Modeling CNS Involvement in Pompe Disease Using Neural Stem Cells Generated from Patient-Derived Induced Pluripotent Stem Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Induced Pluripotent Stem Cell Generation and Culture
2.3. Neural Stem Cell Differentiation and Maintenance
2.4. Immunocytochemistry (ICC) Staining
2.5. LysoTracker and Nile Red Staining
2.6. Western Blot Analysis
2.7. Glycogen Quantification
2.8. Fluorometric GAA Enzyme Assay
2.9. Recombinant Human GAA Treatment
2.10. Treatment with HPβCD and δ-Tocopherol
2.11. Data Analysis and Statistics
3. Results
3.1. Generation and Characterization of Pompe Disease iPSCs from Pompe Patient Fibroblasts
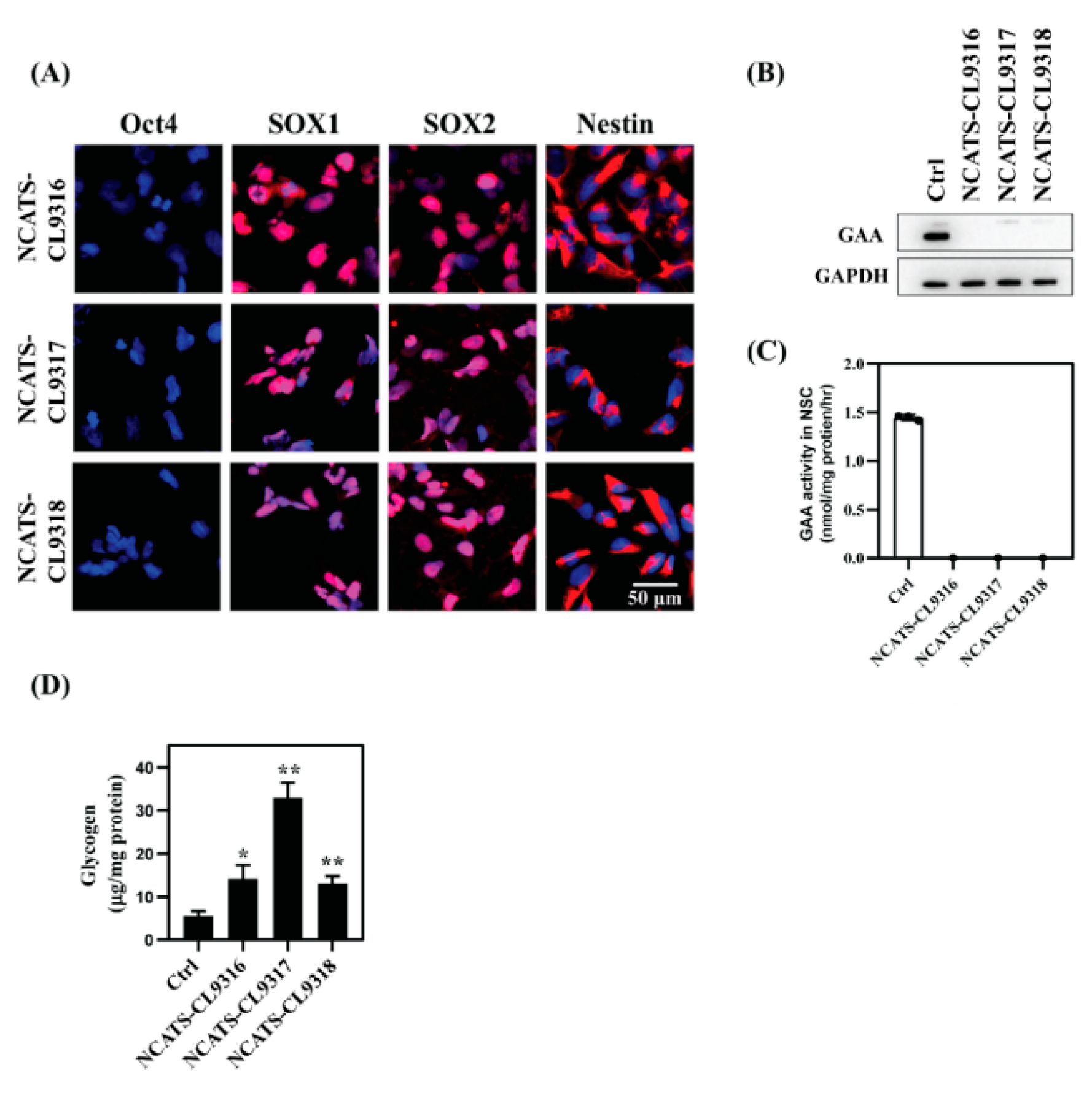
3.2. NSC Differentiation and Characterization
3.3. Enlarged Lysosomes and Neutral Lipid Increase in Pompe Disease NSCs
3.4. Recombinant Human GAA Decreased the Phenotypes in Pompe Disease NSCs
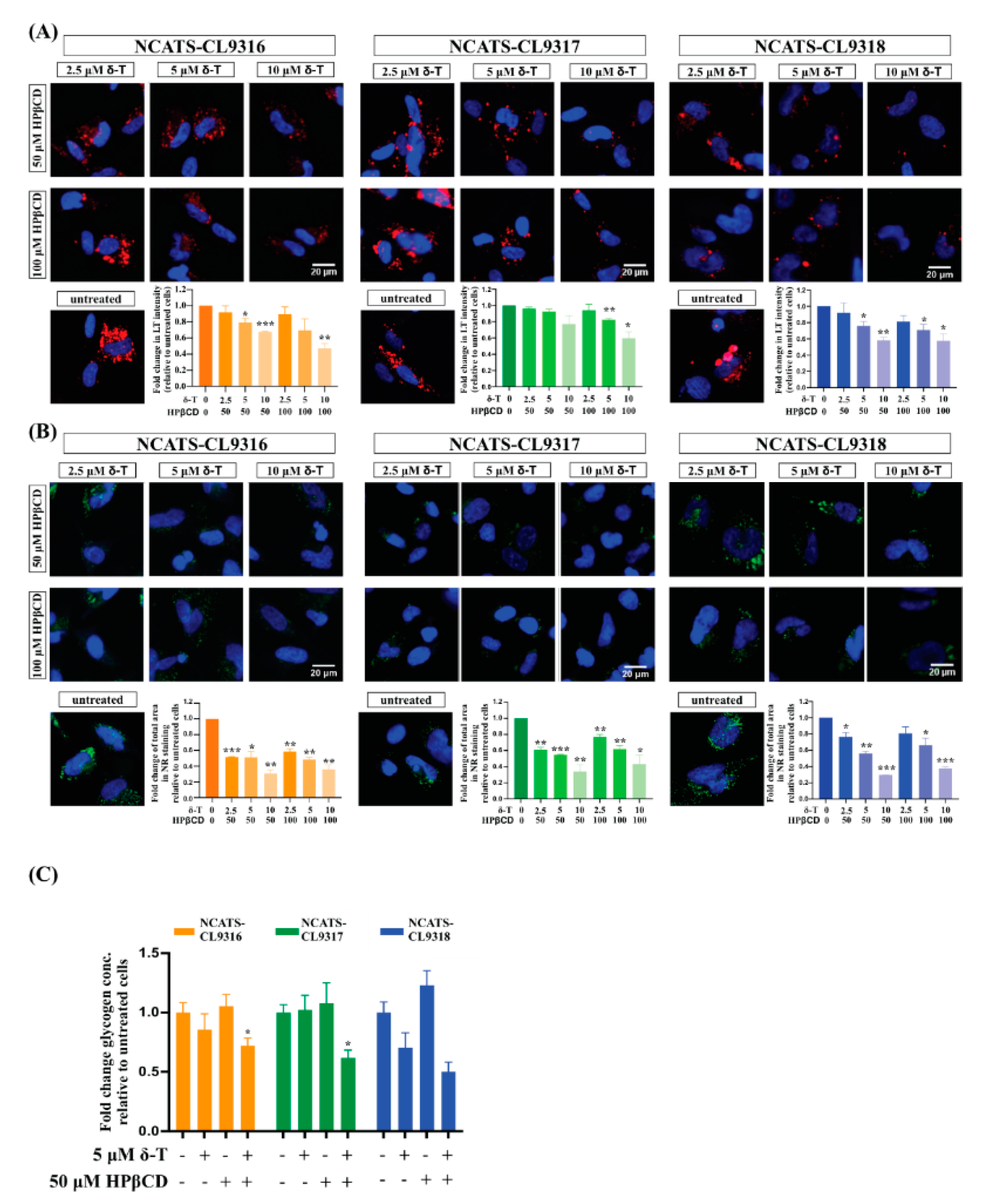
3.5. Delta-Tocopherol Ameliorated Lipid Accumulation in Pompe Disease NSCs
3.6. Combination of δ-Tocopherol and HPβCD Reduced Disease Phenotypes in Pompe Disease NSCs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- van der Ploeg, A.T.; Reuser, A.J. Pompe’s disease. Lancet 2008, 372, 1342–1353. [Google Scholar] [CrossRef]
- Reuser, A.J.J.; Hirschhorn, R.; Kroos, M.A. Pompe Disease: Glycogen Storage Disease Type II, Acid α-Glucosidase (Acid Maltase) Deficiency. In The Online Metabolic and Molecular Bases of Inherited Disease; Valle, D.L., Antonarakis, S., Ballabio, A., Beaudet, A.L., Mitchell, G.A., Eds.; McGraw-Hill Education: New York, NY, USA, 2019. [Google Scholar]
- Kishnani, P.S.; Steiner, R.D.; Bali, D.; Berger, K.; Byrne, B.J.; Case, L.; Crowley, J.F.; Downs, S.; Howell, R.R.; Kravitz, R.M.; et al. Pompe disease diagnosis and management guideline. Genet. Med. 2006, 8, 267–288. [Google Scholar] [CrossRef] [PubMed]
- DeRuisseau, L.R.; Fuller, D.D.; Qiu, K.; DeRuisseau, K.C.; Donnelly, W.H., Jr.; Mah, C.; Reier, P.J.; Byrne, B.J. Neural deficits contribute to respiratory insufficiency in Pompe disease. Proc. Natl. Acad. Sci. USA 2009, 106, 9419–9424. [Google Scholar] [CrossRef] [PubMed]
- Turner, S.M.F.; Falk, D.J.; Byrne, B.J.; Fuller, D.D. Transcriptome assessment of the Pompe (Gaa-/-) mouse spinal cord indicates widespread neuropathology. Physiol. Genom. 2016, 48, 785–794. [Google Scholar] [CrossRef]
