Towards a Functional Cure for Diabetes Using Stem Cell-Derived Beta Cells: Are We There Yet?

 , , and
, , and 
Abstract
1. Introduction
2. Beta Cell Maturation and Heterogeneity
2.1. The Mature Beta Cell
2.2. Heterogeneity in the Adult Beta Cell Population
3. General Properties of Stem Cells
4. Current Status of Human ESC and iPSC Differentiation Protocols
4.1. Generation of Stem Cell-Derived Beta Cells
4.2. Generation of Non-Beta Pancreatic Endocrine Cell Types
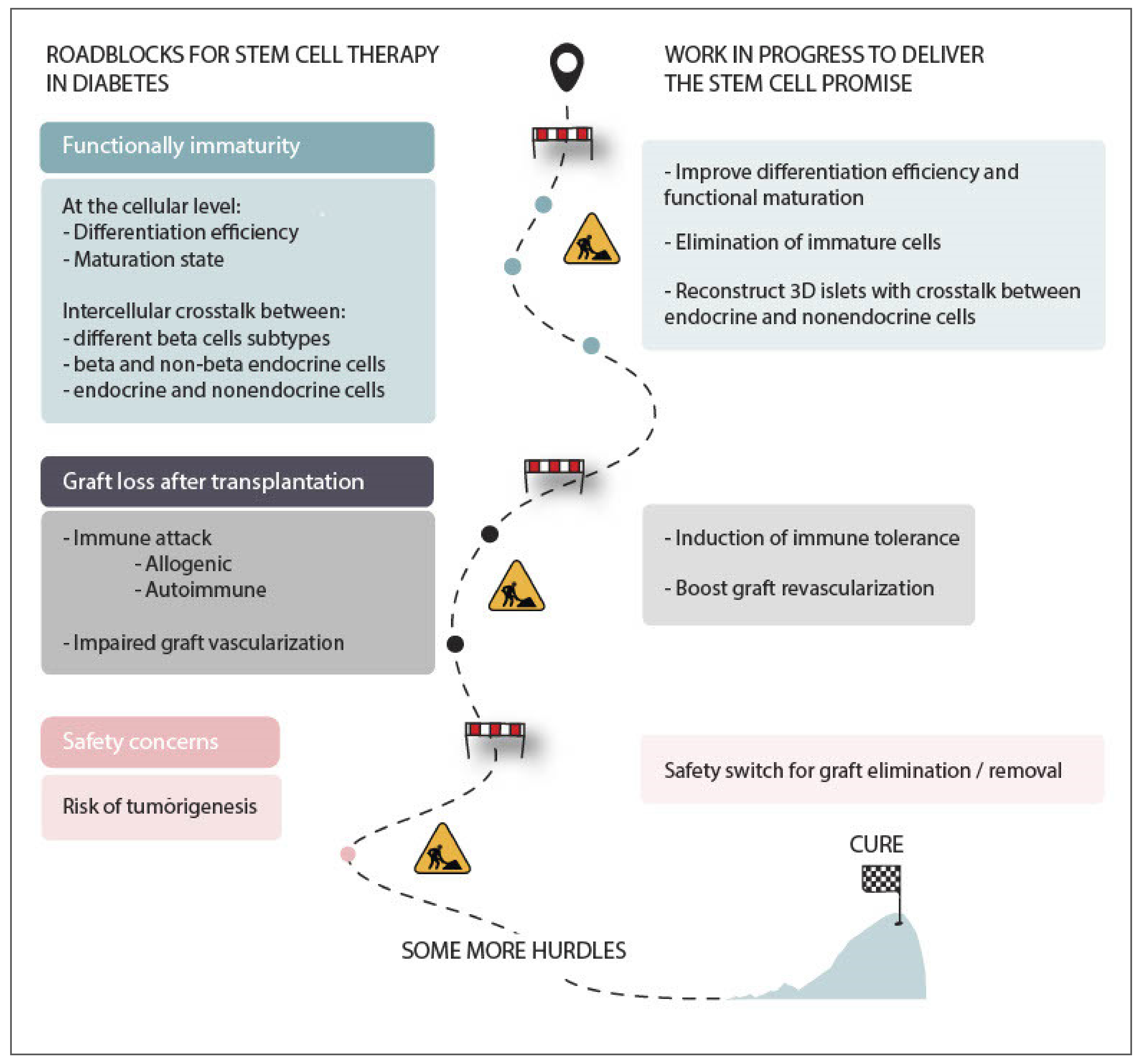
5. Current Hurdles for Stem Cell Therapy and Possible Ways to Tackle Them
5.1. Functional Immaturity of Stem Cell-Derived Beta Cells
5.2. Risk of Tumorigenesis
5.3. Graft Immune Rejection
5.4. The Emergence of Encapsulation Devices
6. Towards a Cure
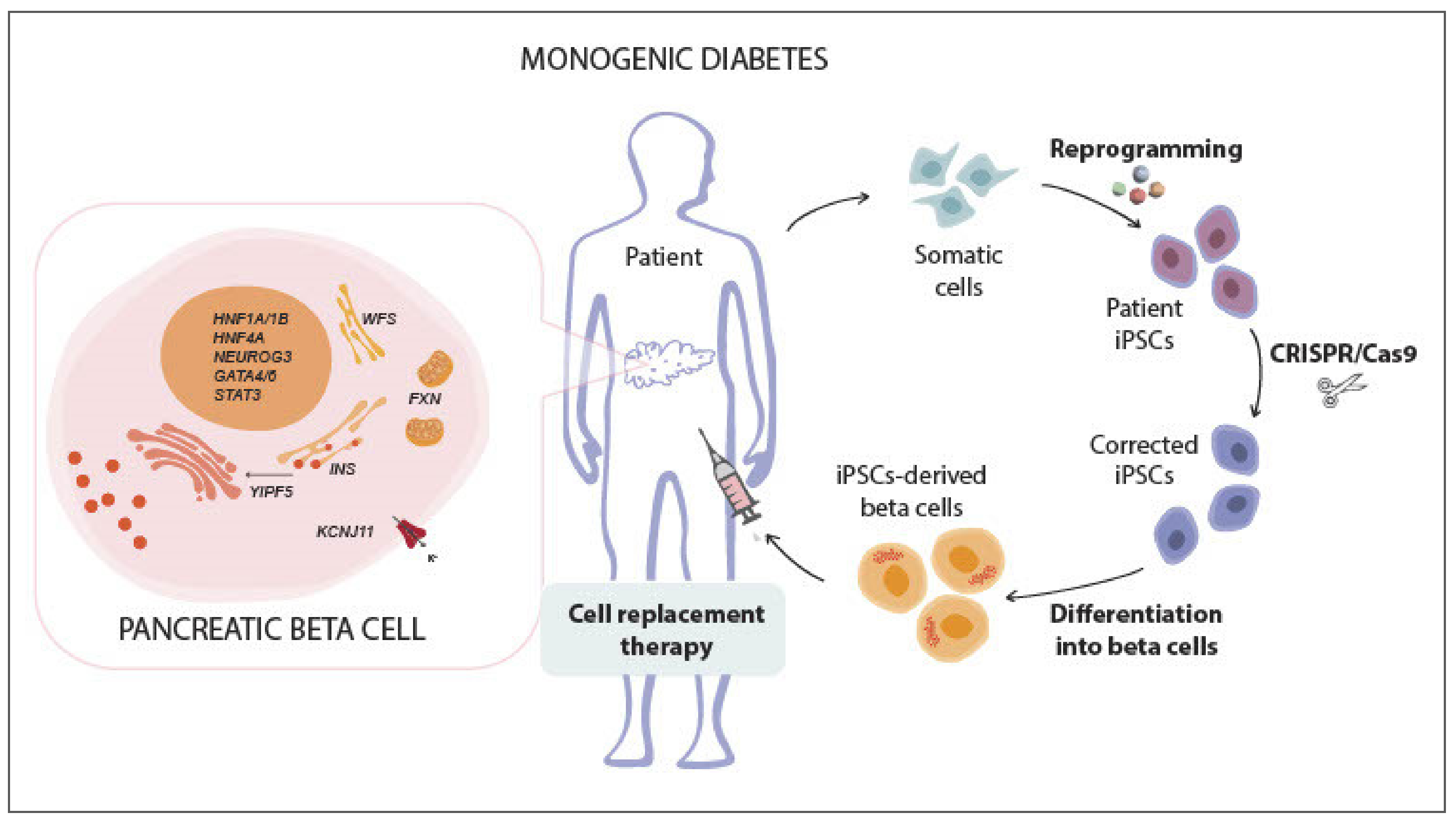
6.1. The Paradigm of Gene-Editing for Monogenic Diabetes
6.2. Where Do the Clinical Trials in T1D Stand?
7. Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- International Diabetes Federation. IDF Diabetes Atlas, 9th ed.; International Diabetes Federation: Brussels, Belgium, 2019; Available online: https://www.diabetesatlas.org (accessed on 1 November 2020).
