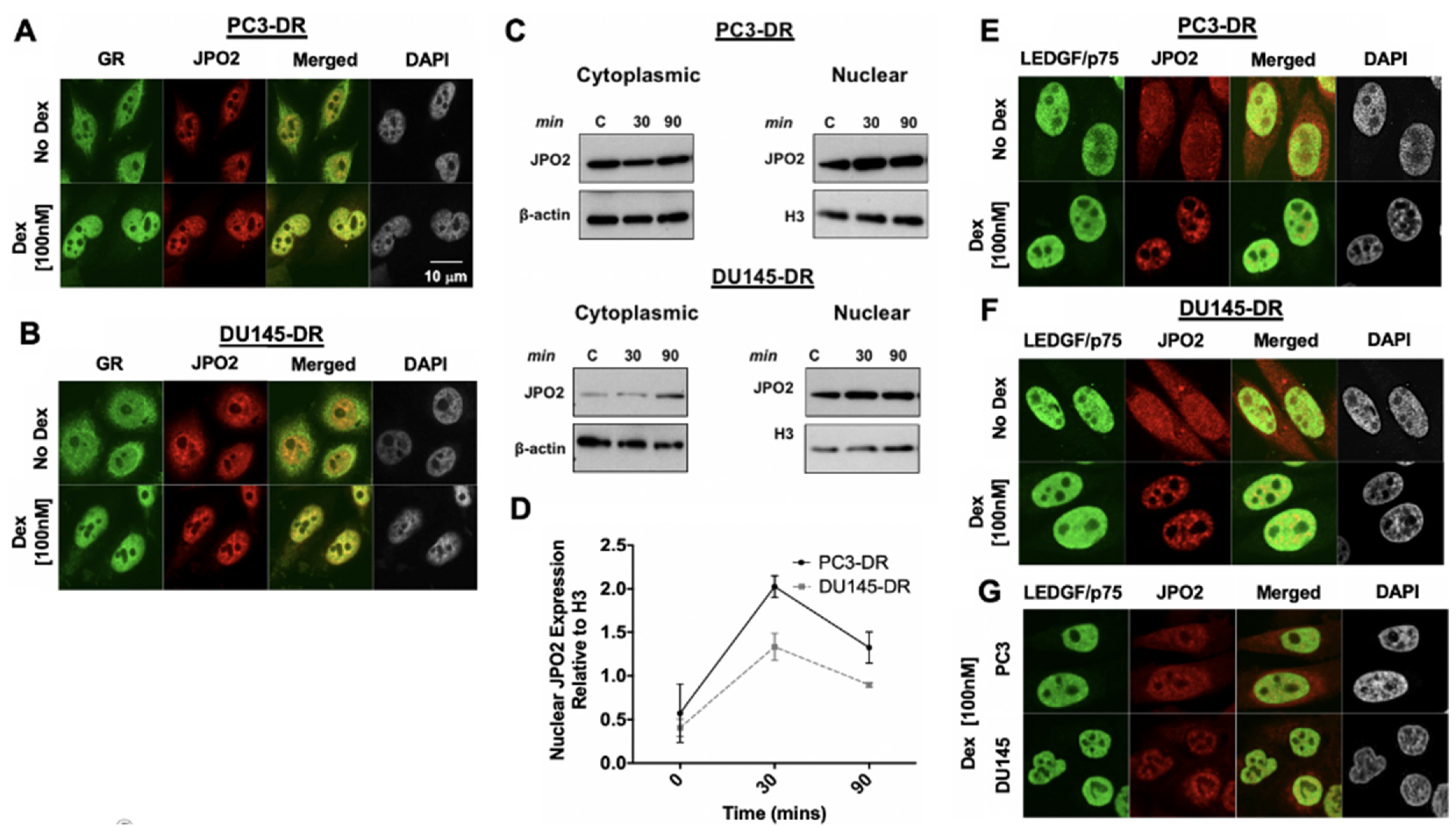
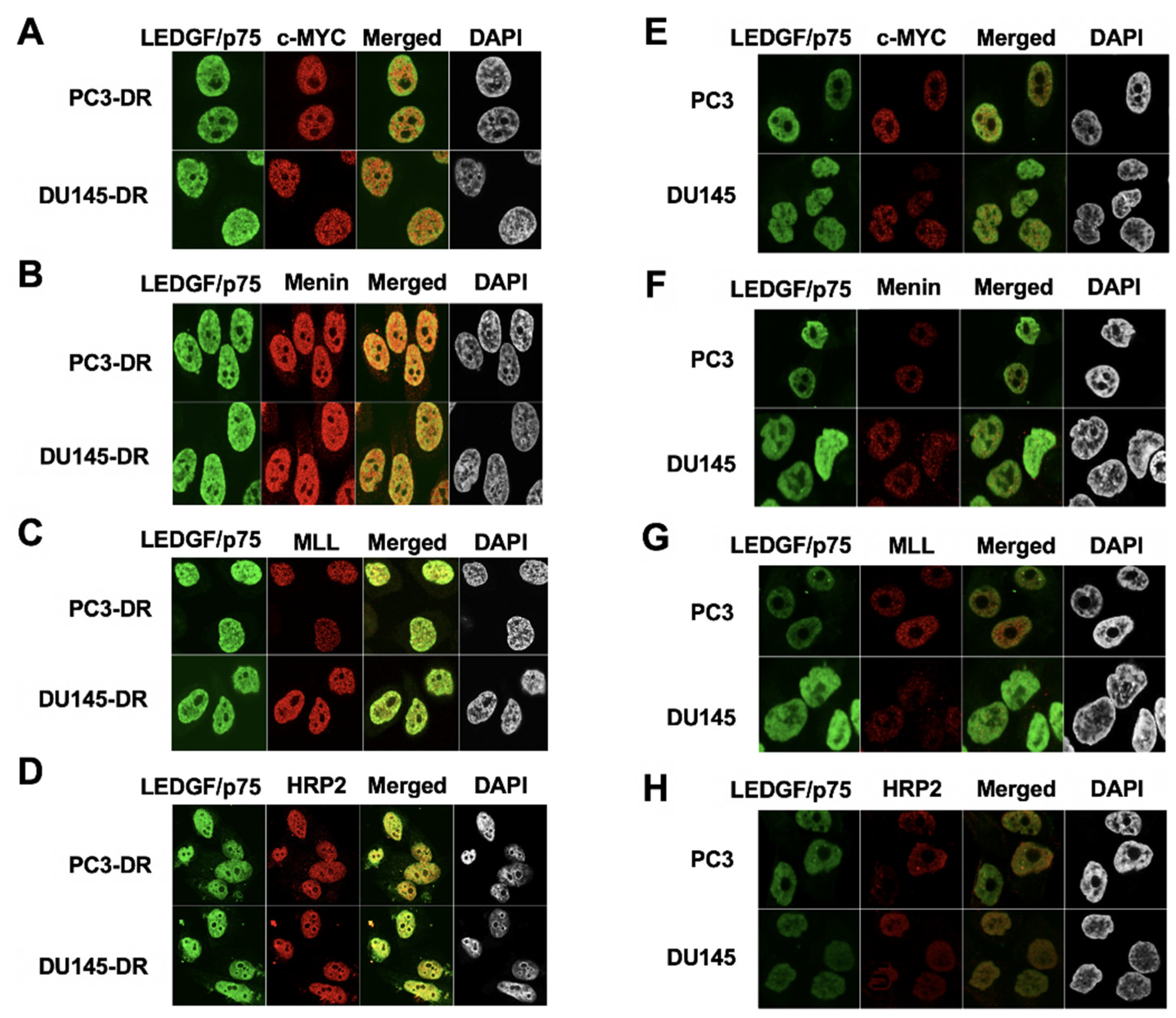
3.3. LEDGF/p75 Colocalizes with Interacting Partners in Nuclei of DTX-Resistant PCa Cells
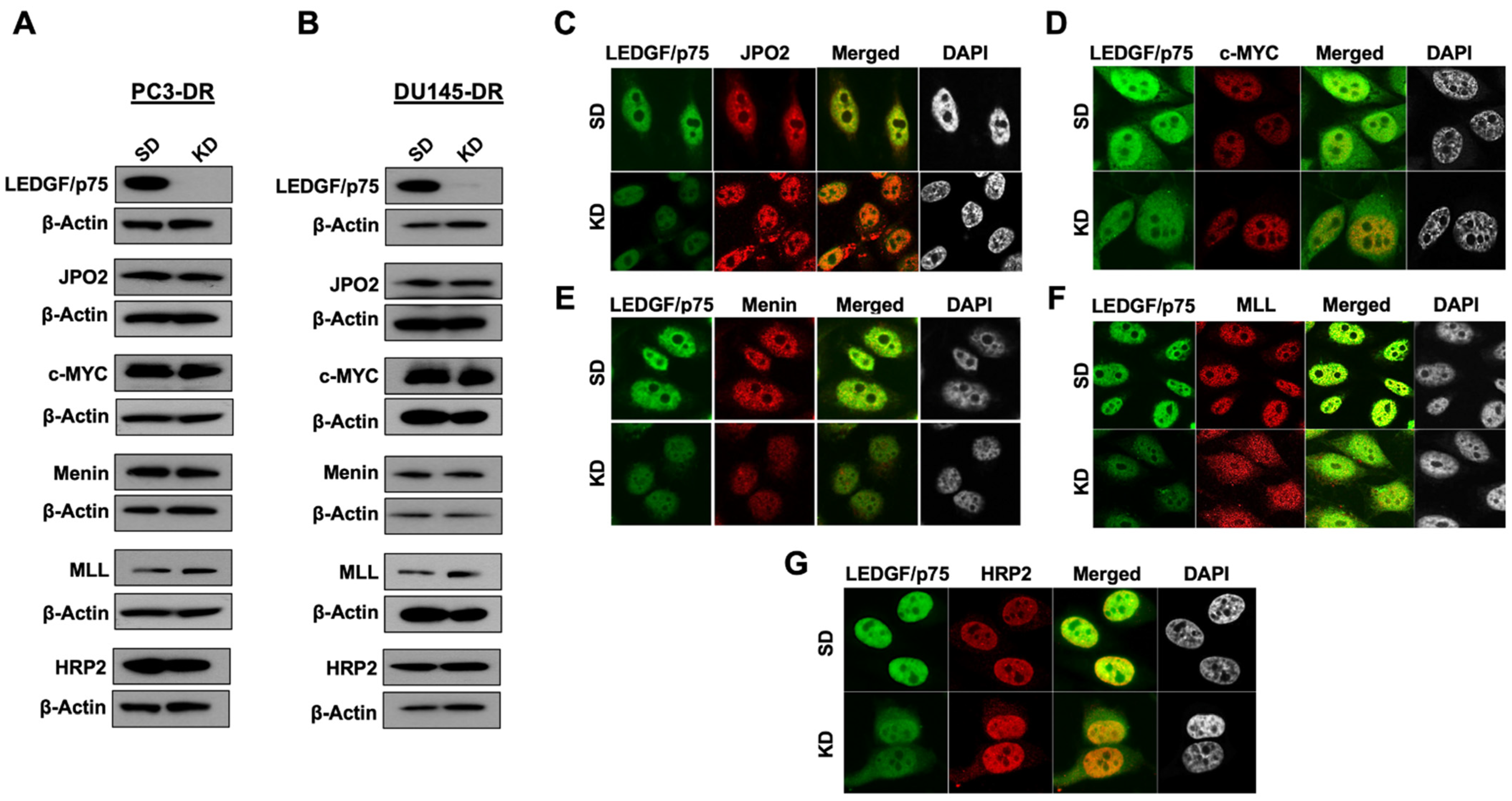
3.4. LEDGF/p75 Depletion Does Not Alter the Protein Expression of Its Interacting Partners but May Influence Their Nuclear Localization
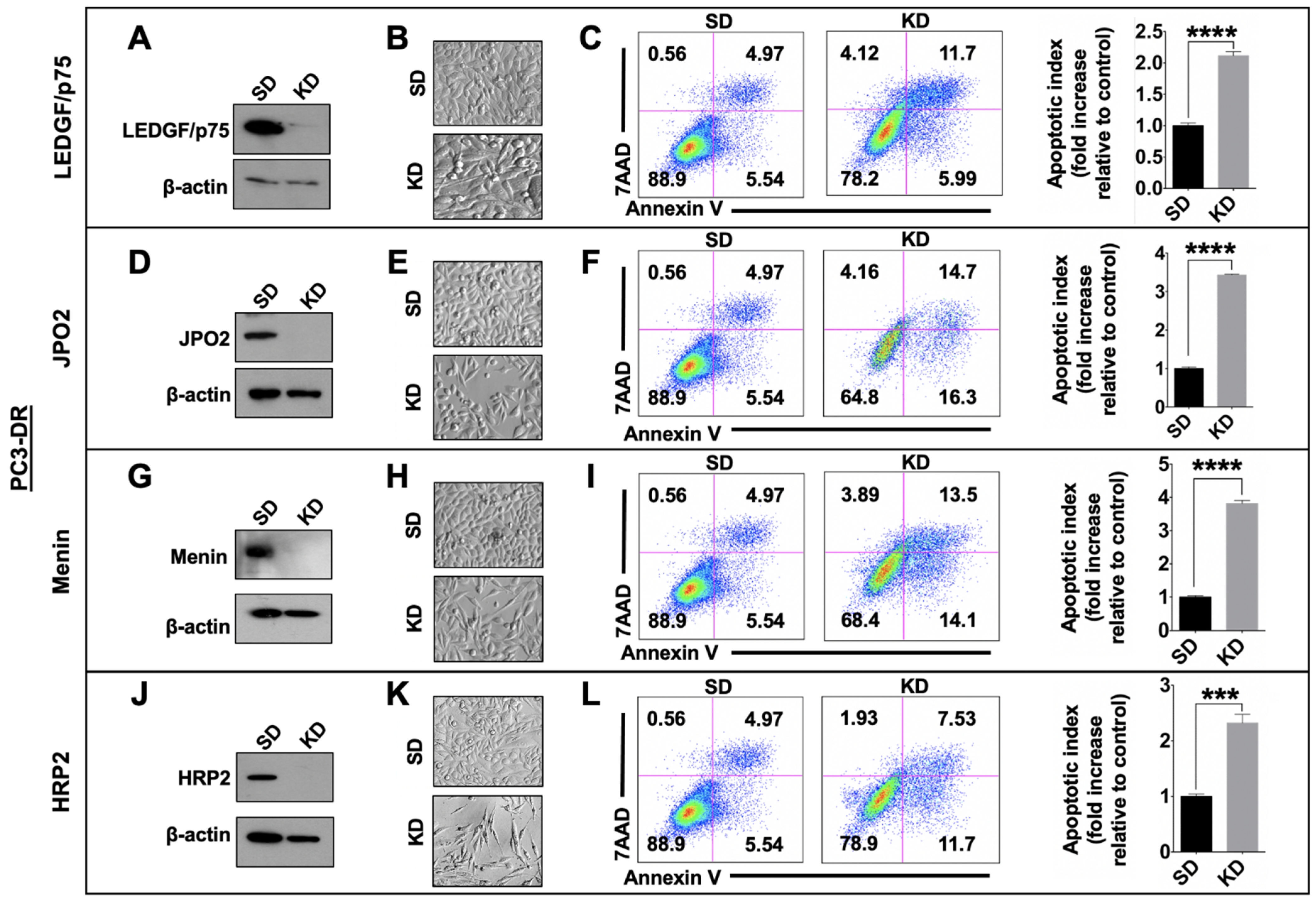
3.5. Depletion of LEDGF/p75, JPO2, Menin, or HRP2 Enhances Apoptosis in DTX-Resistant Cells
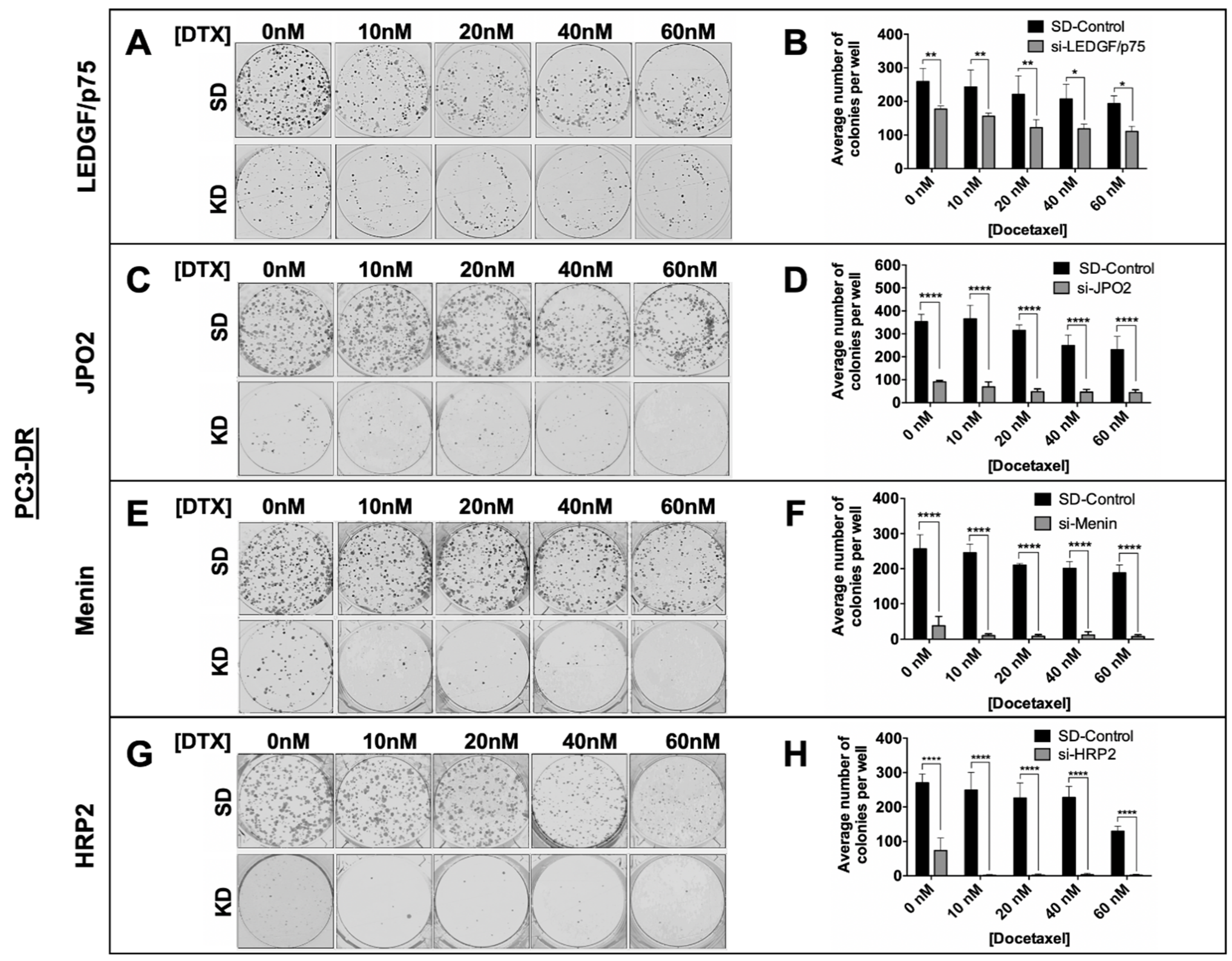
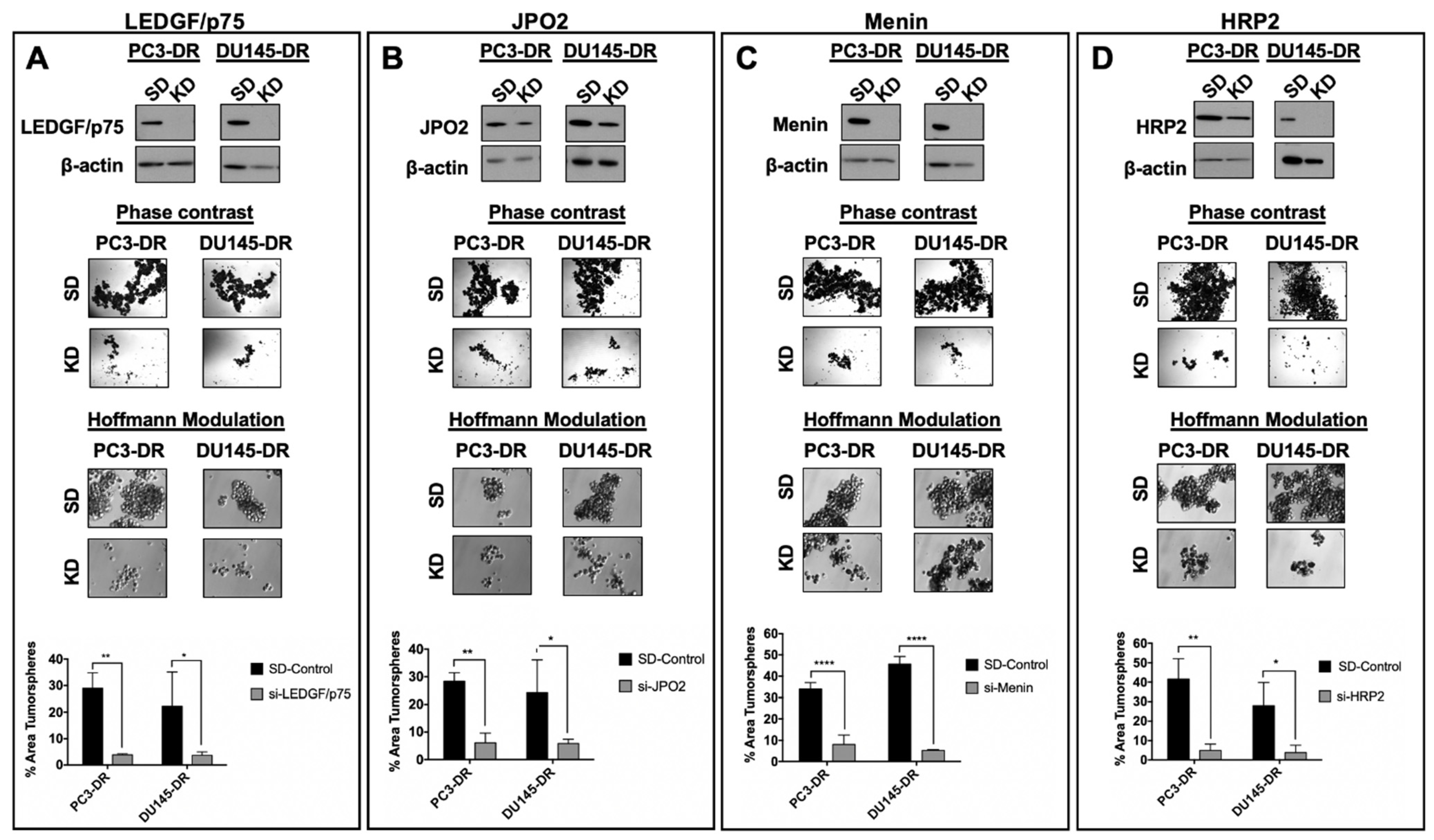
3.6. Depletion of LEDGF/p75, JPO2, Menin, or HRP2 Inhibits the Clonogenicity and Tumorsphere Formation Capacity of DTX-Resistant PCa Cells
3.7. Depletion of LEDGF/p75 in PCa Cells Does Not Lead to Decrease in Lineage-Specific or Stemness Markers
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Gourdin, T. Highlighting recent treatment advances in metastatic prostate cancer: Expanding the treatment arsenal. Curr. Opin. Oncol. 2021, 33, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Zieren, R.C.; Xue, W.; de Reijke, T.M.; Pienta, K.J. Metastatic prostate cancer remains incurable, why? Asian J. Urol. 2019, 6, 26–41. [Google Scholar] [CrossRef] [PubMed]
- Galletti, G.; Leach, B.I.; Lam, L.; Tagawa, S.T. Mechanisms of resistance to systemic therapy in metastatic castration-resistant prostate cancer. Cancer Treat. Rev. 2017, 57, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Däbritz, J.H.M.; Zhao, Z.; Yu, Y.; Dörr, J.R.; Dimitrova, L.; Lenze, D.; Barbosa, I.A.M.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef]
- Cajigas-Du Ross, C.K.; Martinez, S.R.; Woods-Burnham, L.; Durán, A.M.; Roy, S.; Basu, A.; Ramirez, J.A.; Ortiz-Hernández, G.L.; Ríos-Colón, L.; Chirshev, E.; et al. RNA sequencing reveals upregulation of a transcriptomic program associated with stemness in metastatic prostate cancer cells selected for taxane resistance. Oncotarget 2018, 9, 30363. [Google Scholar] [CrossRef]
- Basu, A.; Rojas, H.; Banerjee, H.; Cabrera, I.B.; Perez, K.Y.; De Leon, M.; Casiano, C.A. Expression of the stress onco-protein LEDGF/p75 in major human cancers: A study of 21 tumor types. PLoS ONE 2012, 7, e30132. [Google Scholar] [CrossRef]
- Mediavilla-Varela, M.; Pacheco, F.J.; Almaguel, F.; Perez, J.; Sahakian, E.; Daniels, T.R.; Leoh, L.S.; Padilla, A.; Wall, N.R.; Lilly, M.B.; et al. Docetaxel-induced prostate cancer cell death involves concomitant activation of caspase and lysosomal pathways and is attenuated by LEDGF/p75. Mol. Cancer 2009, 8, 68. [Google Scholar] [CrossRef] [PubMed]
- Rios-Colon, L.; Ross, C.K.C.-D.; Basu, A.; Elix, C.; Alicea-Polanco, I.; Sanchez, T.W.; Radhakrishnan, V.; Chen, C.-S.; Casiano, C.A. Targeting the stress oncoprotein LEDGF/p75 to sensitize chemoresistant prostate cancer cells to taxanes. Oncotarget 2017, 8, 24915–24931. [Google Scholar] [CrossRef]
