
Complete Chloroplast Genome Sequence of Medicago falcata: Comparative Analyses with Other Species of Medicago

Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material, DNA Extraction, and Sequencing
2.2. Chloroplast Genome Assembly and Annotation
2.3. Genome Structure Analysis of Chloroplast Sequence
2.4. Analysis of the Chloroplast Genome Consistency
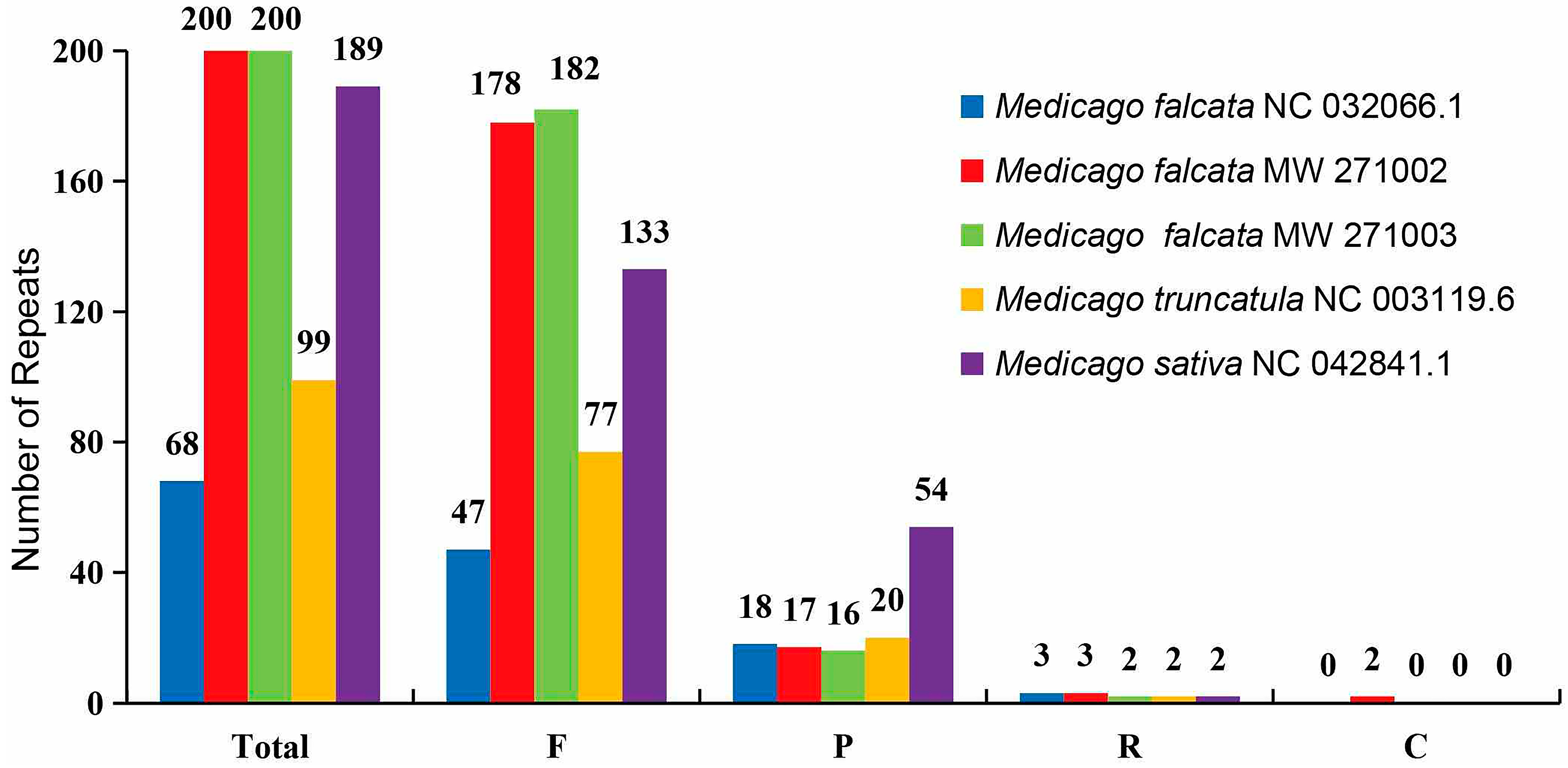
2.5. Analysis of Simple and Complex Repeats
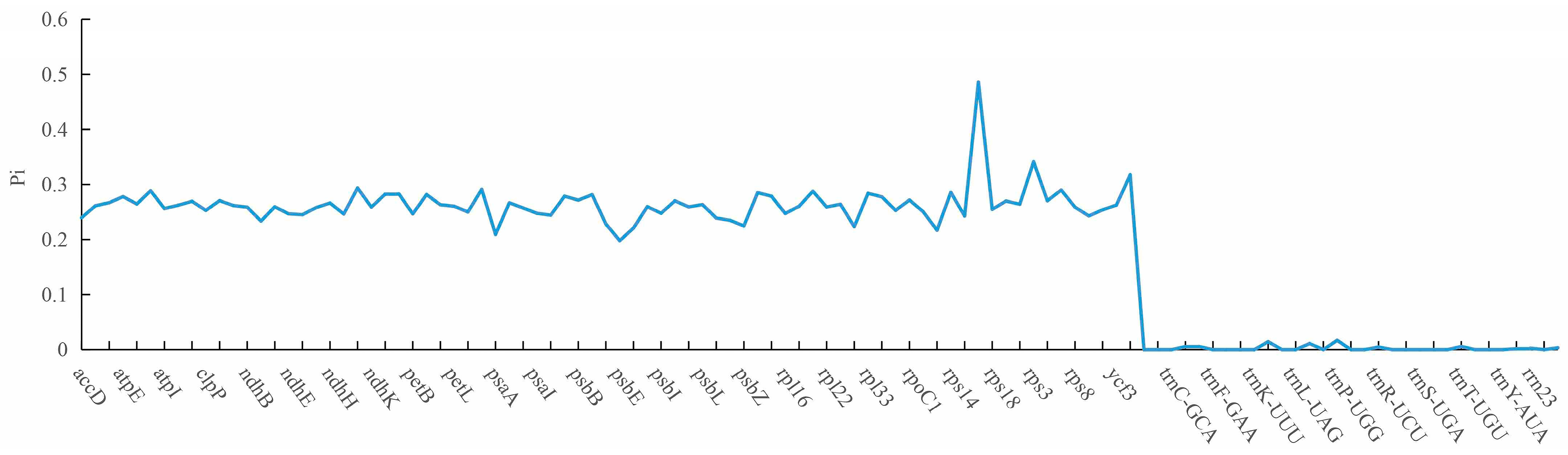
2.6. Analysis of Nucleotide Polymorphism of the Chloroplast Genome
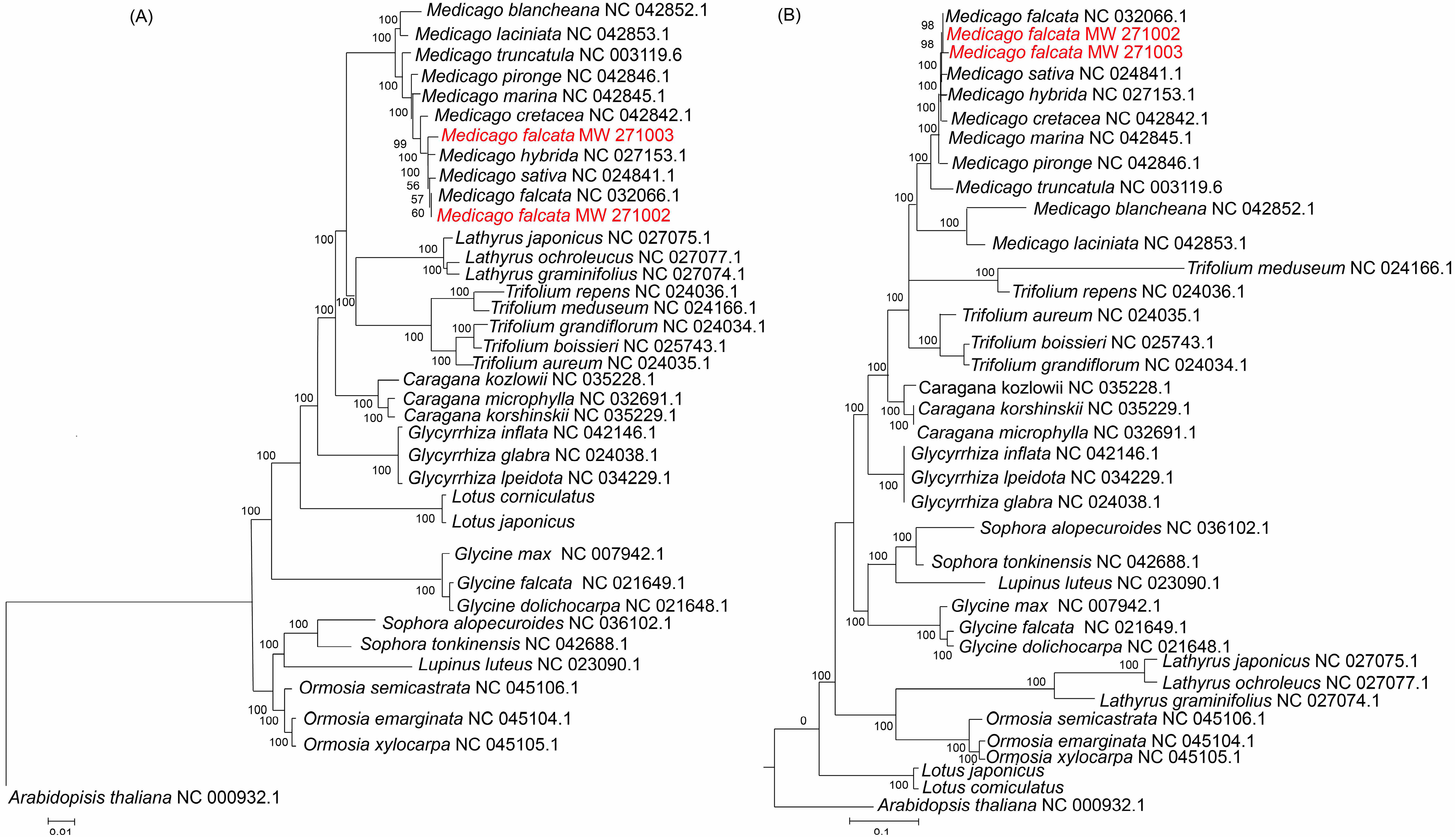
2.7. Phylogenetic Analysis
3. Results
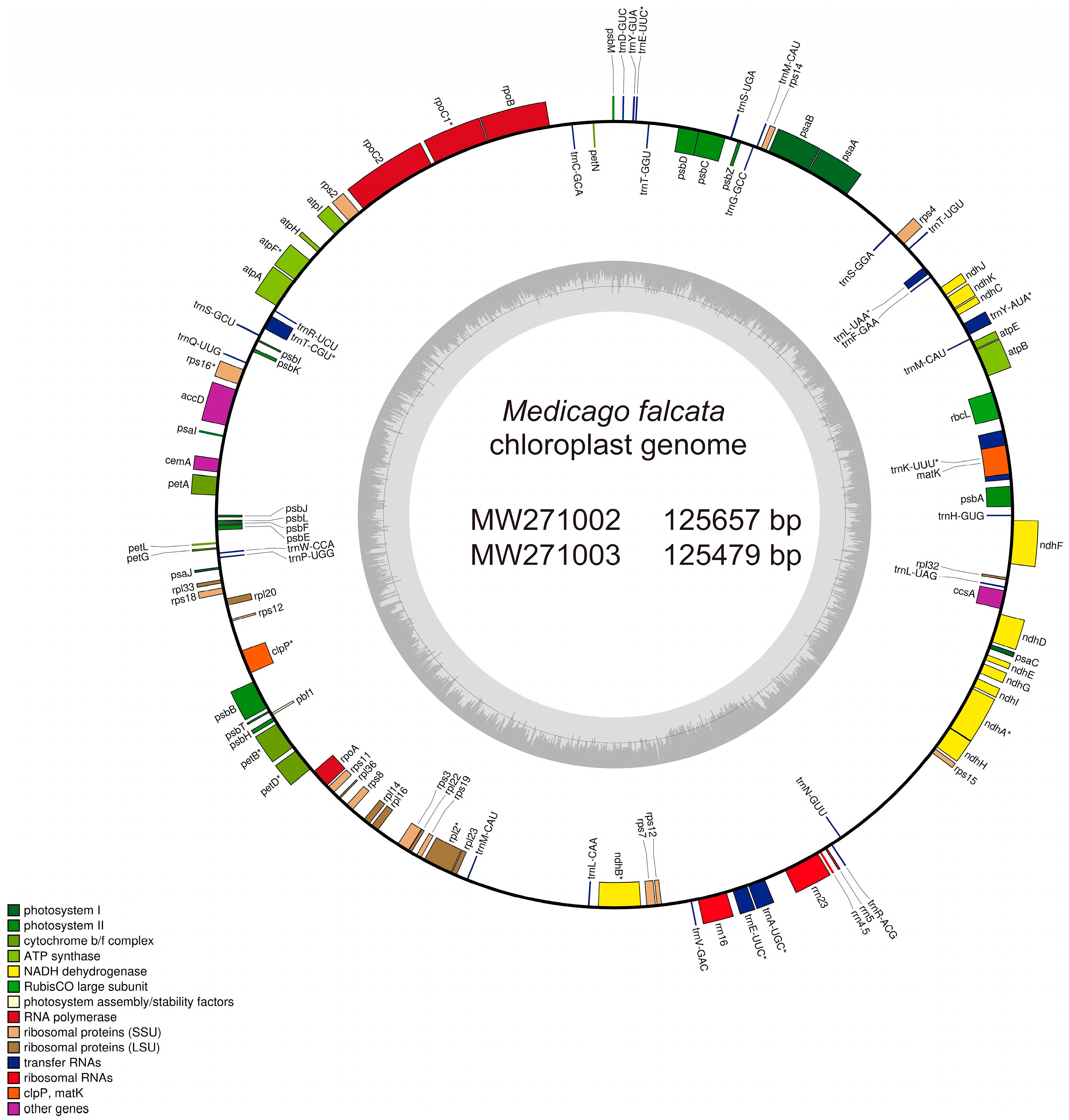
3.1. Characterization of the Chloroplast Genomes of Two M. falcata Ecotypes
3.2. Comparative Analysis of the Chloroplast Genome of Three Medicago Species
3.3. Features of the cpDNA Repeats of Medicago
3.4. Phylogenetic Relationship Between Medicago and Related Species
4. Discussion
4.1. Conservation of Medicago cpDNA
4.2. Simple and Complex Repeats Analysis
4.3. Phylogenetic
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Jansen, R.K.; Cai, Z.; Raubeson, L.A.; Daniell, H.; Depamphilis, C.W.; Leebens-Mack, J.; Müller, K.F.; Guisinger-Bellian, M.; Haberle, R.C.; Hansen, A.K.; et al. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. USA 2007, 104, 19369–19374. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wu, G.; Shrestha, N.; Wu, S.; Guo, W.; Yin, M.; Li, A.; Liu, J.; Ren, G. Phylogeny and Species Delimitation of Chinese Medicago (Leguminosae) and Its Relatives Based on Molecular and Morphological Evidence. Front. Plant Sci. 2021, 11, 619799. [Google Scholar] [CrossRef]