- Herold, K.C.; Vignali, D.A.A.; Cooke, A.; Bluestone, J.A. Type 1 diabetes: Translating mechanistic observations into effective clinical outcomes. Nat. Rev. Immunol. 2013, 13, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Nolan, C.J.; Damm, P.; Prentki, M. Type 2 diabetes across generations: From pathophysiology to prevention and management. Lancet 2011, 378, 169–181. [Google Scholar] [CrossRef]
- Yang, Y.; Chan, L. Monogenic diabetes: What it teaches us on the common forms of type 1 and type 2 diabetes. Endocr. Rev. 2016, 37, 190–222. [Google Scholar] [CrossRef] [PubMed]
- Gamble, A.; Pepper, A.R.; Bruni, A.; Shapiro, A.M.J. The journey of islet cell transplantation and future development. Islets 2018, 10, 80–94. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, A.J.; Lakey, J.R.; Ryan, E.A.; Korbutt, G.S.; Toth, E.; Warnock, G.L.; Kneteman, N.M.; Rajotte, R.V. Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N. Engl. J. Med. 2000, 343, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Fiorina, P.; Shapiro, A.M.J.; Ricordi, C.; Secchi, A. The clinical impact of islet transplantation. Am. J. Transplant. 2008, 8, 1990–1997. [Google Scholar] [CrossRef]
- Thompson, D.M.; Meloche, M.; Ao, Z.; Paty, B.; Keown, P.; Shapiro, R.J.; Ho, S.; Worsley, D.; Fung, M.; Meneilly, G.; et al. Reduced progression of diabetic microvascular complications with islet cell transplantation compared with intensive medical therapy. Transplantation 2011, 91, 373–378. [Google Scholar] [CrossRef]
- Warnock, G.L.; Thompson, D.M.; Meloche, R.M.; Shapiro, R.J.; Ao, Z.; Keown, P.; Johnson, J.D.; Verchere, C.B.; Partovi, N.; Begg, I.S.; et al. A multi-year analysis of islet transplantation compared with intensive medical therapy on progression of complications in type 1 diabetes. Transplantation 2008, 86, 1762–1766. [Google Scholar] [CrossRef]
- Staels, W.; De Groef, S.; Heremans, Y.; Coppens, V.; Van Gassen, N.; Leuckx, G.; Van De Casteele, M.; Van Riet, I.; Luttun, A.; Heimberg, H.; et al. Accessory cells for β-cell transplantation. Diabetes Obes. Metab. 2015, 18, 115–124. [Google Scholar] [CrossRef]
- Carlsson, P.-O.; Palm, F.; Mattsson, G. Low revascularization of experimentally transplanted human pancreatic islets. J. Clin. Endocrinol. Metab. 2002, 87, 5418–5423. [Google Scholar] [CrossRef][Green Version]
- Davalli, A.M.; Scaglia, L.; Zangen, D.H.; Hollister, J.; Bonner-Weir, S.; Weir, G.C. Vulnerability of islets in the immediate posttransplantation period: Dynamic changes in structure and function. Diabetes 1996, 45, 1161–1167. [Google Scholar] [CrossRef] [PubMed]
- Ryan, E.A.; Paty, B.W.; Senior, P.A.; Shapiro, A.M.J. Risks and side effects of islet transplantation. Curr. Diabetes Rep. 2004, 4, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Henry, R.R.; Pettus, J.; Wilensky, J.; Shapiro, A.M.J.; Senior, P.A.; Roep, B.; Wang, R.; Kroon, E.J.; Scott, M.; D’Amour, K.; et al. Initial clinical evaluation of VC-01TM combination product—A stem cell–derived islet replacement for type 1 diabetes (T1D). Diabetes 2018, 67, 138. [Google Scholar] [CrossRef]
- Nicholls, D.G. The pancreatic β-cell: A bioenergetic perspective. Physiol. Rev. 2016, 96, 1385–1447. [Google Scholar] [CrossRef]
- Rorsman, P.; Ashcroft, F.M. Pancreatic β-cell electrical activity and insulin secretion: Of mice and men. Physiol. Rev. 2018, 98, 117–214. [Google Scholar] [CrossRef]
- Lacy, P.E.; Walker, M.M.; Fink, C.J. Perifusion of isolated rat islets in vitro: Participation of the microtubular system in the biphasic release of insulin. Diabetes 1972, 21, 987–998. [Google Scholar] [CrossRef]
- Curry, D.L.; Bennett, L.L.; Grodsky, G.M. Dynamics of insulin secretion by the perfused rat pancreas. Endocrinology 1968, 83, 572–584. [Google Scholar] [CrossRef]
- Gembal, M.; Detimary, P.; Gilon, P.; Gao, Z.Y.; Henquin, J.C. Mechanisms by which glucose can control insulin release independently from its action on adenosine triphosphate-sensitive K+ channels in mouse B cells. J. Clin. Investig. 1993, 91, 871–880. [Google Scholar] [CrossRef]
- Kang, H.S.; Kim, Y.-S.; ZeRuth, G.; Beak, J.Y.; Gerrish, K.; Kilic, G.; Sosa-Pineda, B.; Jensen, J.L.; Foley, J.F.; Jetten, A.M. Transcription factor Glis3, a novel critical player in the regulation of pancreatic β-cell development and insulin gene expression. Mol. Cell. Biol. 2009, 29, 6366–6379. [Google Scholar] [CrossRef]
- Zhu, Y.; Liu, Q.; Zhou, Z.; Ikeda, Y. PDX1, Neurogenin-3, and MAFA: Critical transcription regulators for beta cell development and regeneration. Stem Cell Res. Ther. 2017, 8, 1–7. [Google Scholar] [CrossRef]
- Gu, C.; Stein, G.H.; Pan, N.; Goebbels, S.; Hörnberg, H.; Nave, K.-A.; Herrera, P.L.; White, P.; Kaestner, K.H.; Sussel, L.; et al. Pancreatic β cells require NeuroD to achieve and maintain functional maturity. Cell Metab. 2010, 11, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.L.; Liu, F.-F.; Sander, M. Nkx6.1 is essential for maintaining the functional state of pancreatic beta cells. Cell Rep. 2013, 4, 1262–1275. [Google Scholar] [CrossRef] [PubMed]
- Gosmain, Y.; Katz, L.S.; Masson, M.H.; Cheyssac, C.; Poisson, C.; Philippe, J. Pax6 is crucial for β-cell function, insulin biosynthesis, and glucose-induced insulin secretion. Mol. Endocrinol. 2012, 26, 696–709. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; McKenna, B.; Li, C.; Reichert, M.; Nguyen, J.; Singh, T.; Yang, C.; Pannikar, A.; Doliba, N.M.; Zhang, T.; et al. Pdx1 maintains β cell identity and function by repressing an α cell program. Cell Metab. 2014, 19, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Velazco-Cruz, L.; Goedegebuure, M.M.; Maxwell, K.G.; Augsornworawat, P.; Hogrebe, N.J.; Millman, J.R. SIX2 regulates human β cell differentiation from stem cells and functional maturation in vitro. Cell Rep. 2020, 31, 107687. [Google Scholar] [CrossRef] [PubMed]
- Van Der Meulen, T.; Donaldson, C.J.; Cáceres, E.; Hunter, A.E.; Cowing-Zitron, C.; Pound, L.D.; Adams, M.W.; Zembrzycki, A.; Grove, K.L.; Huising, M.O. Urocortin3 mediates somatostatin-dependent negative feedback control of insulin secretion. Nat. Med. 2015, 21, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Blum, B.; Hrvatin, S.; Schuetz, C.; Bonal, C.; Rezaniaand, A.; Melton, D.A. Functional beta-cell maturation is marked by an increased glucose threshold and by expression of urocortin 3. Nat. Biotechnol. 2012, 30, 261–264. [Google Scholar] [CrossRef] [PubMed]
- Lemaire, K.; Thorrez, L.; Schuit, F. Disallowed and allowed gene expression: Two faces of mature islet beta cells. Annu. Rev. Nutr. 2016, 36, 45–71. [Google Scholar] [CrossRef]
- Ebrahimi, A.; Hollister-Lock, J.; Sullivan, B.A.; Tsuchida, R.; Bonner-Weir, S.; Weir, G.C. Beta cell identity changes with mild hyperglycemia: Implications for function, growth, and vulnerability. Mol. Metab. 2020, 35, 100959. [Google Scholar] [CrossRef]
- Scaglia, L.; Cahill, C.J.; Finegood, D.T.; Bonner-Weir, S. Apoptosis participates in the remodeling of the endocrine pancreas in the neonatal rat. Endocrinology 1997, 138, 1736–1741. [Google Scholar] [CrossRef]