- Daugaard, M.; Kirkegaard-Sørensen, T.; Ostenfeld, M.S.; Aaboe, M.; Høyer-Hansen, M.; Ørntoft, T.F.; Rohde, M.; Jäättelä, M. Lens epithelium-derived growth factor is an Hsp70-2 regulated guardian of lysosomal stability in human cancer. Cancer Res. 2007, 67, 2559–2567. [Google Scholar] [CrossRef]
- Bhargavan, B.; Fatma, N.; Chhunchha, B.; Singh, V.; Kubo, E.; Singh, D.P. LEDGF gene silencing impairs the tumorigenicity of prostate cancer DU145 cells by abating the expression of Hsp27 and activation of the Akt/ERK signaling pathway. Cell Death Dis. 2012, 3, e316. [Google Scholar] [CrossRef] [PubMed]
- Sapoznik, S.; Cohen, B.; Tzuman, Y.; Meir, G.; Ben-Dor, S.; Harmelin, A.; Neeman, M. Gonadotropin-regulated lymphangiogenesis in ovarian cancer is mediated by LEDGF-induced expression of VEGF-C. Cancer Res. 2009, 69, 9306–9314. [Google Scholar] [CrossRef] [PubMed]
- Leitz, J.; Reuschenbach, M.; Lohrey, C.; Honegger, A.; Accardi, R.; Tommasino, M.; Llano, M.; Doeberitz, M.V.K.; Hoppe-Seyler, K.; Hoppe-Seyler, F. Oncogenic human papillomaviruses activate the tumor-associated lens epithelial-derived growth factor (LEDGF) gene. PLoS Pathog. 2014, 10, e1003957. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.-S.; Myklebust, L.M.; Kjarland, E.; Gjertsen, B.T.; Pendino, F.; Bruserud, Ø.; Døskeland, S.O.; Lillehaug, J.R. LEDGF/p75 has increased expression in blasts from chemotherapy-resistant human acute myelogenic leukemia patients and protects leukemia cells from apoptosis in vitro. Mol. Cancer 2007, 6, 31. [Google Scholar] [CrossRef]
- Canella, A.; Van Belle, S.; Brouns, T.; Nigita, G.; Carlon, M.S.; Christ, F.; Debyser, Z. LEDGF/p75-mediated chemoresistance of mixed-lineage leukemia involves cell survival pathways and super enhancer activators. Cancer Gene Ther. 2021. online ahead of print. [Google Scholar] [CrossRef]
- Ortiz-Hernandez, G.L.; Sanchez-Hernandez, E.S.; Casiano, C.A. Twenty years of research on the DFS70/LEDGF autoantibody-autoantigen system: Many lessons learned but still many questions. Autoimmun. Highlights 2020, 11, 3. [Google Scholar] [CrossRef]
- Shinohara, T.; Singh, D.P.; Fatma, N. LEDGF, a survival factor, activates stress-related genes. Prog. Retin. Eye Res. 2002, 21, 341–358. [Google Scholar] [CrossRef]
- Basu, A.; Ross, C.K.C.-D.; Rios-Colon, L.; Mediavilla-Varela, M.; Daniels-Wells, T.R.; Leoh, L.S.; Rojas, H.; Banerjee, H.; Martinez, S.; Acevedo-Martinez, S.; et al. LEDGF/p75 overexpression attenuates oxidative stress-induced necrosis and upregulates the oxidoreductase ERP57/PDIA3/GRP58 in prostate cancer. PLoS ONE 2016, 11, e0146549. [Google Scholar] [CrossRef]