- Benedito, V.A.; Torres-Jerez, I.; Murray, J.D.; Andriankaja, A.; Allen, S.; Kakar, K.; Wandrey, M.; Verdier, J.; Zuber, H.; Ott, T.; et al. A gene expression atlas of the model legume Medicago truncatula. Plant J. 2008, 55, 504–513. [Google Scholar] [CrossRef]
- Small, E.; Jomphe, M. A synopsis of the genus Medicago (Leguminosae). Can. J. Bot. 1989, 67, 3260–3294. [Google Scholar] [CrossRef]
- Liu, Z.; Chen, T.; Ma, L.; Zhao, Z.; Zhao, P.X.; Nan, Z.; Wang, Y. Global transcriptome sequencing using the Illumina platform and the development of EST-SSR markers in autotetraploid alfalfa. PLoS ONE 2013, 8, e83549. [Google Scholar] [CrossRef]
- Shi, H.; He, S.; He, X.; Lu, S.; Guo, Z. An eukaryotic elongation factor 2 from Medicago falcata (MfEF2) confers cold tolerance. BMC Plant Biol. 2019, 19, 218. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.L.; Zhao, M.G.; Tian, Q.Y.; Zhang, W.H. Comparative studies on tolerance of Medicago truncatula and Medicago falcata to freezing. Planta 2011, 234, 445–457. [Google Scholar] [CrossRef]
- Birky, C.W., Jr. The inheritance of genes in mitochondria and chloroplasts: Laws, mechanisms, and models. Annu. Rev. Genet. 2001, 35, 125–148. [Google Scholar] [CrossRef]
- Wu, Z.Q.; Ge, S. The phylogeny of the BEP clade in grasses revisited: Evidence from the whole-genome sequences of chloroplasts. Mol. Phylogenet. Evol. 2012, 62, 573–578. [Google Scholar] [CrossRef]
- Palmer, J.D.; Osorio, B.; Aldrich, J.; Thompson, W.F. Chloroplast DNA evolution among legumes: Loss of a large inverted repeat occurred prior to other sequence rearrangements. Curr. Genet. 1987, 11, 275–286. [Google Scholar] [CrossRef]