- Reers, C.; Erbel, S.; Esposito, I.; Schmied, B.; Büchler, M.W.; Nawroth, P.P.; Ritzel, R.A. Impaired islet turnover in human donor pancreata with aging. Eur. J. Endocrinol. 2009, 160, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Perl, S.; Kushner, J.A.; Buchholz, B.A.; Meeker, A.K.; Stein, G.M.; Hsieh, M.; Kirby, M.; Pechhold, S.; Liu, E.H.; Harlan, D.M.; et al. Significant human β-cell turnover is limited to the first three decades of life as determined by in vivo thymidine analog incorporation and radiocarbon dating. J. Clin. Endocrinol. Metab. 2010, 95, E234–E239. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.A.; Rizza, R.A.; Butler, P.C. β-Cell deficit and increased β-cell apoptosis in humans with type 2 diabetes. Diabetes 2003, 52, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Cnop, M.; Igoillo-Esteve, M.; Hughes, S.; Walker, J.N.; Cnop, I.; Clark, A. Longevity of human islet α- and β-cells. Diabetes Obes. Metab. 2011, 13, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Cnop, M.; Hughes, S.J.; Igoillo-Esteve, M.; Hoppa, M.B.; Sayyed, F.; van de Laar, L.; Gunter, J.H.; de Koning, E.J.P.; Walls, G.V.; Gray, D.W.G.; et al. The long lifespan and low turnover of human islet beta cells estimated by mathematical modelling of lipofuscin accumulation. Diabetologia 2009, 53, 321. [Google Scholar] [CrossRef]
- Pipeleers, D.G. Heterogeneity in pancreatic β-cell population. Diabetes 1992, 41, 777–781. [Google Scholar] [CrossRef]
- Van Schravendijk, C.F.; Kiekens, R.; Pipeleers, D.G. Pancreatic beta cell heterogeneity in glucose-induced insulin secretion. J. Biol. Chem. 1992, 267, 21344–21348. [Google Scholar] [CrossRef]
- Heimberg, H.; De Vos, A.; Vandercammen, A.; Van Schaftingen, E.; Pipeleers, D.; Schuit, F. Heterogeneity in glucose sensitivity among pancreatic beta-cells is correlated to differences in glucose phosphorylation rather than glucose transport. EMBO J. 1993, 12, 2873–2879. [Google Scholar] [CrossRef]
- Johnston, N.R.; Mitchell, R.K.; Haythorne, E.; Pessoa, M.P.; Semplici, F.; Ferrer, J.; Piemonti, L.; Marchetti, P.; Bugliani, M.; Bosco, D.; et al. Beta cell hubs dictate pancreatic islet responses to glucose. Cell Metab. 2016, 24, 389–401. [Google Scholar] [CrossRef]
- Bader, E.; Migliorini, A.; Gegg, M.; Moruzzi, N.; Gerdes, J.; Roscioni, S.S.; Bakhti, M.; Brandl, E.; Irmler, M.; Beckers, J.; et al. Identification of proliferative and mature β-cells in the islets of Langerhans. Nature 2016, 535, 430–434. [Google Scholar] [CrossRef]
- Van Der Meulen, T.; Mawla, A.M.; DiGruccio, M.R.; Adams, M.W.; Nies, V.; Dólleman, S.; Liu, S.; Ackermann, A.M.; Cáceres, E.; Hunter, A.E.; et al. Virgin beta cells persist throughout life at a neogenic niche within pancreatic islets. Cell Metab. 2017, 25, 911–926.e6. [Google Scholar] [CrossRef]
- Weissman, I.L.; Anderson, D.J.; Gage, F. Stem and progenitor cells: Origins, phenotypes, lineage commitments, and transdifferentiations. Annu. Rev. Cell Dev. Biol. 2001, 17, 387–403. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.J.; Kaufman, M.H. Establishment in culture of pluripotential cells from mouse embryos. Nat. Cell Biol. 1981, 292, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- D’Amour, K.A.; Agulnick, A.D.; Eliazer, S.; Kelly, O.G.; Kroon, E.; Baetge, E.E. Efficient differentiation of human embryonic stem cells to definitive endoderm. Nat. Biotechnol. 2005, 23, 1534–1541. [Google Scholar] [CrossRef] [PubMed]
- D’Amour, K.A.; Bang, A.G.; Eliazer, S.; Kelly, O.G.; Agulnick, A.D.; Smart, N.G.; Moorman, M.A.; Kroon, E.; Carpenter, M.K.; Baetge, E.E. Production of pancreatic hormone–expressing endocrine cells from human embryonic stem cells. Nat. Biotechnol. 2006, 24, 1392–1401. [Google Scholar] [CrossRef]
- Riedel, M.J.; Asadi, A.; Wang, R.; Ao, Z.; Warnock, G.L.; Kieffer, T.J. Immunohistochemical characterisation of cells co-producing insulin and glucagon in the developing human pancreas. Diabetologia 2011, 55, 372–381. [Google Scholar] [CrossRef]
- Jennings, R.E.; Scharfmann, R.; Staels, W. Transcription factors that shape the mammalian pancreas. Diabetologia 2020, 63, 1974–1980. [Google Scholar] [CrossRef]
- Russ, H.A.; Parent, A.V.; Ringler, J.J.; Hennings, T.G.; Nair, G.G.; Shveygert, M.; Guo, T.; Puri, S.; Haataja, L.; Cirulli, V.; et al. Controlled induction of human pancreatic progenitors produces functional beta-like cells in vitro. EMBO J. 2015, 34, 1759–1772. [Google Scholar] [CrossRef]
- Pagliuca, F.W.; Millman, J.R.; Gürtler, M.; Segel, M.; van Dervort, A.; Ryu, J.H.; Peterson, Q.P.; Greiner, D.; Melton, D.A. Generation of functional human pancreatic β cells in vitro. Cell 2014, 159, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Rezania, A.; Bruin, J.E.; Arora, P.; Rubin, A.; Batushansky, I.; Asadi, A.; O’Dwyer, S.; Quiskamp, N.; Mojibian, M.; Albrecht, T.; et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat. Biotechnol. 2014, 32, 1121–1133. [Google Scholar] [CrossRef] [PubMed]
- Ghazizadeh, Z.; Kao, D.-I.; Amin, S.; Cook, B.; Rao, S.; Zhou, T.; Zhang, T.; Xiang, Z.; Kenyon, R.; Kaymakcalan, O.; et al. ROCKII inhibition promotes the maturation of human pancreatic beta-like cells. Nat. Commun. 2017, 8, 298. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Russ, H.A.; Wang, X.; Zhang, M.; Ma, T.; Xu, T.; Tang, S.; Hebrok, M.; Ding, S. Human pancreatic beta-like cells converted from fibroblasts. Nat. Commun. 2016, 7, 10080. [Google Scholar] [CrossRef] [PubMed]
- Millman, J.R.; Xie, C.; Van Dervort, A.; Gürtler, M.; Pagliuca, F.W.; Melton, D.A. Generation of stem cell-derived β-cells from patients with type 1 diabetes. Nat. Commun. 2016, 7, 11463. [Google Scholar] [CrossRef] [PubMed]
- Nair, G.G.; Liu, J.S.; Russ, H.A.; Tran, S.; Saxton, M.S.; Chen, R.; Juang, C.; Li, M.-L.; Nguyen, V.Q.; Giacometti, S.; et al. Recapitulating endocrine cell clustering in culture promotes maturation of human stem-cell-derived β cells. Nat. Cell Biol. 2019, 21, 263–274. [Google Scholar] [CrossRef]
- Velazco-Cruz, L.; Song, J.; Maxwell, K.G.; Goedegebuure, M.M.; Augsornworawat, P.; Hogrebe, N.J.; Millman, J.R. Acquisition of dynamic function in human stem cell-derived β cells. Stem Cell Rep. 2019, 12, 351–365. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, R.; Dai, S.; Zhang, X.; Li, X.; Zhu, Z. Role of TGF-β/smad pathway in the transcription of pancreas-specific genes during beta cell differentiation. Front. Cell Dev. Biol. 2019, 7, 351. [Google Scholar] [CrossRef]
- Lin, H.-M.; Lee, J.-H.; Yadav, H.; Kamaraju, A.K.; Liu, E.; Zhigang, D.; Vieira, A.; Kim, S.-J.; Collins, H.W.; Matschinsky, F.M.; et al. Transforming growth factor-β/smad3 signaling regulates insulin gene transcription and pancreatic islet β-cell function. J. Biol. Chem. 2009, 284, 12246–12257. [Google Scholar] [CrossRef]
- Totsuka, Y.; Tabuchi, M.; Kojima, I.; Eto, Y.; Shibai, H.; Ogata, E. Stimulation of insulin secretion by transforming growth factor-β. Biochem. Biophys. Res. Commun. 1989, 158, 1060–1065. [Google Scholar] [CrossRef]