- Yokoyama, A.; Cleary, M.L. Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell 2008, 14, 36–46. [Google Scholar] [CrossRef]
- Mahler, M.; Andrade, L.E.; Casiano, C.A.; Malyavantham, K.; Fritzler, M.J. Anti-DFS70 antibodies: An update on our current un-derstanding and their clinical usefulness. Expert Rev. Clin. Immunol. 2019, 15, 241–250. [Google Scholar] [CrossRef]
- Debyser, Z.; Christ, F.; De Rijck, J.; Gijsbers, R. Host factors for retroviral integration site selection. Trends Biochem. Sci. 2015, 40, 108–116. [Google Scholar] [CrossRef]
- Engelman, A.N.; Singh, P.K. Cellular and molecular mechanisms of HIV-1 integration targeting. Cell. Mol. Life Sci. 2018, 75, 2491–2507. [Google Scholar] [CrossRef] [PubMed]
- Dietz, F.; Franken, S.; Yoshida, K.; Nakamura, H.; Kappler, J.; Gieselmann, V. The family of hepatoma-derived growth factor proteins: Characterization of a new member HRP-4 and classification of its subfamilies. Biochem. J. 2002, 366, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Bao, C.; Wang, J.; Ma, W.; Wang, X.; Cheng, Y. HDGF: A novel jack-of-all-trades in cancer. Futur. Oncol. 2014, 10, 2675–2685. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Farnung, L.; Dienemann, C.; Cramer, P. Structure of H3K36-methylated nucleosome-PWWP complex reveals multi-valent cross-gyre binding. Nat. Struct. Mol. Biol. 2020, 27, 8–13. [Google Scholar] [CrossRef]
- Singh, D.P.; Kubo, E.; Takamura, Y.; Shinohara, T.; Kumar, A.; Chylack, L.T., Jr.; Fatma, N. DNA binding domains and nuclear local-ization signal of LEDGF: Contribution of two helix-turn-helix (HTH)-like domains and a stretch of 58 amino acids of the N-terminal to the trans-activation potential of LEDGF. J. Mol. Biol. 2006, 355, 379–394. [Google Scholar] [CrossRef] [PubMed]
- Leoh, L.S.; Van Heertum, B.; De Rijck, J.; Filippova, M.; Rios-Colon, L.; Basu, A.; Martinez, S.; Tungteakkhun, S.S.; Filippov, V.; Christ, F.; et al. The stress oncoprotein LEDGF/p75 interacts with the methyl CpG binding Protein MeCP2 and influences its transcriptional activity. Mol. Cancer Res. 2012, 10, 378–391. [Google Scholar] [CrossRef]
- Singh, P.K.; Plumb, M.R.; Ferris, A.L.; Iben, J.R.; Wu, X.; Fadel, H.J.; Luke, B.T.; Esnault, C.; Poeschla, E.M.; Hughes, S.H.; et al. LEDGF/p75 interacts with mRNA splicing factors and targets HIV-1 integration to highly spliced genes. Genes Dev. 2015, 29, 2287–2297. [Google Scholar] [CrossRef] [PubMed]
- Daugaard, M.; Baude, A.; Fugger, K.; Povlsen, L.K.; Beck, H.; Sorensen, C.; Petersen, N.H.T.; Sorensen, P.H.B.; Lukas, C.; Bartek, J.; et al. LEDGF (p75) promotes DNA-end resection and homologous recombination. Nat. Struct. Mol. Biol. 2012, 19, 803–810. [Google Scholar] [CrossRef]
- Liedtke, V.; Schröder, C.; Roggenbuck, D.; Weiss, R.; Stohwasser, R.; Schierack, P.; Rödiger, S.; Schenk, L. LEDGF/p75 is required for an efficient DNA damage response. Int. J. Mol. Sci. 2021, 22, 5866. [Google Scholar] [CrossRef]