- Saski, C.; Lee, S.B.; Daniell, H.; Wood, T.C.; Tomkins, J.; Kim, H.G.; Jansen, R.K. Complete Chloroplast Genome Sequence of Glycine max and Comparative Analyses with other Legume Genomes. Plant Mol. Biol. 2005, 59, 309–322. [Google Scholar] [CrossRef]
- Palmer, J.D.; Thompson, W.F. Rearrangements in the chloroplast genomes of mung bean and pea. Proc. Natl. Acad. Sci. USA 1981, 78, 5533–5537. [Google Scholar] [CrossRef]
- Bruneau, A.; Palmer, D.J.D. A Chloroplast DNA Inversion as a Subtribal Character in the Phaseoleae (Leguminosae). Syst. Bot. 1990, 15, 378–386. [Google Scholar] [CrossRef]
- Wojciechowski, M.F.; Lavin, M.; Sanderson, M.J. A phylogeny of legumes (Leguminosae) based on analysis of the plastid matK gene resolves many well-supported subclades within the family. Am. J. Bot. 2004, 91, 1846–1862. [Google Scholar] [CrossRef]
- Tao, X.; Ma, L.; Zhang, Z.; Liu, W.; Liu, Z. Characterization of the complete chloroplast genome of alfalfa (Medicago sativa) (Leguminosae). Gene Rep. 2017, 6, 67–73. [Google Scholar] [CrossRef]
- Ren, Y.L.; Ma, Y.J.; Li, X.; Li, X.A.; Yang, G.Z.; Li, P. Complete chloroplast genome sequence and characteristics analysis of Qingda no.1 alfalfa (Medicago sativa L. cv. Qingda no.1). Czech J. Genet. Plant Breed. 2023, 59, 160–168. [Google Scholar] [CrossRef]
- Quiros, C.F.; Bauchan, G.R. The genus Medicago and the origin of the Medicago sativa complex. Agronomy 1988, 93–124. [Google Scholar] [CrossRef]
- Allen, G.C.; Flores-Vergara, M.A.; Krasynanski, S.; Kumar, S.; Thompson, W.F. A modified protocol for rapid DNA isolation from plant tissues using cetyltrimethylammonium bromide. Nat. Protoc. 2006, 1, 2320–2325. [Google Scholar] [CrossRef]