- Falk, D.J.; Todd, A.G.; Lee, S.; Soustek, M.S.; ElMallah, M.K.; Fuller, D.D.; Notterpek, L.; Byrne, B.J. Peripheral nerve and neuromuscular junction pathology in Pompe disease. Hum. Mol. Genet. 2015, 24, 625–636. [Google Scholar] [CrossRef]
- Korlimarla, A.; Lim, J.A.; Kishnani, P.S.; Sun, B. An emerging phenotype of central nervous system involvement in Pompe disease: From bench to bedside and beyond. Ann. Transl. Med. 2019, 7, 289. [Google Scholar] [CrossRef]
- Fuller, D.D.; ElMallah, M.K.; Smith, B.K.; Corti, M.; Lawson, L.A.; Falk, D.J.; Byrne, B.J. The respiratory neuromuscular system in Pompe disease. Respir. Physiol. Neurobiol. 2013, 189, 241–249. [Google Scholar] [CrossRef]
- McIntosh, P.T.; Hobson-Webb, L.D.; Kazi, Z.B.; Prater, S.N.; Banugaria, S.G.; Austin, S.; Wang, R.; Enterline, D.S.; Frush, D.P.; Kishnani, P.S. Neuroimaging findings in infantile Pompe patients treated with enzyme replacement therapy. Mol. Genet. Metab. 2018, 123, 85–91. [Google Scholar] [CrossRef]
- Ebbink, B.J.; Poelman, E.; Aarsen, F.K.; Plug, I.; Régal, L.; Muentjes, C.; van der Beek, N.; Lequin, M.H.; van der Ploeg, A.T.; van den Hout, J.M.; et al. Classic infantile Pompe patients approaching adulthood: A cohort study on consequences for the brain. Dev. Med. Child Neurol. 2018, 60. [Google Scholar] [CrossRef]
- Huang, H.P.; Chen, P.H.; Hwu, W.L.; Chuang, C.Y.; Chien, Y.H.; Stone, L.; Chien, C.L.; Li, L.T.; Chiang, S.C.; Chen, H.F.; et al. Human Pompe disease-induced pluripotent stem cells for pathogenesis modeling, drug testing and disease marker identification. Hum. Mol. Genet. 2011, 20, 4851–4864. [Google Scholar] [CrossRef]
- Sato, Y.; Kobayashi, H.; Higuchi, T.; Shimada, Y.; Era, T.; Kimura, S.; Eto, Y.; Ida, H.; Ohashi, T. Disease modeling and lentiviral gene transfer in patient-specific induced pluripotent stem cells from late-onset Pompe disease patient. Mol. Ther. Methods Clin. Dev. 2015, 2, 15023. [Google Scholar] [CrossRef] [PubMed]
- Raval, K.K.; Tao, R.; White, B.E.; De Lange, W.J.; Koonce, C.H.; Yu, J.; Kishnani, P.S.; Thomson, J.A.; Mosher, D.F.; Ralphe, J.C.; et al. Pompe disease results in a Golgi-based glycosylation deficit in human induced pluripotent stem cell-derived cardiomyocytes. J. Biol. Chem. 2015, 290, 3121–3136. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Kobayashi, H.; Higuchi, T.; Shimada, Y.; Ida, H.; Ohashi, T. Metabolomic Profiling of Pompe Disease-Induced Pluripotent Stem Cell-Derived Cardiomyocytes Reveals That Oxidative Stress Is Associated with Cardiac and Skeletal Muscle Pathology. Stem Cells Transl. Med. 2017, 6, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Kobayashi, H.; Higuchi, T.; Shimada, Y.; Ida, H.; Ohashi, T. TFEB overexpression promotes glycogen clearance of Pompe disease iPSC-derived skeletal muscle. Mol. Ther. Methods Clin. Dev. 2016, 3, 16054. [Google Scholar] [CrossRef]