- Nomura, M.; Zhu, H.-L.; Wang, L.; Morinaga, H.; Takayanagi, R.; Teramoto, N. SMAD2 disruption in mouse pancreatic beta cells leads to islet hyperplasia and impaired insulin secretion due to the attenuation of ATP-sensitive K+ channel activity. Diabetologia 2013, 57, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, E.; O’Connor, C.; Gasser, E.; Wei, Z.; Oh, T.G.; Tseng, T.W.; Wang, D.; Cayabyab, F.; Dai, Y.; Yu, R.T.; et al. Immune-evasive human islet-like organoids ameliorate diabetes. Nature 2020, 586, 606–611. [Google Scholar] [CrossRef] [PubMed]
- Nostro, M.C.; Sarangi, F.; Yang, C.; Holland, A.M.; Elefanty, A.G.; Stanley, E.G.; Greiner, D.L.; Keller, G. Efficient generation of NKX6-1+ pancreatic progenitors from multiple human pluripotent stem cell lines. Stem Cell Rep. 2015, 4, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, K.Y.; Chan, V.W.; Leung, K.T.; Zhang, X.-B.; Wong, A.S.; Chong, C.C.; Wang, C.C.; Ku, M.; Lui, K.O. Single-cell RNA-seq reveals that CD9 is a negative marker of glucose-responsive pancreatic β-like cells derived from human pluripotent stem cells. Stem Cell Rep. 2020, 15, 1111–1126. [Google Scholar] [CrossRef] [PubMed]
- Veres, A.; Faust, A.L.; Bushnell, H.L.; Engquist, E.N.; Kenty, J.H.-R.; Harb, G.; Poh, Y.-C.; Sintov, E.; Gürtler, M.; Pagliuca, F.W.; et al. Charting cellular identity during human in vitro β-cell differentiation. Nature 2019, 569, 368–373. [Google Scholar] [CrossRef]
- Brissova, M.; Haliyur, R.; Saunders, D.; Shrestha, S.; Dai, C.; Blodgett, D.M.; Bottino, R.; Campbell-Thompson, M.; Aramandla, R.; Poffenberger, G.; et al. α cell function and gene expression are compromised in type 1 diabetes. Cell Rep. 2018, 22, 2667–2676. [Google Scholar] [CrossRef]
- Unger, R.H.; Cherrington, A.D. Glucagonocentric restructuring of diabetes: A pathophysiologic and therapeutic makeover. J. Clin. Investig. 2012, 122, 4–12. [Google Scholar] [CrossRef]
- Siafarikas, A.; Johnston, R.J.; Bulsara, M.K.; O’Leary, P.; Jones, T.W.; Davis, E.A. Early loss of the glucagon response to hypoglycemia in adolescents with type 1 diabetes. Diabetes Care 2012, 35, 1757–1762. [Google Scholar] [CrossRef]
- Rezania, A.; Bruin, J.E.; Riedel, M.J.; Mojibian, M.; Asadi, A.; Xu, J.; Gauvin, R.; Narayan, K.; Karanu, F.; O’Neil, J.J.; et al. Maturation of human embryonic stem cell-derived pancreatic progenitors into functional islets capable of treating pre-existing diabetes in mice. Diabetes 2012, 61, 2016–2029. [Google Scholar] [CrossRef]
- Schulz, T.C.; Young, H.Y.; Agulnick, A.D.; Babin, M.J.; Baetge, E.E.; Bang, A.G.; Bhoumik, A.; Cepa, I.; Cesario, R.M.; Haakmeester, C.; et al. A scalable system for production of functional pancreatic progenitors from human embryonic stem cells. PLoS ONE 2012, 7, e37004. [Google Scholar] [CrossRef]
- Hrvatin, S.; O’Donnell, C.W.; Deng, F.; Millman, J.R.; Pagliuca, F.W.; DiIorio, P.; Rezania, A.; Gifford, D.K.; Melton, D.A. Differentiated human stem cells resemble fetal, not adult, β cells. Proc. Natl. Acad. Sci. USA 2014, 111, 3038–3043. [Google Scholar] [CrossRef] [PubMed]
- Rezania, A.; Riedel, M.J.; Wideman, R.D.; Karanu, F.; Ao, Z.; Warnock, G.L.; Kieffer, T.J. Production of functional glucagon-secreting α-cells from human embryonic stem cells. Diabetes 2010, 60, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Peterson, Q.P.; Veres, A.; Chen, L.; Slama, M.Q.; Kenty, J.H.; Hassoun, S.; Brown, M.R.; Dou, H.; Duffy, C.D.; Zhou, Q.; et al. A method for the generation of human stem cell-derived alpha cells. Nat. Commun. 2020, 11, 2241. [Google Scholar] [CrossRef] [PubMed]
- Ravier, M.A.; Güldenagel, M.; Charollais, A.; Gjinovci, A.; Caille, D.; Söhl, G.; Wollheim, C.B.; Willecke, K.; Henquin, J.-C.; Meda, P. Loss of Connexin36 channels alters β-cell coupling, islet synchronization of glucose-induced Ca2+ and insulin oscillations, and basal insulin release. Diabetes 2005, 54, 1798–1807. [Google Scholar] [CrossRef] [PubMed]
- Charollais, A.; Gjinovci, A.; Huarte, J.; Bauquis, J.; Nadal, A.; Martín, F.; Andreu, E.; Sánchez-Andrés, J.V.; Calabrese, A.; Bosco, D.; et al. Junctional communication of pancreatic β cells contributes to the control of insulin secretion and glucose tolerance. J. Clin. Investig. 2000, 106, 235–243. [Google Scholar] [CrossRef][Green Version]
- Bavamian, S.; Klee, P.; Britan, A.; Populaire, C.; Caille, D.; Cancela, J.; Charollais, A.; Meda, P. Islet-cell-to-cell communication as basis for normal insulin secretion. Diabetes Obes. Metab. 2007, 9, 118–132. [Google Scholar] [CrossRef]
- Svendsen, B.; Larsen, O.; Gabe, M.B.N.; Christiansen, C.B.; Rosenkilde, M.M.; Drucker, D.J.; Holst, J.J. Insulin secretion depends on intra-islet glucagon signaling. Cell Rep. 2018, 25, 1127–1134.e2. [Google Scholar] [CrossRef]
- Svendsen, B.; Holst, J.J. Paracrine regulation of somatostatin secretion by insulin and glucagon in mouse pancreatic islets. Diabetologia 2020, 1–10. [Google Scholar] [CrossRef]
- Townsend, S.E.; Gannon, M. Extracellular matrix–associated factors play critical roles in regulating pancreatic β-cell proliferation and survival. Endocrinology 2019, 160, 1885–1894. [Google Scholar] [CrossRef]
- Staels, W.; Heremans, Y.; Heimberg, H.; De Leu, N. VEGF-A and blood vessels: A beta cell perspective. Diabetologia 2019, 62, 1961–1968. [Google Scholar] [CrossRef]
- Sasson, A.; Rachi, E.; Sakhneny, L.; Baer, D.; Lisnyansky, M.; Epshtein, A.; Landsman, L. Islet pericytes are required for β-cell maturity. Diabetes 2016, 65, 3008–3014. [Google Scholar] [CrossRef]
- Borden, P.; Houtz, J.; Leach, S.D.; Kuruvilla, R. Sympathetic innervation during development is necessary for pancreatic islet architecture and functional maturation. Cell Rep. 2013, 4, 287–301. [Google Scholar] [CrossRef] [PubMed]
- Ahrén, B. Autonomic regulation of islet hormone secretion—Implications for health and disease. Diabetologia 2000, 43, 393–410. [Google Scholar] [CrossRef] [PubMed]
- Van Gassen, N.; Staels, W.; Van Overmeire, E.; De Groef, S.; Sojoodi, M.; Heremans, Y.; Leuckx, G.; Van de Casteele, M.; Van Ginderachter, J.A.; Heimberg, H.; et al. Concise review: Macrophages: Versatile gatekeepers during pancreatic β-cell development, injury, and regeneration. Stem Cells Transl. Med. 2015, 4, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Potten, C.S.; Loeffler, M. Stem cells: Attributes, cycles, spirals, pitfalls and uncertainties. Lessons for and from the crypt. Development 1990, 110, 1001–1020. [Google Scholar] [PubMed]
- Bruin, J.E.; Asadi, A.; Fox, J.K.; Erener, S.; Rezania, A.; Kieffer, T.J. Accelerated maturation of human stem cell-derived pancreatic progenitor cells into insulin-secreting cells in immunodeficient rats relative to mice. Stem Cell Rep. 2015, 5, 1081–1096. [Google Scholar] [CrossRef] [PubMed]
- Kroon, E.; Martinson, L.A.; Kadoya, K.; Bang, A.G.; Kelly, O.G.; Eliazer, S.; Young, H.; Richardson, M.K.; Smart, N.G.; Cunningham, J.J.; et al. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat. Biotechnol. 2008, 26, 443–452. [Google Scholar] [CrossRef]
- Hogrebe, N.J.; Augsornworawat, P.; Maxwell, K.G.; Velazco-Cruz, L.; Millman, J.R. Targeting the cytoskeleton to direct pancreatic differentiation of human pluripotent stem cells. Nat. Biotechnol. 2020, 38, 460–470. [Google Scholar] [CrossRef]