- Wu, X.; Daniels, T.; Molinaro, C.; Lilly, M.B.; Casiano, C.A. Caspase cleavage of the nuclear autoantigen LEDGF/p75 abrogates its pro-survival function: Implications for autoimmunity in atopic disorders. Cell Death Differ. 2002, 9, 915–925. [Google Scholar] [CrossRef][Green Version]
- Brown-Bryan, T.A.; Leoh, L.S.; Ganapathy, V.; Pacheco, F.J.; Mediavilla-Varela, M.; Filippova, M.; Linkhart, T.A.; Gijsbers, R.; Debyser, Z.; Casiano, C.A. Alternative splicing and caspase-mediated cleavage generate antagonistic variants of the stress oncoprotein LEDGF/p75. Mol. Cancer Res. 2008, 6, 1293–1307. [Google Scholar] [CrossRef] [PubMed]
- Blokken, J.; De Rijck, J.; Christ, F.; Debyser, Z. Protein–protein and protein–chromatin interactions of LEDGF/p75 as novel drug targets. Drug Discov. Today Technol. 2017, 24, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Tesina, P.; Čermáková, K.; Hořejší, M.; Procházková, K.; Fábry, M.; Sharma, S.; Christ, F.; Demeulemeester, J.; Debyser, Z.; De Rijck, J.; et al. Multiple cellular proteins interact with LEDGF/p75 through a conserved unstructured consensus motif. Nat. Commun. 2015, 6, 7968. [Google Scholar] [CrossRef] [PubMed]
- Schrijvers, R.; De Rijck, J.; Demeulemeester, J.; Adachi, N.; Vets, S.; Ronen, K.; Christ, F.; Bushman, F.D.; Debyser, Z.; Gijsbers, R. LEDGF/p75-independent HIV-1 replication demonstrates a role for HRP-2 and remains sensitive to inhibition by LEDGINs. PLoS Pathog. 2012, 8, e1002558. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Jurado, K.A.; Wu, X.; Shun, M.-C.; Li, X.; Ferris, A.L.; Smith, S.J.; Patel, P.A.; Fuchs, J.R.; Cherepanov, P.; et al. HRP2 determines the efficiency and specificity of HIV-1 integration in LEDGF/p75 knockout cells but does not contribute to the antiviral activity of a potent LEDGF/p75-binding site integrase inhibitor. Nucleic Acids Res. 2012, 40, 11518–11530. [Google Scholar] [CrossRef]
- Van Belle, S.; El Ashkar, S.; Čermáková, K.; Matthijssens, F.; Goossens, S.; Canella, A.; Hodges, C.H.; Christ, F.; De Rijck, J.; Van Vlierberghe, P.; et al. Unlike its paralog LEDGF/p75, HRP-2 is dispensable for MLL-R leukemogenesis but important for leukemic cell survival. Cells 2021, 10, 192. [Google Scholar] [CrossRef] [PubMed]
- LeRoy, G.; Oksuz, O.; Descostes, N.; Aoi, Y.; Ganai, R.A.; Kara, H.O.; Yu, J.-R.; Lee, C.-H.; Stafford, J.; Shilatifard, A.; et al. LEDGF and HDGF2 relieve the nucleosome-induced barrier to transcription in differentiated cells. Sci. Adv. 2019, 5, eaay3068. [Google Scholar] [CrossRef]