- van der Wal, E.; Bergsma, A.J.; van Gestel, T.J.; in ‘t Groen, S.L.; Zaehres, H.; Araúzo-Bravo, M.J.; Schöler, H.R.; van der Ploeg, A.T.; Pijnappel, W.P. GAA Deficiency in Pompe Disease Is Alleviated by Exon Inclusion in iPSC-Derived Skeletal Muscle Cells. Mol. Ther. Nucleic Acids 2017, 7, 101–115. [Google Scholar] [CrossRef]
- Yoshida, T.; Awaya, T.; Jonouchi, T.; Kimura, R.; Kimura, S.; Era, T.; Heike, T.; Sakurai, H. A Skeletal Muscle Model of Infantile-onset Pompe Disease with Patient-specific iPS Cells. Sci. Rep. 2017, 7, 13473. [Google Scholar] [CrossRef]
- Aguisanda, F.; Yeh, C.D.; Chen, C.Z.; Li, R.; Beers, J.; Zou, J.; Thorne, N.; Zheng, W. Neural stem cells for disease modeling of Wolman disease and evaluation of therapeutics. Orphanet J. Rare Dis. 2017, 12, 120. [Google Scholar] [CrossRef]
- Sima, N.; Li, R.; Huang, W.; Xu, M.; Beers, J.; Zou, J.; Titus, S.; Ottinger, E.A.; Marugan, J.J.; Xie, X.; et al. Neural Stem Cells for Disease Modeling and Evaluation of Therapeutics for Infantile (CLN1/PPT1) and Late Infantile (CLN2/TPP1) Neuronal Ceroid Lipofuscinoses. Orphanet J. Rare Dis. 2018, 13. [Google Scholar] [CrossRef]
- Xu, M.; Liu, K.; Swaroop, M.; Porter, F.D.; Sidhu, R.; Finkes, S.; Ory, D.S.; Marugan, J.J.; Xiao, J.; Southall, N.; et al. δ-Tocopherol Reduces Lipid Accumulation in Niemann-Pick Type C1 and Wolman Cholesterol Storage Disorders*. J. Biol. Chem. 2012, 287, 39349–39360. [Google Scholar] [CrossRef]
- Yu, D.; Swaroop, M.; Wang, M.; Baxa, U.; Yang, R.; Yan, Y.; Coksaygan, T.; DeTolla, L.; Marugan, J.J.; Austin, C.P.; et al. Niemann-Pick Disease Type C: Induced Pluripotent Stem Cell-Derived Neuronal Cells for Modeling Neural Disease and Evaluating Drug Efficacy. J. Biomol. Screen. 2014, 19, 1164–1173. [Google Scholar] [CrossRef]
- Long, Y.; Xu, M.; Li, R.; Dai, S.; Beers, J.; Chen, G.; Soheilian, F.; Baxa, U.; Wang, M.; Marugan, J.J.; et al. Induced Pluripotent Stem Cells for Disease Modeling and Evaluation of Therapeutics for Niemann-Pick Disease Type A. Stem Cells Transl. Med. 2016, 5, 1644–1655. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.W.; Li, C.; Ioannou, Y.A. Cyclodextrin induces calcium-dependent lysosomal exocytosis. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Beers, J.; Linask, K.L.; Chen, J.A.; Siniscalchi, L.I.; Lin, Y.; Zheng, W.; Rao, M.; Chen, G. A cost-effective and efficient reprogramming platform for large-scale production of integration-free human induced pluripotent stem cells in chemically defined culture. Sci. Rep. 2015, 5, 11319. [Google Scholar] [CrossRef] [PubMed]