- Augsornworawat, P.; Maxwell, K.G.; Velazco-Cruz, L.; Millman, J.R. Single-cell transcriptome profiling reveals β cell maturation in stem cell-derived islets after transplantation. Cell Rep. 2020, 32, 108067. [Google Scholar] [CrossRef]
- Lammert, E.; Cleaver, O.; Melton, D. Induction of pancreatic differentiation by signals from blood vessels. Science 2001, 294, 564–567. [Google Scholar] [CrossRef]
- Anderson, S.J.; White, M.G.; Armour, S.L.; Maheshwari, R.; Tiniakos, D.; Muller, Y.D.; Berishvili, E.; Berney, T.; Shaw, J.A.M. Loss of end-differentiated β-cell phenotype following pancreatic islet transplantation. Am. J. Transplant. 2017, 18, 750–755. [Google Scholar] [CrossRef] [PubMed]
- De Leu, N.; Heremans, Y.; Coppens, V.; Van Gassen, N.; Cai, Y.; D’Hoker, J.; Magenheim, J.; Salpeter, S.; Swisa, A.; Khalaileh, A.; et al. Short-term overexpression of VEGF-A in mouse beta cells indirectly stimulates their proliferation and protects against diabetes. Diabetologia 2013, 57, 140–147. [Google Scholar] [CrossRef]
- Brissova, M.; Aamodt, K.; Brahmachary, P.; Prasad, N.; Hong, J.-Y.; Dai, C.; Mellati, M.; Shostak, A.; Poffenberger, G.; Aramandla, R.; et al. Islet microenvironment, modulated by vascular endothelial growth factor-a signaling, promotes β cell regeneration. Cell Metab. 2014, 19, 498–511. [Google Scholar] [CrossRef] [PubMed]
- Staels, W.; Verdonck, Y.; Heremans, Y.; Leuckx, G.; De Groef, S.; Heirman, C.; De Koning, E.; Gysemans, C.; Thielemans, K.; Baeyens, L.; et al. Vegf-A mRNA transfection as a novel approach to improve mouse and human islet graft revascularisation. Diabetologia 2018, 61, 1804–1810. [Google Scholar] [CrossRef] [PubMed]
- Ben-David, U.; Gan, Q.-F.; Golan-Lev, T.; Arora, P.; Yanuka, O.; Oren, Y.S.; Leikin-Frenkel, A.; Graf, M.; Garippa, R.; Boehringer, M.; et al. Selective elimination of human pluripotent stem cells by an oleate synthesis inhibitor discovered in a high-throughput screen. Cell Stem Cell 2013, 12, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Lee, A.S.; Volkmer, J.-P.; Sahoo, D.; Nag, D.; Mosley, A.R.; Inlay, M.A.; Ardehali, R.; Chavez, S.L.; Pera, R.R.; et al. An antibody against SSEA-5 glycan on human pluripotent stem cells enables removal of teratoma-forming cells. Nat. Biotechnol. 2011, 29, 829–834. [Google Scholar] [CrossRef]
- Fong, C.Y.; Peh, G.S.L.; Gauthaman, K.; Bongso, A. Separation of SSEA-4 and TRA-1–60 labelled undifferentiated human embryonic stem cells from a heterogeneous cell population using magnetic-activated cell sorting (MACS) and fluorescence-activated cell sorting (FACS). Stem Cell Rev. Rep. 2009, 5, 72–80. [Google Scholar] [CrossRef]
- Ben-David, U.; Nudel, N.; Benvenisty, N. Immunologic and chemical targeting of the tight-junction protein Claudin-6 eliminates tumorigenic human pluripotent stem cells. Nat. Commun. 2013, 4, 1992. [Google Scholar] [CrossRef]
- Qadir, M.M.F.; Álvarez-Cubela, S.; Belle, K.; Sapir, T.; Messaggio, F.; Johnson, K.B.; Umland, O.; Hardin, D.; Klein, D.; Pérez-Álvarez, I.; et al. A double fail-safe approach to prevent tumorigenesis and select pancreatic β cells from human embryonic stem cells. Stem Cell Rep. 2019, 12, 611–623. [Google Scholar] [CrossRef]
- Yagyu, S.; Hoyos, V.; Del Bufalo, F.; Brenner, M.K. An inducible Caspase-9 suicide gene to improve the safety of therapy using human induced pluripotent stem cells. Mol. Ther. 2015, 23, 1475–1485. [Google Scholar] [CrossRef]
- Oshima, M.; Pechberty, S.; Bellini, L.; Göpel, S.O.; Campana, M.; Rouch, C.; Dairou, J.; Cosentino, C.; Fantuzzi, F.; Toivonen, S.; et al. Stearoyl CoA desaturase is a gatekeeper that protects human beta cells against lipotoxicity and maintains their identity. Diabetologia 2019, 63, 395–409. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.J.; Peacock, S.; Chaudhry, A.N.; Bradley, J.A.; Bolton, E.M. Generating an iPSC bank for HLA-matched tissue transplantation based on known donor and recipient HLA types. Cell Stem Cell 2012, 11, 147–152. [Google Scholar] [CrossRef] [PubMed]
- De Rham, C.; Villard, J. Potential and limitation of HLA-based banking of human pluripotent stem cells for cell therapy. J. Immunol. Res. 2014, 2014, 518135. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.J.; Bolton, E.M.; Bradley, J.A. Immunological considerations for embryonic and induced pluripotent stem cell banking. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2011, 366, 2312–2322. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zhang, W.; Ng, T.W.; Wang, Y.; Liu, Q.; Gorantla, V.; Lakkis, F.; Zheng, X.X. Adoptive cell therapy using antigen-specific CD4−CD8−T regulatory cells to prevent autoimmune diabetes and promote islet allograft survival in NOD mice. Diabetologia 2011, 54, 2082–2092. [Google Scholar] [CrossRef]
- Marek-Trzonkowska, N.; Myśliwiec, M.; Dobyszuk, A.; Grabowska, M.; Techmańska, I.; Juścińska, J.; Wujtewicz, M.A.; Witkowski, P.; Młynarski, W.; Balcerska, A.; et al. Administration of CD4+CD25highCD127− regulatory T cells preserves β-cell function in type 1 diabetes in children. Diabetes Care 2012, 35, 1817–1820. [Google Scholar] [CrossRef]
- Marek-Trzonkowska, N.; Myśliwiec, M.; Dobyszuk, A.; Grabowska, M.; Derkowska, I.; Juścińska, J.; Owczuk, R.; Szadkowska, A.; Witkowski, P.; Młynarski, W.; et al. Therapy of type 1 diabetes with CD4+CD25highCD127− regulatory T cells prolongs survival of pancreatic islets—Results of one year follow-up. Clin. Immunol. 2014, 153, 23–30. [Google Scholar] [CrossRef]
- Tang, Q.; Bluestone, J.A. Regulatory T-cell therapy in transplantation: Moving to the clinic. Cold Spring Harb. Perspect. Med. 2013, 3, a015552. [Google Scholar] [CrossRef]
- Ferreira, L.M.R.; Muller, Y.D.; Bluestone, J.A.; Tang, Q. Next-generation regulatory T cell therapy. Nat. Rev. Drug Discov. 2019, 18, 749–769. [Google Scholar] [CrossRef]
- Lim, D.; Sreekanth, V.; Cox, K.J.; Law, B.K.; Wagner, B.K.; Karp, J.M.; Choudhary, A. Engineering designer beta cells with a CRISPR-Cas9 conjugation platform. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Carter, J.D.; Ellett, J.D.; Chen, M.; Smith, K.M.; Fialkow, L.B.; McDuffie, M.J.; Tung, K.S.; Nadler, J.L.; Yang, Z. Viral IL-10-mediated immune regulation in pancreatic islet transplantation. Mol. Ther. 2005, 12, 360–368. [Google Scholar] [CrossRef] [PubMed]
- English, K. Mesenchymal stem cells to promote islet transplant survival. Curr. Opin. Organ. Transplant. 2016, 21, 568–573. [Google Scholar] [CrossRef] [PubMed]
- Ben Nasr, M.; Vergani, A.; Avruch, J.; Liu, L.; Kefaloyianni, E.; D’Addio, F.; Tezza, S.; Corradi, D.; Bassi, R.; Valderrama-Vasquez, A.; et al. Co-transplantation of autologous MSCs delays islet allograft rejection and generates a local immunoprivileged site. Acta Diabetol. 2015, 52, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.M.; Yu, X.F.; Zhang, D.; Zhang, M.X.; Zhou, J.F.; Tan, P.H.; Ding, Y. Mesenchymal stem cells differentially mediate regulatory T cells and conventional effector T cells to protect fully allogeneic islet grafts in mice. Diabetologia 2012, 55, 1091–1102. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Longoni, B.; Szilagyi, E.; Quaranta, P.; Paoli, G.T.; Tripodi, S.; Urbani, S.; Mazzanti, B.; Rossi, B.; Fanci, R.; Demontis, G.C.; et al. Mesenchymal stem cells prevent acute rejection and prolong graft function in pancreatic islet transplantation. Diabetes Technol. Ther. 2010, 12, 435–446. [Google Scholar] [CrossRef]