- Jin, J.J.; Yu, W.B.; Yang, J.B.; Song, Y.; dePamphilis, C.W.; Yi, T.S.; Li, D.Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq—Versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef]
- Laslett, D.; Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef]
- Brudno, M.; Do, C.B.; Cooper, G.M.; Kim, M.F.; Davydov, E.; NISC Comparative Sequencing Program; Green, E.D.; Sidow, A.; Batzoglou, S. LAGAN and Multi-LAGAN: Efficient tools for large-scale multiple alignment of genomic DNA. Genome Res. 2003, 13, 721–731. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Schleiermacher, C. REPuter: Fast computation of maximal repeats in complete genomes. Bioinformatics 1999, 15, 426–427. [Google Scholar] [CrossRef]
- Beier, S.; Himmelbach, A.; Colmsee, C.; Zhang, X.Q.; Barrero, R.A.; Zhang, Q.; Li, L.; Bayer, M.; Bolser, D.; Taudien, S.; et al. Construction of a map-based reference genome sequence for barley, Hordeum vulgare L. Sci. Data 2017, 4, 170044. [Google Scholar] [CrossRef]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, M. The chloroplast genome. Plant Mol. Biol. 1992, 19, 149–168. [Google Scholar] [CrossRef]
- Raveendar, S.; Na, Y.W.; Lee, J.R.; Shim, D.; Ma, K.H.; Lee, S.Y.; Chung, J.W. The complete chloroplast genome of Capsicum annuum var. glabriusculum using Illumina sequencing. Molecules 2015, 20, 13080–13088. [Google Scholar] [CrossRef]
- Yan, W.; Shi, W.; Liu, L.; Ma, Y.; Chen, L.; Wang, Z.; Hou, X. Complete sequencing of the chloroplast genomes of two Medicago species. Mitochondrial DNA Part B Resour. 2017, 2, 302–303. [Google Scholar] [CrossRef]