- Beers, J.; Gulbranson, D.R.; George, N.; Siniscalchi, L.I.; Jones, J.; Thomson, J.A.; Chen, G. Passaging and colony expansion of human pluripotent stem cells by enzyme-free dissociation in chemically defined culture conditions. Nat. Protoc. 2012, 7, 2029–2040. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Kallwass, H.; Young, S.P.; Carr, C.; Dai, J.; Kishnani, P.S.; Millington, D.S.; Keutzer, J.; Chen, Y.T.; Bali, D. Comparison of maltose and acarbose as inhibitors of maltase-glucoamylase activity in assaying acid alpha-glucosidase activity in dried blood spots for the diagnosis of infantile Pompe disease. Genet. Med. 2006, 8, 302–306. [Google Scholar] [CrossRef]
- NHLBI GO Exome Sequencing Project (ESP). Exome Variant Server. Available online: http://evs.gs.washington.edu/EVS/ (accessed on 20 February 2020).
- Cheng, Y.S.; Li, R.; Baskfield, A.; Beers, J.; Zou, J.; Liu, C.; Zheng, W. A human induced pluripotent stem cell line (TRNDi007-B) from an infantile onset Pompe patient carrying p.R854X mutation in the GAA gene. Stem Cell Res. 2019, 37, 101435. [Google Scholar] [CrossRef]
- Vu, M.; Li, R.; Baskfield, A.; Lu, B.; Farkhondeh, A.; Gorshkov, K.; Motabar, O.; Beers, J.; Chen, G.; Zou, J.; et al. Neural stem cells for disease modeling and evaluation of therapeutics for Tay-Sachs disease. Orphanet J. Rare Dis. 2018, 13, 152. [Google Scholar] [CrossRef]
- Wang, Z.; Okamoto, P.; Keutzer, J. A new assay for fast, reliable CRIM status determination in infantile-onset Pompe disease. Mol. Genet. Metab. 2014, 111, 92–100. [Google Scholar] [CrossRef]
- Martiniuk, F.; Mehler, M.; Tzall, S.; Meredith, G.; Hirschhorn, R. Extensive genetic heterogeneity in patients with acid alpha glucosidase deficiency as detected by abnormalities of DNA and mRNA. Am. J. Hum. Genet. 1990, 47, 73–78. [Google Scholar]
- Xu, M.; Liu, K.; Swaroop, M.; Sun, W.; Dehdashti, S.J.; McKew, J.C.; Zheng, W. A phenotypic compound screening assay for lysosomal storage diseases. J. Biomol. Screen. 2014, 19, 168–175. [Google Scholar] [CrossRef]