- Berman, D.M.; Willman, M.A.; Han, D.; Kleiner, G.; Kenyon, N.M.; Cabrera, O.; Karl, J.A.; Wiseman, R.W.; O’Connor, D.H.; Bartholomew, A.M. Mesenchymal stem cells enhance allogeneic islet engraftment in nonhuman primates. Diabetes 2010, 59, 2558–2568. [Google Scholar] [CrossRef]
- Arzouni, A.A.; Vargas-Seymour, A.; Nardi, N.; King, A.J.F.; Jones, P. Using mesenchymal stromal cells in islet transplantation. Stem Cells Transl. Med. 2018, 7, 559–563. [Google Scholar] [CrossRef]
- Arzouni, A.A.; Vargas-Seymour, A.; Rackham, C.L.; Dhadda, P.; Huang, G.-C.; Choudhary, P.; Nardi, N.; King, A.J.; Jones, P. Mesenchymal stromal cells improve human islet function through released products and extracellular matrix. Clin. Sci. 2017, 131, 2835–2845. [Google Scholar] [CrossRef]
- Rackham, C.L.; Amisten, S.; Persaud, S.; King, A.J.; Jones, P. Mesenchymal stromal cell secretory factors induce sustained improvements in islet function pre- and post-transplantation. Cytotherapy 2018, 20, 1427–1436. [Google Scholar] [CrossRef]
- Mandal, P.K.; Ferreira, L.M.R.; Collins, R.; Meissner, T.B.; Boutwell, C.L.; Friesen, M.; Vrbanac, V.; Garrison, B.S.; Stortchevoi, A.; Bryder, D.; et al. Efficient ablation of genes in human hematopoietic stem and effector cells using CRISPR/Cas9. Cell Stem Cell 2014, 15, 643–652. [Google Scholar] [CrossRef]
- Mattapally, S.; Pawlik, K.M.; Fast, V.G.; Zumaquero, E.; Lund, F.E.; Randall, T.D.; Townes, T.M.; Zhang, J. Human leukocyte antigen class I and II knockout human induced pluripotent stem cell–derived cells: Universal donor for cell therapy. J. Am. Heart Assoc. 2018, 7, e010239. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wang, M.; Duan, S.; Franco, P.J.; Kenty, J.H.-R.; Hedrick, P.; Xia, Y.; Allen, A.; Ferreira, L.M.R.; Strominger, J.L.; et al. Generation of hypoimmunogenic human pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2019, 116, 10441–10446. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Quan, Y.; Yan, Q.; Morales, J.E.; Wetsel, R.A. Targeted disruption of the β2-microglobulin gene minimizes the immunogenicity of human embryonic stem cells. Stem Cells Transl. Med. 2015, 4, 1234–1245. [Google Scholar] [CrossRef] [PubMed]
- Batra, L.; Shrestha, P.; Zhao, H.; Woodward, K.B.; Togay, A.; Tan, M.; Grimany-Nuno, O.; Malik, M.T.; Coronel, M.M.; García, A.J.; et al. Localized Immunomodulation with PD-L1 results in sustained survival and function of allogeneic islets without chronic immunosuppression. J. Immunol. 2020, 204, 2840–2851. [Google Scholar] [CrossRef] [PubMed]
- Rong, Z.; Wang, M.; Hu, Z.; Stradner, M.; Zhu, S.; Kong, H.; Yi, H.; Goldrath, A.; Yang, Y.-G.; Xu, Y.; et al. An effective approach to prevent immune rejection of human ESC-derived allografts. Cell Stem Cell 2014, 14, 121–130. [Google Scholar] [CrossRef]
- Coronel, M.M.; Martin, K.E.; Hunckler, M.D.; Barber, G.; O’Neill, E.B.; Medina, J.D.; Opri, E.; McClain, C.A.; Batra, L.; Weaver, J.D.; et al. Immunotherapy via PD-L1–presenting biomaterials leads to long-term islet graft survival. Sci. Adv. 2020, 6, eaba5573. [Google Scholar] [CrossRef] [PubMed]
- Cabello-Kindelan, C.; Mackey, S.; Sands, A.; Rodriguez, J.; Vazquez, C.; Pugliese, A.; Bayer, A.L. Immunomodulation followed by antigen-specific treg infusion controls islet autoimmunity. Diabetes 2019, 69, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Szot, G.L.; Yadav, M.; Lang, J.; Kroon, E.; Kerr, J.; Kadoya, K.; Brandon, E.P.; Baetge, E.E.; Bour-Jordan, H.; Bluestone, J.A. Tolerance induction and reversal of diabetes in mice transplanted with human embryonic stem cell-derived pancreatic endoderm. Cell Stem Cell 2015, 16, 148–157. [Google Scholar] [CrossRef]
- Desai, T.; Shea, L.D. Advances in islet encapsulation technologies. Nat. Rev. Drug Discov. 2017, 16, 338–350. [Google Scholar] [CrossRef]
- Song, S.; Roy, S. Progress and challenges in macroencapsulation approaches for type 1 diabetes (T1D) treatment: Cells, biomaterials, and devices. Biotechnol. Bioeng. 2016, 113, 1381–1402. [Google Scholar] [CrossRef]
- Smithies, O.; Gregg, R.G.; Boggs, S.S.; Koralewski, M.A.; Kucherlapati, R.S. Insertion of DNA sequences into the human chromosomal β-globin locus by homologous recombination. Nature 1985, 317, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Hattersley, A.T.; Patel, K.A. Precision diabetes: Learning from monogenic diabetes. Diabetologia 2017, 60, 769–777. [Google Scholar] [CrossRef]
- Vethe, H.; Bjørlykke, Y.; Ghila, L.; Paulo, J.A.; Scholz, H.; Gygi, S.P.; Chera, S.; Ræder, H. Probing the missing mature β-cell proteomic landscape in differentiating patient iPSC-derived cells. Sci. Rep. 2017, 7, 4780. [Google Scholar] [CrossRef] [PubMed]
- Edghill, E.L.; Bingham, C.; Ellard, S.; Hattersley, A.T. Mutations in hepatocyte nuclear factor-1 and their related phenotypes. J. Med. Genet. 2005, 43, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Teo, A.K.; Lau, H.H.; Valdez, I.A.; Dirice, E.; Tjora, E.; Raeder, H.; Kulkarni, R.N. Early developmental perturbations in a human stem cell model of MODY5/HNF1B pancreatic hypoplasia. Stem Cell Rep. 2016, 6, 357–367. [Google Scholar] [CrossRef]
- Cardenas-Diaz, F.L.; Osorio-Quintero, C.; Diaz-Miranda, M.A.; Kishore, S.; Leavens, K.; Jobaliya, C.; Stanescu, D.; Ortiz-Gonzalez, X.R.; Yoon, C.; Chen, C.S.; et al. Modeling monogenic diabetes using human ESCs reveals developmental and metabolic deficiencies caused by mutations in HNF1A. Cell Stem Cell 2019, 25, 273–289.e5. [Google Scholar] [CrossRef]
- Rubio-Cabezas, O.; Codner, E.; Flanagan, S.E.; Gómez, J.L.; Ellard, S.; Hattersley, A.T. Neurogenin 3 is important but not essential for pancreatic islet development in humans. Diabetologia 2014, 57, 2421–2424. [Google Scholar] [CrossRef]
- Sayar, E.; Islek, A.; Yilmaz, A.; Akçam, M.; Flanagan, S.E.; Artan, R. Extremely rare cause of congenital diarrhea: Enteric anendocrinosis. Pediatr. Int. 2013, 55, 661–663. [Google Scholar] [CrossRef]
- Pinney, S.E.; Oliverkrasinski, J.M.; Ernst, L.M.; Hughes, N.; Patel, P.; Stoffers, D.A.; Russo, P.; De León, D.D. Neonatal diabetes and congenital malabsorptive diarrhea attributable to a novel mutation in the human Neurogenin-3 gene coding sequence. J. Clin. Endocrinol. Metab. 2011, 96, 1960–1965. [Google Scholar] [CrossRef]
- Wang, J.; Cortina, G.; Wu, S.V.; Tran, R.; Cho, J.-H.; Tsai, M.-J.; Bailey, T.J.; Jamrich, M.; Ament, M.E.; Treem, W.R.; et al. Mutant Neurogenin-3 in congenital malabsorptive diarrhea. N. Engl. J. Med. 2006, 355, 270–280. [Google Scholar] [CrossRef]
- McGrath, P.S.; Watson, C.L.; Ingram, C.; Helmrath, M.A.; Wells, J.M. The basic helix-loop-helix transcription factor NEUROG3 Is required for development of the human endocrine pancreas. Diabetes 2015, 64, 2497–2505. [Google Scholar] [CrossRef] [PubMed]
- De Franco, E.; Ellard, S. Genome, exome, and targeted next-generation sequencing in neonatal diabetes. Pediatr. Clin. North Am. 2015, 62, 1037–1053. [Google Scholar] [CrossRef] [PubMed]