- Sharma, S.; Čermáková, K.; De Rijck, J.; Demeulemeester, J.; Fábry, M.; El Ashkar, S.; Van Belle, S.; Lepsik, M.; Tesina, P.; Duchoslav, V.; et al. Affinity switching of the LEDGF/p75 IBD interactome is governed by kinase-dependent phosphorylation. Proc. Natl. Acad. Sci. USA 2018, 115, E7053–E7062. [Google Scholar] [CrossRef] [PubMed]
- Malik, R.; Khan, A.P.; Asangani, I.; Cieślik, M.; Prensner, J.; Wang, X.; Iyer, M.K.; Jiang, X.; Borkin, D.; Escara-Wilke, J.; et al. Targeting the MLL complex in castration-resistant prostate cancer. Nat. Med. 2015, 21, 344–352. [Google Scholar] [CrossRef]
- Li, X.; Wu, J.B.; Li, Q.; Shigemura, K.; Chung, L.W.; Huang, W.-C. SREBP-2 promotes stem cell-like properties and metastasis by transcriptional activation of c-Myc in prostate cancer. Oncotarget 2016, 7, 12869–12884. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Woods-Burnham, L.; Ortiz, G.; Rios-Colon, L.; Figueroa, J.; Albesa, R.; Andrade, L.E.; Mahler, M.; Casiano, C.A. Specificity of antinuclear auto-antibodies recognizing the dense fine speckled nuclear pattern: Preferential targeting of DFS70/LEDGFp75 over its interacting partner MeCP2. Clin. Immunol. 2015, 161, 241–250. [Google Scholar] [CrossRef][Green Version]
- Scacheri, P.C.; Rozenblatt-Rosen, O.; Caplen, N.; Wolfsberg, T.G.; Umayam, L.; Lee, J.C.; Hughes, C.M.; Shanmugam, K.S.; Bhattacharjee, A.; Meyerson, M.; et al. Short interfering RNAs can induce unexpected and divergent changes in the levels of untargeted proteins in mammalian cells. Proc. Natl. Acad. Sci. USA 2004, 101, 1892–1897. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.P.; Li, J.; Li, Q.; Li, X.; Liu, C.; Zeng, N.; Huang, J.M.; Chu, G.C.Y.; Lin, C.H.; Zhau, H.E.; et al. R1 regulates prostate tumor growth and progression by transcriptional sup-pression of the E3 ligase HUWE1 to stabilize c-Myc. Mol. Cancer Res. 2018, 16, 1940–1951. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, Y.; Sugiura, K.; Watanabe, A.; Kunimatsu, M.; Mishima, M.; Tomita, Y.; Muro, Y. Autoantigenicity of DFS70 is restricted to the conformational epitope of C-terminal alpha-helical domain. J. Autoimmun. 2004, 23, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Ou, X.-M.; Wu, J.B.; Shih, J.C. Transcription factor E2F-associated phosphoprotein (EAPP), RAM2/CDCA7L/JPO2 (R1), and simian virus 40 promoter factor 1 (Sp1) cooperatively regulate glucocorticoid activation of monoamine oxidase, B. Mol. Pharmacol. 2010, 79, 308–317. [Google Scholar] [CrossRef]
- Ishiguro, T.; Ohata, H.; Sato, A.; Yamawaki, K.; Enomoto, T.; Okamoto, K. Tumor-derived spheroids: Relevance to cancer stem cells and clinical applications. Cancer Sci. 2017, 108, 283–289. [Google Scholar] [CrossRef]
- Kroon, J.; Puhr, M.; Buijs, J.; van der Horst, G.; Hemmer, D.M.; Marijt, K.; Hwang, M.S.; Masood, M.; Grimm, S.; Storm, G.; et al. Glucocorticoid receptor antagonism reverts docetaxel resistance in human prostate cancer. Endocr. Relat. Cancer 2015, 23, 35–45. [Google Scholar] [CrossRef]