- Raubeson, L.A.; Peery, R.; Chumley, T.W.; Dziubek, C.; Fourcade, H.M.; Boore, J.L.; Jansen, R.K. Comparative chloroplast genomics: Analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genom. 2007, 8, 174. [Google Scholar] [CrossRef] [PubMed]
- Strauss, S.H.; Palmer, J.D.; Howe, G.T.; Doerksen, A.H. Chloroplast genomes of two conifers lack a large inverted repeat and are extensively rearranged. Proc. Natl. Acad. Sci. USA 1988, 85, 3898–3902. [Google Scholar] [CrossRef]
- Kim, S.; Park, J.Y.; Yang, T. Comparative analysis of the complete chloroplast genome sequences of a normal male-fertile cytoplasm and two different cytoplasms conferring cytoplasmic male sterility in onion (Allium cepa L). J. Hortic. Sci. Biotechnol. 2015, 90, 459–468. [Google Scholar] [CrossRef]
- Du, Y.P.; Bi, Y.; Yang, F.P.; Zhang, M.F.; Chen, X.Q.; Xue, J.; Zhang, X.H. Complete chloroplast genome sequences of Lilium: Insights into evolutionary dynamics and phylogenetic analyses. Sci. Rep. 2017, 7, 5751. [Google Scholar] [CrossRef]
- Huo, Y.; Gao, L.; Liu, B.; Yang, Y.; Kong, S.; Sun, Y.; Yang, Y.; Wu, X. Complete chloroplast genome sequences of four Allium species: Comparative and phylogenetic analyses. Sci. Rep. 2019, 9, 12250. [Google Scholar] [CrossRef]
- Timme, R.E.; Kuehl, J.V.; Boore, J.L.; Jansen, R.K. A comparative analysis of the Lactuca and Helianthus (Asteraceae) plastid genomes: Identification of divergent regions and categorization of shared repeats. Am. J. Bot. 2007, 94, 302–312. [Google Scholar] [CrossRef]
- Weng, M.L.; Blazier, J.C.; Govindu, M.; Jansen, R.K. Reconstruction of the ancestral plastid genome in Geraniaceae reveals a correlation between genome rearrangements, repeats, and nucleotide substitution rates. Mol. Biol. Evol. 2014, 31, 645–659. [Google Scholar] [CrossRef]