- Saarimäki-Vire, J.; Balboa, D.; Russell, M.A.; Saarikettu, J.; Kinnunen, M.; Keskitalo, S.; Malhi, A.; Valensisi, C.; Andrus, C.; Eurola, S.; et al. An activating STAT3 mutation causes neonatal diabetes through premature induction of pancreatic differentiation. Cell Rep. 2017, 19, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Shimomura, K.; Girard, C.A.; Proks, P.; Nazim, J.; Lippiat, J.D.; Cerutti, F.; Lorini, R.; Ellard, S.; Hattersley, A.T.; Barbetti, F.; et al. Mutations at the same residue (R50) of Kir6.2 (KCNJ11) that cause neonatal diabetes produce different functional effects. Diabetes 2006, 55, 1705–1712. [Google Scholar] [CrossRef][Green Version]
- Massa, O.; Iafusco, D.; D’Amato, E.; Gloyn, A.L.; Hattersley, A.T.; Pasquino, B.; Tonini, G.; Dammacco, F.; Zanette, G.; Meschi, F.; et al. KCNJ11 activating mutations in Italian patients with permanent neonatal diabetes. Hum. Mutat. 2004, 25, 22–27. [Google Scholar] [CrossRef]
- Proks, P.; Antcliff, J.F.; Lippiat, J.D.; Gloyn, A.L.; Hattersley, A.T.; Ashcroft, F.M. Molecular basis of Kir6.2 mutations associated with neonatal diabetes or neonatal diabetes plus neurological features. Proc. Natl. Acad. Sci. USA 2004, 101, 17539–17544. [Google Scholar] [CrossRef]
- Zeng, H.; Guo, M.; Zhou, T.; Tan, L.; Chong, C.N.; Zhang, T.; Dong, X.; Xiang, J.Z.; Yu, A.S.; Yue, L.; et al. An isogenic human ESC platform for functional evaluation of genome-wide-association-study-identified diabetes genes and drug discovery. Cell Stem Cell 2016, 19, 326–340. [Google Scholar] [CrossRef]
- Allen, H.L.; The International Pancreatic Agenesis Consortium; Flanagan, S.E.; Shaw-Smith, C.; De Franco, E.; Akerman, I.; Caswell, R.; Ferrer, J.; Hattersley, A.T.; Ellard, S. GATA6 haploinsufficiency causes pancreatic agenesis in humans. Nat. Genet. 2012, 44, 20–22. [Google Scholar] [CrossRef]
- Shaw-Smith, C.; De Franco, E.; Allen, H.L.; Batlle, M.; Flanagan, S.E.; Borowiec, M.; Taplin, C.E.; Velden, J.V.A.-V.D.; Cruz-Rojo, J.; De Nanclares, G.P.; et al. GATA4 mutations are a cause of neonatal and childhood-onset diabetes. Diabetes 2014, 63, 2888–2894. [Google Scholar] [CrossRef]
- Tiyaboonchai, A.; Cardenas-Diaz, F.L.; Ying, L.; Maguire, J.A.; Sim, X.; Jobaliya, C.; Gagne, A.L.; Kishore, S.; Stanescu, D.E.; Hughes, N.; et al. GATA6 plays an important role in the induction of human definitive endoderm, development of the pancreas, and functionality of pancreatic β cells. Stem Cell Rep. 2017, 8, 589–604. [Google Scholar] [CrossRef]
- Shi, Z.-D.; Lee, K.; Yang, D.; Amin, S.; Verma, N.; Li, Q.V.; Zhu, Z.; Soh, C.-L.; Kumar, R.; Evans, T.; et al. Genome editing in hPSCs reveals GATA6 haploinsufficiency and a genetic interaction with GATA4 in human pancreatic development. Cell Stem Cell 2017, 20, 675–688.e6. [Google Scholar] [CrossRef] [PubMed]
- Huopio, H.; Miettinen, P.J.; Ilonen, J.; Nykänen, P.; Veijola, R.; Keskinen, P.; Näntö-Salonen, K.; Vangipurapu, J.; Raivo, J.; Stančáková, A.; et al. Clinical, genetic, and biochemical characteristics of early-onset diabetes in the Finnish population. J. Clin. Endocrinol. Metab. 2016, 101, 3018–3026. [Google Scholar] [CrossRef] [PubMed]
- Støy, J.; Steiner, D.F.; Park, S.-Y.; Ye, H.; Philipson, L.H.; Bell, G.I. Clinical and molecular genetics of neonatal diabetes due to mutations in the insulin gene. Rev. Endocr. Metab. Disord. 2010, 11, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Colombo, C.; Porzio, O.; Liu, M.; Massa, O.; Vasta, M.; Salardi, S.; Beccaria, L.; Monciotti, C.; Toni, S.; Pedersen, O.; et al. Seven mutations in the human insulin gene linked to permanent neonatal/infancy-onset diabetes mellitus. J. Clin. Investig. 2008, 118, 2148–2156. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Hodish, I.; Haataja, L.; Lara-Lemus, R.; Rajpal, G.; Wright, J.; Arvan, P. Proinsulin misfolding and diabetes: Mutant INS gene-induced diabetes of youth. Trends Endocrinol. Metab. 2010, 21, 652–659. [Google Scholar] [CrossRef]
- Balboa, D.; Saarimäki-Vire, J.; Borshagovski, D.; Survila, M.; Lindholm, P.; Galli, E.; Eurola, S.; Ustinov, J.; Grym, H.; Huopio, H.; et al. Insulin mutations impair beta-cell development in a patient-derived iPSC model of neonatal diabetes. eLife 2018, 7. [Google Scholar] [CrossRef]
- De Franco, E.; Lytrivi, M.; Ibrahim, H.; Montaser, H.; Wakeling, M.N.; Fantuzzi, F.; Patel, K.; Demarez, C.; Cai, Y.; Igoillo-Esteve, M.; et al. YIPF5 mutations cause neonatal diabetes and microcephaly through endoplasmic reticulum stress. J. Clin. Invest. 2020, 130, 6338–6353. [Google Scholar] [CrossRef]
- Urano, F. Wolfram syndrome: Diagnosis, management, and treatment. Curr. Diabetes Rep. 2016, 16, 1–8. [Google Scholar] [CrossRef]
- Marshall, B.; Permutt, M.A.; Paciorkowski, A.R.; Hoekel, J.; Karzon, R.; Wasson, J.; Viehover, A.; White, N.H.; Shimony, J.; Manwaring, L.; et al. Phenotypic characteristics of early Wolfram syndrome. Orphanet J. Rare Dis. 2013, 8, 64. [Google Scholar] [CrossRef]
- Maxwell, K.G.; Augsornworawat, P.; Velazco-Cruz, L.; Kim, M.H.; Asada, R.; Hogrebe, N.J.; Morikawa, S.; Urano, F.; Millman, J.R. Gene-edited human stem cell–derived β cells from a patient with monogenic diabetes reverse preexisting diabetes in mice. Sci. Transl. Med. 2020, 12, eaax9106. [Google Scholar] [CrossRef]
- Campuzano, V.; Montermini, L.; Moltò, M.D.; Pianese, L.; Cossée, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; et al. Friedreich’s ataxia: Autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996, 271, 1423–1427. [Google Scholar] [CrossRef]
- Al-Mahdawi, S.; Pinto, R.M.; Ismail, O.; Varshney, D.; Lymperi, S.; Sandi, C.; Trabzuni, D.; Pook, M.A. The Friedreich ataxia GAA repeat expansion mutation induces comparable epigenetic changes in human and transgenic mouse brain and heart tissues. Hum. Mol. Genet. 2007, 17, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Babcock, M.; Yang, J.; Liu, X.; Bhalla, K.; Kim, C.N.; Ibrado, A.M.; Cai, J.; Peng, T.-I.; Jones, D.P.; Wang, X. Regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of frataxin. Science 1997, 276, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Rötig, A.; De Lonlay, P.; Chretien, D.; Foury, F.; Koenig, M.; Sidi, D.; Munnich, A.; Rustin, P. Aconitase and mitochondrial iron–sulphur protein deficiency in Friedreich ataxia. Nat. Genet. 1997, 17, 215–217. [Google Scholar] [CrossRef]
- Igoillo-Esteve, M.; Oliveira, A.F.; Cosentino, C.; Fantuzzi, F.; Demarez, C.; Toivonen, S.; Hu, A.; Chintawar, S.; Lopes, M.; Pachera, N.; et al. Exenatide induces frataxin expression and improves mitochondrial function in Friedreich ataxia. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Report | Approach | Outcome to Beta-Cell Function | Reference |
|---|---|---|---|
| Pagliuca et al., 2014 | Protocol for generation of beta-like cells by modifying and combining three previous protocols [70,71,72] | Expression of key markers of mature pancreatic beta-cells, glucose-induced Ca2+ influx, insulin secretion in response to multiple sequential glucose challenges | [52] |