- Rizzo, M. Mechanisms of docetaxel resistance in prostate cancer: The key role played by miRNAs. Biochim. Biophys. Acta BBA Rev. Cancer 2021, 1875, 188481. [Google Scholar] [CrossRef]
- Stope, M.B.; Weiss, M.; Preuss, M.; Streitbörger, A.; Ritter, C.A.; Zimmermann, U.; Walther, R.; Burchardt, M. Immediate and transient phosphorylation of the heat shock protein 27 initiates chemoresistance in prostate cancer cells. Oncol. Rep. 2014, 32, 2380–2386. [Google Scholar] [CrossRef]
- Christ, F.; Debyser, Z. The LEDGF/p75 integrase interaction, a novel target for anti-HIV therapy. Virology 2013, 435, 102–109. [Google Scholar] [CrossRef]
- Sanchez, T.W.; Debnath, B.; Christ, F.; Otake, H.; Debyser, Z.; Neamati, N. Discovery of novel inhibitors of LEDGF/p75-IN pro-tein-protein interactions. Bioorg. Med. Chem. 2013, 21, 957–963. [Google Scholar] [CrossRef]
- Čermáková, K.; Tesina, P.; Demeulemeester, J.; El Ashkar, S.; Méreau, H.; Schwaller, J.; Řezáčová, P.; Veverka, V.; De Rijck, J. Validation and structural character-ization of the LEDGF/p75-MLL interface as a new target for the treatment of MLL-dependent leukemia. Cancer Res. 2014, 74, 5139–5151. [Google Scholar] [CrossRef]
- Chan, T.S.; Hawkins, C.; Krieger, J.R.; McGlade, C.J.; Huang, A. JPO2/CDCA7L and LEDGF/p75 are novel mediators of PI3K/AKT signaling and aggressive phenotypes in medulloblastoma. Cancer Res. 2016, 76, 2802–2812. [Google Scholar] [CrossRef]
- Sanidas, I.; Polytarchou, C.; Hatziapostolou, M.; Ezell, S.A.; Kottakis, F.; Hu, L.; Guo, A.; Xie, J.; Comb, M.J.; Iliopoulos, D.; et al. Phosphoproteomics screen reveals akt iso-form-specific signals linking RNA processing to lung cancer. Mol. Cell 2014, 53, 577–590. [Google Scholar] [CrossRef]
- Dávila-González, D.; Choi, D.S.; Rosato, R.R.; Granados-Principal, S.; Kuhn, J.G.; Li, W.-F.; Qian, W.; Chen, W.; Kozielski, A.J.; Wong, H.H.; et al. Pharmacological inhibition of NOS activates ASK1/JNK pathway augmenting docetaxel-mediated apoptosis in triple-negative breast cancer. Clin. Cancer Res. 2018, 24, 1152–1162. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, J.; van Heertum, B.; Vanstreels, E.; Daelemans, D.; De Rijck, J. Dynamics of the ternary complex formed by c-Myc interactor JPO2, transcriptional co-activator LEDGF/p75, and chromatin. J. Biol. Chem. 2014, 289, 12494–12506. [Google Scholar] [CrossRef]
- Hughes, S.; Jenkins, V.; Dar, M.J.; Engelman, A.; Cherepanov, P. Transcriptional co-activator LEDGF interacts with Cdc7-Activator of s-phase kinase (ASK) and stimulates its enzymatic activity. J. Biol. Chem. 2010, 285, 541–554. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Ruiz, S.; Urióstegui-Arcos, M.; Zurita, M. The transcriptional stress response and its implications in cancer treatment. Biochim. Biophys. Acta BBA Rev. Cancer 2021, 1876, 188620. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R. Emerging role of glucocorticoid receptor in castration resistant prostate cancer: A potential therapeutic target. J. Cancer 2020, 11, 696–701. [Google Scholar] [CrossRef] [PubMed]