- Wang, L.; Guo, Z.H. The complete chloroplast genome of Tamarix ramosissima and comparative analysis of Tamaricaceae species. Biol. Plant. 2021, 65, 237–245. [Google Scholar] [CrossRef]
- Kaila, T.; Chaduvla, P.K.; Rawal, H.C.; Saxena, S.; Tyagi, A.; Mithra, S.V.A.; Solanke, A.U.; Kalia, P.; Sharma, T.R.; Singh, N.K.; et al. Chloroplast Genome Sequence of Clusterbean (Cyamopsis tetragonoloba L.): Genome Structure and Comparative Analysis. Genes 2017, 8, 212. [Google Scholar] [CrossRef]
- Li, X.; Tan, W.; Sun, J.; Du, J.; Zheng, C.; Tian, X.; Zheng, M.; Xiang, B.; Wang, Y. Author Correction: Comparison of Four Complete Chloroplast Genomes of Medicinal and Ornamental Meconopsis Species: Genome Organization and Species Discrimination. Sci. Rep. 2019, 9, 15163. [Google Scholar] [CrossRef] [PubMed]
- Somaratne, Y.; Guan, D.L.; Wang, W.Q.; Zhao, L.; Xu, S.Q. Complete chloroplast genome sequence of Xanthium sibiricum provides useful DNA barcodes for future species identification and phylogeny. Plant Syst. Evol. 2019, 305, 949–960. [Google Scholar] [CrossRef]
- Yang, Y.; Zhou, T.; Duan, D.; Yang, J.; Feng, L.; Zhao, G. Comparative Analysis of the Complete Chloroplast Genomes of Five Quercus Species. Front. Plant Sci. 2016, 7, 959. [Google Scholar] [CrossRef]
- Jung, J.; Do, H.D.K.; Hyun, J.; Kim, C.; Kim, J.H. Comparative analysis and implications of the chloroplast genomes of three thistles (Carduus L., Asteraceae). PeerJ 2021, 9, e10687. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Wu, X.Y.; Guo, X.; Bao, P.J.; Chu, M.; Zhou, X.L.; Liang, Z.Y.; Ding, X.Z.; Yan, P. The complete chloroplast genome of Medicago sativa cv. Hangmu No.1, a plant of space mutation breeding. Mitochondrial DNA Part B Resour. 