- Verity, M.A. Infantile Pompe’s disease, lipid storage, and partial carnitine deficiency. Muscle Nerve 1991, 14, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Sarnat, H.B.; Roth, S.I.; Carroll, J.E.; Brown, B.I.; Dungan, W.T. Lipid storage myopathy in infantile Pompe’s disease. Arch. Neurol. 1982, 39, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Gorba, T.; Conti, L. Neural Stem Cells as Tools for Drug Discovery: Novel Platforms and Approaches. Expert Opin. Drug Discov. 2013, 8. [Google Scholar] [CrossRef]
- Borger, D.K.; McMahon, B.; Roshan Lal, T.; Serra-Vinardell, J.; Aflaki, E.; Sidransky, E. Induced pluripotent stem cell models of lysosomal storage disorders. Dis. Model. Mech. 2017, 10, 691–704. [Google Scholar] [CrossRef]
- Li, L.; Chao, J.; Shi, Y. Modeling neurological diseases using iPSC-derived neural cells. Cell Tissue Res. 2017, 371, 143–151. [Google Scholar] [CrossRef]
- Swaroop, M.; Brooks, M.J.; Gieser, L.; Swaroop, A.; Zheng, W. Patient iPSC-derived Neural Stem Cells Exhibit Phenotypes in Concordance With the Clinical Severity of Mucopolysaccharidosis I. Hum. Mol. Genet. 2018, 27. [Google Scholar] [CrossRef]
- Mathur, P.; Ding, Z.; Saldeen, T.; Mehta, J.L. Tocopherols in the Prevention and Treatment of Atherosclerosis and Related Cardiovascular Disease. Clin. Cardiol. 2015, 38, 570–576. [Google Scholar] [CrossRef] [PubMed]
- Smolarek, A.K.; So, J.Y.; Burgess, B.; Kong, A.-N.T.; Reuhl, K.; Lin, Y.; Shih, W.J.; Li, G.; Lee, M.-J.; Chen, Y.-K.; et al. Dietary Administration of δ- and γ-Tocopherol Inhibits Tumorigenesis in the Animal Model of Estrogen Receptor-Positive, but not HER-2 Breast Cancer. Cancer Prev. Res. 2012, 5, 1310–1320. [Google Scholar] [CrossRef]
- Guan, F.; Li, G.; Liu, A.B.; Lee, M.-J.; Yang, Z.; Chen, Y.-K.; Lin, Y.; Shih, W.; Yang, C.S. δ- and γ-tocopherols, but not α-tocopherol, inhibit colon carcinogenesis in azoxymethane-treated F344 rats. Cancer Prev. Res. 2012, 5, 644–654. [Google Scholar] [CrossRef]
- Li, G.-X.; Lee, M.-J.; Liu, A.B.; Yang, Z.; Lin, Y.; Shih, W.J.; Yang, C.S. δ-tocopherol is more active than α- or γ-tocopherol in inhibiting lung tumorigenesis In Vivo. Cancer Prev. Res. 2011, 4, 404–413. [Google Scholar] [CrossRef]
- Wang, H.; Hong, J.; Yang, C.S. δ-Tocopherol inhibits receptor tyrosine kinase-induced AKT activation in prostate cancer cells. Mol. Carcinog. 2015, 55, 1728–1738. [Google Scholar] [CrossRef] [PubMed]