- Woods-Burnham, L.; Ross, C.K.C.-D.; Love, A.; Basu, A.; Sanchez-Hernandez, E.S.; Martinez, S.; Ortiz-Hernández, G.L.; Stiel, L.; Durán, A.M.; Wilson, C.; et al. Glucocorticoids induce stress oncoproteins associated with therapy-resistance in African American and European American prostate cancer cells. Sci. Rep. 2018, 8, 15063. [Google Scholar] [CrossRef]
- Yu, J.R.; LeRoy, G.; Bready, D.; Frenster, J.D.; Saldaña-Meyer, R.; Jin, Y.; Descostes, N.; Stafford, J.M.; Placantonakis, D.G.; Reinberg, D. The H3K36me2 writer-reader dependency in H3K27M-DIPG. Sci. Adv. 2021, 7, eabg7444. [Google Scholar] [CrossRef]
- Baude, A.; Aaes, T.L.; Zhai, B.; Al-Nakouzi, N.; Oo, H.Z.; Daugaard, M.; Rohde, M.; Jäättelä, M. Hepatoma-derived growth factor-related protein 2 promotes DNA repair by homologous recombination. Nucleic Acids Res. 2016, 44, 2214–2226. [Google Scholar] [CrossRef]
- Dong, J.; Li, J.; Li, Y.; Ma, Z.; Yu, Y.; Wang, C.-Y. Transcriptional super-enhancers control cancer stemness and metastasis genes in squamous cell carcinoma. Nat. Commun. 2021, 12, 3974. [Google Scholar] [CrossRef]
- Civenni, G.; Malek, A.; Albino, D.; Garcia-Escudero, R.; Napoli, S.; Di Marco, S.; Pinton, S.; Sarti, M.; Carbone, G.M.; Catapano, C.V. RNAi-mediated silencing of Myc transcription inhibits stem-like cell maintenance and tumorigenicity in prostate cancer. Cancer Res. 2013, 73, 6816–6827. [Google Scholar] [CrossRef] [PubMed]
- Fatma, H.; Maurya, S.K.; Siddique, H.R. Epigenetic modifications of c-MYC: Role in cancer cell reprogramming, progression and chemoresistance. Semin. Cancer Biol. 2020. online ahead of print. [Google Scholar] [CrossRef]
- Han, H.; Lee, H.H.; Choi, K.; Moon, Y.J.; Heo, J.E.; Ham, W.S.; Jang, W.S.; Rha, K.H.; Cho, N.H.; Giancotti, F.G.; et al. Prostate epithelial genes define therapy-relevant prostate cancer molecular subtype. Prostate Cancer Prostatic Dis. 2021. online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Hatano, K.; Yamaguchi, S.; Nimura, K.; Murakami, K.; Nagahara, A.; Fujita, K.; Uemura, M.; Nakai, Y.; Tsuchiya, M.; Nakayama, M.; et al. Residual prostate cancer cells after docetaxel therapy increase the tumorigenic potential via constitutive signaling of CXCR4, ERK1/2 and c-Myc. Mol. Cancer Res. 2013, 11, 1088–1100. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Approximate MW | Other Common Names | Functions | PC3-DR * | DU145-DR * |
|---|---|---|---|---|---|