2019, 4, 603–604. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Length (bp) | Gene Number | Protein-Coding Gene Number | Protein Coding Gene (%) | rRNA_Gene Number | rRNA (%) | tRNA Gene Number | tRNA (%) | GC Content (%) | IR Length/bp |
|---|---|---|---|---|---|---|---|---|---|---|
| Medicago falcata MW 271002 | 125,657 | 112 | 78 | 69.64 | 4 | 3.57 | 30 | 26.79 | 33.85 | N/A |
| Medicago falcata MW 271003 | 125,479 | 112 | 78 | 69.64 | 4 | 3.57 | 30 | 26.79 | 33.84 | N/A |
| Medicago falcata NC 032066.1 | 124,430 | 110 | 78 | 70.91 | 4 | 3.64 | 28 | 25.45 | 33.96 | N/A |
| Medicago truncatula NC 003119.6 | 124,033 | 111 | 77 | 69.37 | 4 | 3.6 | 30 | 27.03 | 33.97 | N/A |
| Medicago sativa NC 042841.1 | 125,330 | 112 | 78 | 69.64 | 4 | 3.57 | 30 | 26.79 | 33.87 | N/A |
| Gene Category | Gene Group | Gene Names |
|---|---|---|
| Other genes | Envelope membrane protein (1) | cemA |
| Maturase (1) | matK | |
| Protease (1) | ClpP a | |
| Subunit of acetyl-CoA carboxylase (1) | accD | |
| c-type cytochrome synthesis gene (1) | ccsA | |
| Photosynthesis | Others (3) | pbf1, ycf3 b, ycf4 |
| Subunits of ATP synthase (6) | atpA, atpB, atpE, atpF a, atpH, atpI | |
| Subunits of NADH dehydrogenase (11) | NdhA a, ndhB a, ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK | |
| Subunits of cytochrome (6) | petA, petB a, petD a, petG, petL, petN | |
| Subunits of photosystem II (14) | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbT, psbZ | |
| Subunits of photosystem (5) | psaA, psaB, psaC, psaI, psaJ | |
| Subunits of rubisco (1) | rbcL | |