- Loftsson, T.; Duchêne, D. Cyclodextrins and their pharmaceutical applications. Int. J. Pharm. 2007, 329, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Vite, C.H.; Bagel, J.H.; Swain, G.P.; Prociuk, M.; Sikora, T.U.; Stein, V.M.; O’Donnell, P.; Ruane, T.; Ward, S.; Crooks, A.; et al. Intracisternal cyclodextrin prevents cerebellar dysfunction and Purkinje cell death in feline Niemann-Pick type C1 disease. Sci. Transl. Med. 2015, 7, 276ra26. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Tokumaru, H.; Ishitsuka, Y.; Matsumoto, T.; Taguchi, M.; Motoyama, K.; Higashi, T.; Arima, H.; Matsuo, M.; Higaki, K.; et al. In vitro evaluation of 2-hydroxyalkylated β-cyclodextrins as potential therapeutic agents for Niemann-Pick Type C disease. Mol. Genet. Metab. 2016, 118, 214–219. [Google Scholar] [CrossRef]
- Davidson, C.D.; Ali, N.F.; Micsenyi, M.C.; Stephney, G.; Renault, S.; Dobrenis, K.; Ory, D.S.; Vanier, M.T.; Walkley, S.U. Chronic Cyclodextrin Treatment of Murine Niemann-Pick C Disease Ameliorates Neuronal Cholesterol and Glycosphingolipid Storage and Disease Progression. PLoS ONE 2009, 4, e6951. [Google Scholar] [CrossRef]
- Liu, B.; Turley, S.D.; Burns, D.K.; Miller, A.M.; Repa, J.J.; Dietschy, J.M. Reversal of defective lysosomal transport in NPC disease ameliorates liver dysfunction and neurodegeneration in the npc1-/- mouse. Proc. Natl. Acad. Sci. USA 2009, 106, 2377–2382. [Google Scholar] [CrossRef]
- Camargo, F.; Erickson, R.P.; Garver, W.S.; Hossain, G.; Carbone, P.N.; Heidenreich, R.A.; Blanchard, J. Cyclodextrins in the treatment of a mouse model of Niemann-Pick C disease. Life Sci. 2001, 70, 131–142. [Google Scholar] [CrossRef]
- van der Ploeg, A.T.; Barohn, R.; Carlson, L.; Charrow, J.; Clemens, P.R.; Hopkin, R.J.; Kishnani, P.S.; Laforet, P.; Morgan, C.; Nations, S.; et al. Open-label extension study following the Late-Onset Treatment Study (LOTS) of alglucosidase alfa. Mol. Genet. Metab. 2012, 107, 456–461. [Google Scholar] [CrossRef]
- Prater, S.N.; Patel, T.T.; Buckley, A.F.; Mandel, H.; Vlodavski, E.; Banugaria, S.G.; Feeney, E.J.; Raben, N.; Kishnani, P.S. Skeletal muscle pathology of infantile Pompe disease during long-term enzyme replacement therapy. Orphanet J. Rare Dis. 2013, 8, 90. [Google Scholar] [CrossRef]
- Toscano, A.; Schoser, B. Enzyme replacement therapy in late-onset Pompe disease: A systematic literature review. J. Neurol. 2013, 260, 951–959. [Google Scholar] [CrossRef]