| Rezania et al., 2014 | 7-stage protocol for generation of beta-like cells based on previous own protocol [70] | Expression of key markers of mature pancreatic beta-cells, insulin secretion in response to high glucose | [53] |
| Russ et al., 2015 | Protocol for generation of beta-like cells based on two previous protocols [70] by culture without additional growth factors after endocrine progenitor stage | Expression of key markers of mature pancreatic beta-cells, insulin secretion in response to high glucose | [51] |
| Millman et al., 2016 | Addition of ROCK inhibitor and Activin A based on [52] at pancreatic progenitor stage | Similar to their previous studies [52], beneficial effect on insulin expression and secretion | [56] |
| Zhu et al., 2016 | Addition of vitamin C and BayK-8644 at final stage | Increased insulin expression and secretion | [55] |
| Ghazizadeh et al., 2017 | ROCKII inhibition at pancreatic progenitor stage | Generation and maturation of glucose-responsive cells | [54] |
| Nair et al., 2019 | Reaggregation/clustering after FACS at final stage | Robust dynamic insulin secretion, metabolic maturation by driving mitochondrial oxidative respiration | [57] |
| Velazco-Cruz et al., 2019 | Allowing TGF-β signaling during the final stage and reaggregation/clustering | Pure populations of beta-like cells that secrete high levels of insulin and express key beta cell markers | [58] |
| Yoshihara et al., 2020 | Allowing WNT4 signaling during the final stage | Metabolic maturation with robust insulin secretion and high mitochondrial oxidative respiration | [63] |
| Li et al., 2020 | Combination of three previous protocols [52], and reaggregation/clustering after negative sorting by CD9 | High glucose-responsiveness and insulin production | [65] |
| Approach | Target | Intervention | Reference |
|---|---|---|---|
| Optimization of differentiation | Oxygen supply | 7-stage protocol including culture at air-liquid interface | [53] |
| Signaling pathways | Sequential modulation of signaling pathways in a 3D cell culture system | [52] | |
| Removal of BMP inhibitors in combination with retinoic acid and EGF/KGF addition | [51] | ||
| Modulation of TGF-β signaling | [58] | ||
| Cell clustering | Isolation and reaggregation of immature beta-like cells to form islet-sized beta cell-enriched clusters | [57] | |
| Elimination of remaining undifferentiated cells | Chemical methods | Addition of PluriSln1 | [96] |
| Immunological methods | Antibodies against SSEA-5 glycan | [97] | |
| Antibodies against beta cell surface marker CD49a followed by MACS | [66] | ||
| Removal of Claudin-6-positive cells | [99] | ||
| Separation of SSEA-4 and TRA-1-60 undifferentiated cells by MACS and FACS | [98] | ||
| Genetic methods | Double suicide cassette: HSV-TK and NTR | [100] | |
| Inducible Caspase-9 suicide gene | [101] |
| Point of Action | Approach | Intervention | Species (Graft to Host) | Outcome | Reference |
|---|---|---|---|---|---|
| Graft | Hypo-immunity | Deletion of β2M | B2M knock-out ESCs transplanted in NK-cell depleted mice | (+) Prevents immune rejection, confers resistance to T-cell mediated killing | [124] |
| (−) Loss of NK-tolerance | |||||
| Deletion of β2M and CIITA | HLA-1/2 knock-out iPSCs in vitro | (+) Eliminates immunogenicity, universal donor cell therapy potential | [122] | ||
| (−) Loss of NK-tolerance | |||||
| Deletion of HLA-A/-B/-C and CIITA + PD-L1, HLA-G and CD47 upregulation | Engineered human stem cells transplanted in mice | (+) Less immune activation, decreases T–& NK-cell mediated killing and macrophage engulfment | [123] | ||
| (−) Complex genetic engineering, risk of off-target events | |||||
| Immune-checkpoint modulation | SA-PD-L1 engineered islet grafts | SA-PDL1 mouse islets transplanted in mice | (+) Sustains graft survival, prevents allograft immune rejection, confers localized immunomodulation | [125] | |
| (−) Short rapamycin treatment for long-term graft survival | |||||
| CTLA-4Ig and PD-L1 knock-in | Knock-in human ESCs transplanted in humanized mice | (+) Protects from allogeneic immune response | [126] | ||
| (−) Escape from immune surveillance (by grafted tumorigenic cells and viral infections) | |||||
| Graft supplementation | PD-L1 microgels | Allogeneic mouse islets co-transplanted with PD-L1 microgels in mice | (+) Prevents graft rejection, promotes tolerance, confers local immuno-protective response, off-the-shelf immunomodulation strategy | [127] | |
| (−) Some recipients only partial benefit, short low-dose rapamycin treatment to fully prevent rejection | |||||
| MSCs | Allograft islets in combination with MSCs transplanted in mice, rats and non-human primates | (+) Downregulates pro-inflammatory cytokines, prevents allograft immune rejection, promotes graft survival | [114,115,116,117] | ||
| (−) Logistical and regulatory concerns–clinical transplantation via hepatic portal vein limits co-engraftment | |||||
| MSC-derived cell-free cocktail | Pre-treated mouse islets transplanted in diabetic mice | (+) Protects from cytokine-mediated cell death in vitro, improves functional graft survival in vivo | [120] | ||
| Host | Treg therapy | Ex vivo expanded polyclonal or antigen-specific Tregs | Tregs injected systemically in mice and T1D patients | (+) Prolongs islet allograft survival, inhibits alloimmune response in mice, clinical trials demonstrated feasibility and safety in human | [106,107,108,128] |
| (−) Poor engraftment, Treg manufacturing is challenging | |||||
| Production of anti-inflammatory cytokines | IL-10 secretion by designer beta cells | Glucose-dependent secretion of IL-10 by murine beta cells | (+) Protects from pro-inflammatory cytokine-induced cell death, minimal systemic effects on host immune system, efficient engineering | [111] | |
| Immune-checkpoint modulation | Administration of CTLA-4Ig and anti CD40L mAbs | ESC-PE transplanted in (humanized) mice | (+) Prevents immune rejection, induces immune tolerance, prolongs graft survival | [129] | |
| (−) Testing only possible in immunocompromised animals (limited to rodents) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bourgeois, S.; Sawatani, T.; Van Mulders, A.; De Leu, N.; Heremans, Y.; Heimberg, H.; Cnop, M.; Staels, W. Towards a Functional Cure for Diabetes Using Stem Cell-Derived Beta Cells: Are We There Yet? Cells 2021, 10, 191. https://doi.org/10.3390/cells10010191
Bourgeois S, Sawatani T, Van Mulders A, De Leu N, Heremans Y, Heimberg H, Cnop M, Staels W. Towards a Functional Cure for Diabetes Using Stem Cell-Derived Beta Cells: Are We There Yet? Cells. 2021; 10(1):191. https://doi.org/10.3390/cells10010191
Chicago/Turabian StyleBourgeois, Stephanie, Toshiaki Sawatani, Annelore Van Mulders, Nico De Leu, Yves Heremans, Harry Heimberg, Miriam Cnop, and Willem Staels. 2021. "Towards a Functional Cure for Diabetes Using Stem Cell-Derived Beta Cells: Are We There Yet?" Cells 10, no. 1: 191. https://doi.org/10.3390/cells10010191
APA StyleBourgeois, S., Sawatani, T., Van Mulders, A., De Leu, N., Heremans, Y., Heimberg, H., Cnop, M., & Staels, W. (2021). Towards a Functional Cure for Diabetes Using Stem Cell-Derived Beta Cells: Are We There Yet? Cells, 10(1), 191. https://doi.org/10.3390/cells10010191