| Self-replication | DNA dependent RNA polymerase (4) | rpoA, rpoB, rpoC1 a, rpoC2 |
| Large subunits of ribosome (9) | rpl14, rpl16, rpl2 a, rpl20, rpl22, rpl23, rpl32, rpl33, rpl36 | |
| Small subunits of ribosome (12) | rps11, rps12 a, rps14, rps15, rps1 6 a, rps18, rps19, rps2, rps3, rps4, rps7, rps8 | |
| rRNA genes (4) | rrn16, rrn23, rrn4.5, rrn5 | |
| tRNA genes (30) | trnA-UGC, trnC-GCA, trnD-GUC, trnE-UUC(×2), trnF-GAA, trnG-GCC, trnH-GUG, trnK-UUU, trnL-CAA, trnL-UAA, trnL-UAG, trnM-CAU(×3), trnN-GUU, trnP-UGG, trnQ-UUG, trnR-ACG, trnR-UCU, trnS-GCU, trnS-GGA, trnS-UGA, trnT-CGU, trnT-GGU, trnT-UGU, trnV-GAC, trnW-CCA, trnY-AUA, trnY-GUA | |
| Unknown function | Conserved open reading frames (2) | ycf1, ycf2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duan, W.; Zhang, X.; Wang, Y.; Li, Q. Complete Chloroplast Genome Sequence of Medicago falcata: Comparative Analyses with Other Species of Medicago. Agronomy 2025, 15, 1856. https://doi.org/10.3390/agronomy15081856
Duan W, Zhang X, Wang Y, Li Q. Complete Chloroplast Genome Sequence of Medicago falcata: Comparative Analyses with Other Species of Medicago. Agronomy. 2025; 15(8):1856. https://doi.org/10.3390/agronomy15081856
Chicago/Turabian StyleDuan, Wei, Xueli Zhang, Yuxiang Wang, and Qian Li. 2025. "Complete Chloroplast Genome Sequence of Medicago falcata: Comparative Analyses with Other Species of Medicago" Agronomy 15, no. 8: 1856. https://doi.org/10.3390/agronomy15081856
APA StyleDuan, W., Zhang, X., Wang, Y., & Li, Q. (2025). Complete Chloroplast Genome Sequence of Medicago falcata: Comparative Analyses with Other Species of Medicago. Agronomy, 15(8), 1856. https://doi.org/10.3390/agronomy15081856