- Hahn, A.; Praetorius, S.; Karabul, N.; Diessel, J.; Schmidt, D.; Motz, R.; Haase, C.; Baethmann, M.; Hennermann, J.B.; Smitka, M.; et al. Outcome of patients with classical infantile pompe disease receiving enzyme replacement therapy in Germany. JIMD Rep. 2015, 20, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Schoser, B.; Stewart, A.; Kanters, S.; Hamed, A.; Jansen, J.; Chan, K.; Karamouzian, M.; Toscano, A. Survival and long-term outcomes in late-onset Pompe disease following alglucosidase alfa treatment: A systematic review and meta-analysis. J. Neurol. 2017, 264, 621–630. [Google Scholar] [CrossRef]
- Raben, N.; Roberts, A.; Plotz, P.H. Role of autophagy in the pathogenesis of Pompe disease. Acta Myol. 2007, 26, 45–48. [Google Scholar]
- Raben, N.; Takikita, S.; Pittis, M.G.; Bembi, B.; Marie, S.K.; Roberts, A.; Page, L.; Kishnani, P.S.; Schoser, B.G.; Chien, Y.H.; et al. Deconstructing Pompe disease by analyzing single muscle fibers: To see a world in a grain of sand. Autophagy 2007, 3, 546–552. [Google Scholar] [CrossRef] [PubMed]
- Do, H.V.; Raben, N.; Khanna, R.; Xu, S.; Lun, Y.; Frascella, M.; Garcia, A.; Soska, R.; Nair, A.; Ponery, A.S.; et al. Improved efficacy of a next-generation ERT in murine Pompe disease. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Huang, H.P.; Chiang, W.; Stone, L.; Kang, C.K.; Chuang, C.Y.; Kuo, H.C. Using human Pompe disease-induced pluripotent stem cell-derived neural cells to identify compounds with therapeutic potential. Hum. Mol. Genet. 2019. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, Y.-S.; Yang, S.; Hong, J.; Li, R.; Beers, J.; Zou, J.; Huang, W.; Zheng, W. Modeling CNS Involvement in Pompe Disease Using Neural Stem Cells Generated from Patient-Derived Induced Pluripotent Stem Cells. Cells 2021, 10, 8. https://doi.org/10.3390/cells10010008
Cheng Y-S, Yang S, Hong J, Li R, Beers J, Zou J, Huang W, Zheng W. Modeling CNS Involvement in Pompe Disease Using Neural Stem Cells Generated from Patient-Derived Induced Pluripotent Stem Cells. Cells. 2021; 10(1):8. https://doi.org/10.3390/cells10010008
Chicago/Turabian StyleCheng, Yu-Shan, Shu Yang, Junjie Hong, Rong Li, Jeanette Beers, Jizhong Zou, Wenwei Huang, and Wei Zheng. 2021. "Modeling CNS Involvement in Pompe Disease Using Neural Stem Cells Generated from Patient-Derived Induced Pluripotent Stem Cells" Cells 10, no. 1: 8. https://doi.org/10.3390/cells10010008
APA StyleCheng, Y.-S., Yang, S., Hong, J., Li, R., Beers, J., Zou, J., Huang, W., & Zheng, W. (2021). Modeling CNS Involvement in Pompe Disease Using Neural Stem Cells Generated from Patient-Derived Induced Pluripotent Stem Cells. Cells, 10(1), 8. https://doi.org/10.3390/cells10010008