| LEDGF/p75 | 70 kD | DFS70, PSIP1 | Stress survival, transcription coactivator of RNAPII, chromatin binding | 3.35 | 4.50 |
| JPO2 | 52 kD | CDCA7L, R1 | PI3K regulator, c-MYC binding protein and potentiator | 10.32 | 3.92 |
| c-MYC | 50 kD | Oncogenic transcription factor; cancer stemness marker | 4.44 | 3.71 | |
| Menin | 83 kD | MEN1 | Histone methyltransferase, transcription factor, hematopoiesis, leukemogenesis | 1.45 | 1.90 |
| MLL | 432 kD | KMT2A | Transcription factor, early development, hematopoiesis, leukemogenesis | 3.13 | 2.05 |
| IWS1 | 56 kD | RNAPII elongation, transcription regulator | 2.18 | 2.15 | |
| ASK1 | 70 kD | MAP3K5 | Stress-activated cell cycle regulating mitogen activated kinase | 7.40 | 4.35 |
| PogZ | 155 kD | Pogo transposable element, mitosis, chromatin remodeling | 4.21 | 3.93 | |
| Med-1 | 168 kD | TRAPP220 | Mediator of RNAPII transcription subunit 1 | ||
| HRP2 | 74 kD | HDGF2, HDGFL2, HDGFRP2 | RNAPII transcription regulator, relieves nucleosome-induced barrier to transcription | 2.44 | 2.42 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ortiz-Hernandez, G.L.; Sanchez-Hernandez, E.S.; Ochoa, P.T.; Elix, C.C.; Alkashgari, H.R.; McMullen, J.R.W.; Soto, U.; Martinez, S.R.; Diaz Osterman, C.J.; Mahler, M.; et al. The LEDGF/p75 Integrase Binding Domain Interactome Contributes to the Survival, Clonogenicity, and Tumorsphere Formation of Docetaxel-Resistant Prostate Cancer Cells. Cells 2021, 10, 2723. https://doi.org/10.3390/cells10102723
Ortiz-Hernandez GL, Sanchez-Hernandez ES, Ochoa PT, Elix CC, Alkashgari HR, McMullen JRW, Soto U, Martinez SR, Diaz Osterman CJ, Mahler M, et al. The LEDGF/p75 Integrase Binding Domain Interactome Contributes to the Survival, Clonogenicity, and Tumorsphere Formation of Docetaxel-Resistant Prostate Cancer Cells. Cells. 2021; 10(10):2723. https://doi.org/10.3390/cells10102723
Chicago/Turabian StyleOrtiz-Hernandez, Greisha L., Evelyn S. Sanchez-Hernandez, Pedro T. Ochoa, Catherine C. Elix, Hossam R. Alkashgari, James R. W. McMullen, Ubaldo Soto, Shannalee R. Martinez, Carlos J. Diaz Osterman, Michael Mahler, and et al. 2021. "The LEDGF/p75 Integrase Binding Domain Interactome Contributes to the Survival, Clonogenicity, and Tumorsphere Formation of Docetaxel-Resistant Prostate Cancer Cells" Cells 10, no. 10: 2723. https://doi.org/10.3390/cells10102723
APA StyleOrtiz-Hernandez, G. L., Sanchez-Hernandez, E. S., Ochoa, P. T., Elix, C. C., Alkashgari, H. R., McMullen, J. R. W., Soto, U., Martinez, S. R., Diaz Osterman, C. J., Mahler, M., Roy, S., & Casiano, C. A. (2021). The LEDGF/p75 Integrase Binding Domain Interactome Contributes to the Survival, Clonogenicity, and Tumorsphere Formation of Docetaxel-Resistant Prostate Cancer Cells. Cells, 10(10), 2723. https://doi.org/10.3390/cells10102723