
Molecular Elucidation of Anthocyanin Accumulation Mechanisms in Hippeastrum hybridum Cultivars

 , ,
, ,  , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Material and Method
2.1. Materials
2.2. Content Determination of Total Anthocyanins and Flavonoids
2.3. Total RNA Extraction and Quality Control
2.4. cDNA Synthesis and Library Construction
2.5. The Analyzes of DEGs
2.6. qRT-PCR (Quantitative Real-Time Polymerase Chain Reaction) Assay
2.7. Extraction of Metabolites
2.8. LC-MS Parameter Description
2.9. Mass Spectrometry Parameter Description
2.10. Metabolite Data Analyses
2.11. Co-Expression Analysis
2.12. Y2H Assay
2.13. Heatmap Generation and Statistical Analysis
3. Results
3.1. Phenotypic Characterization and Determination of Total Anthocyanins and Total Flavonoids
3.2. Metabolomics Data Analysis
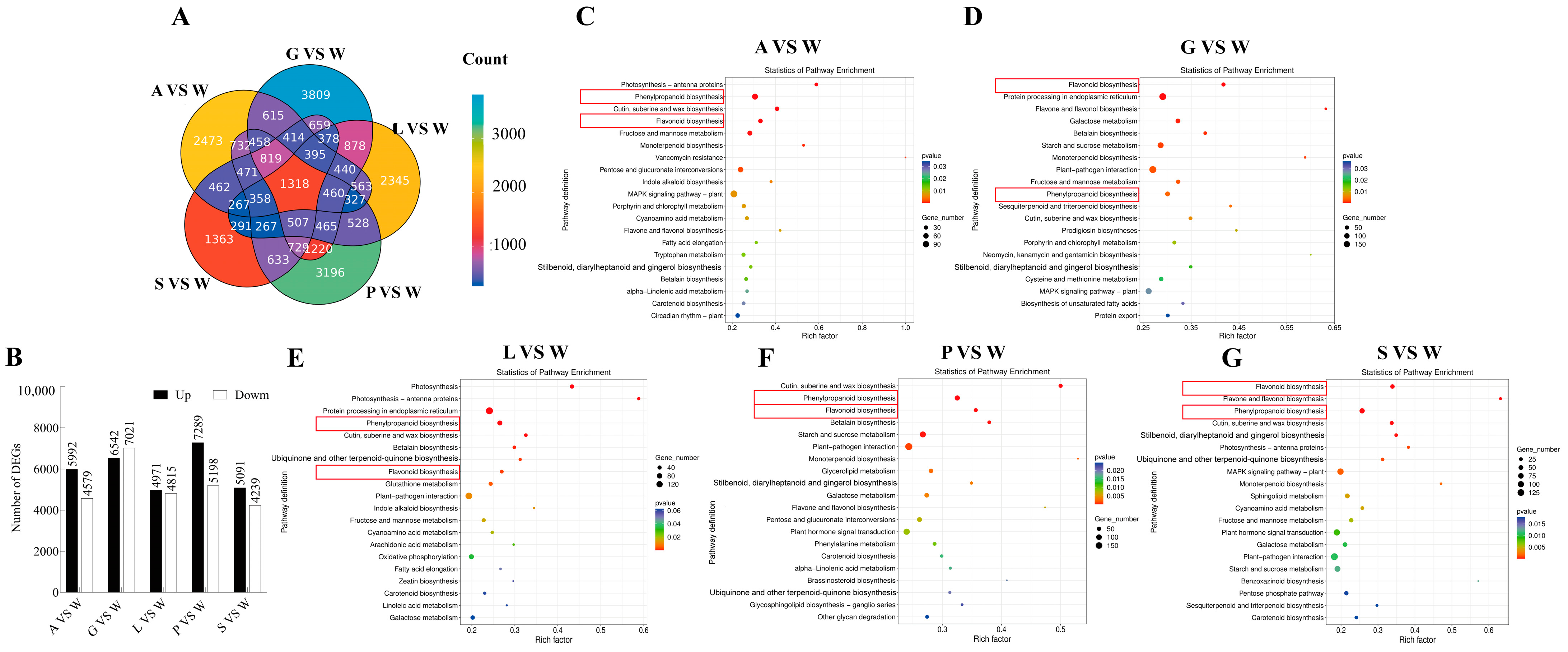
3.3. RNA High-Throughput Sequencing Data Analysis
3.4. Analysis of the Expression Profiles of Genes Involved in the Anthocyanin Biosynthesis Pathway
3.5. Integrated Analysis of Transcriptomics and Metabolomics
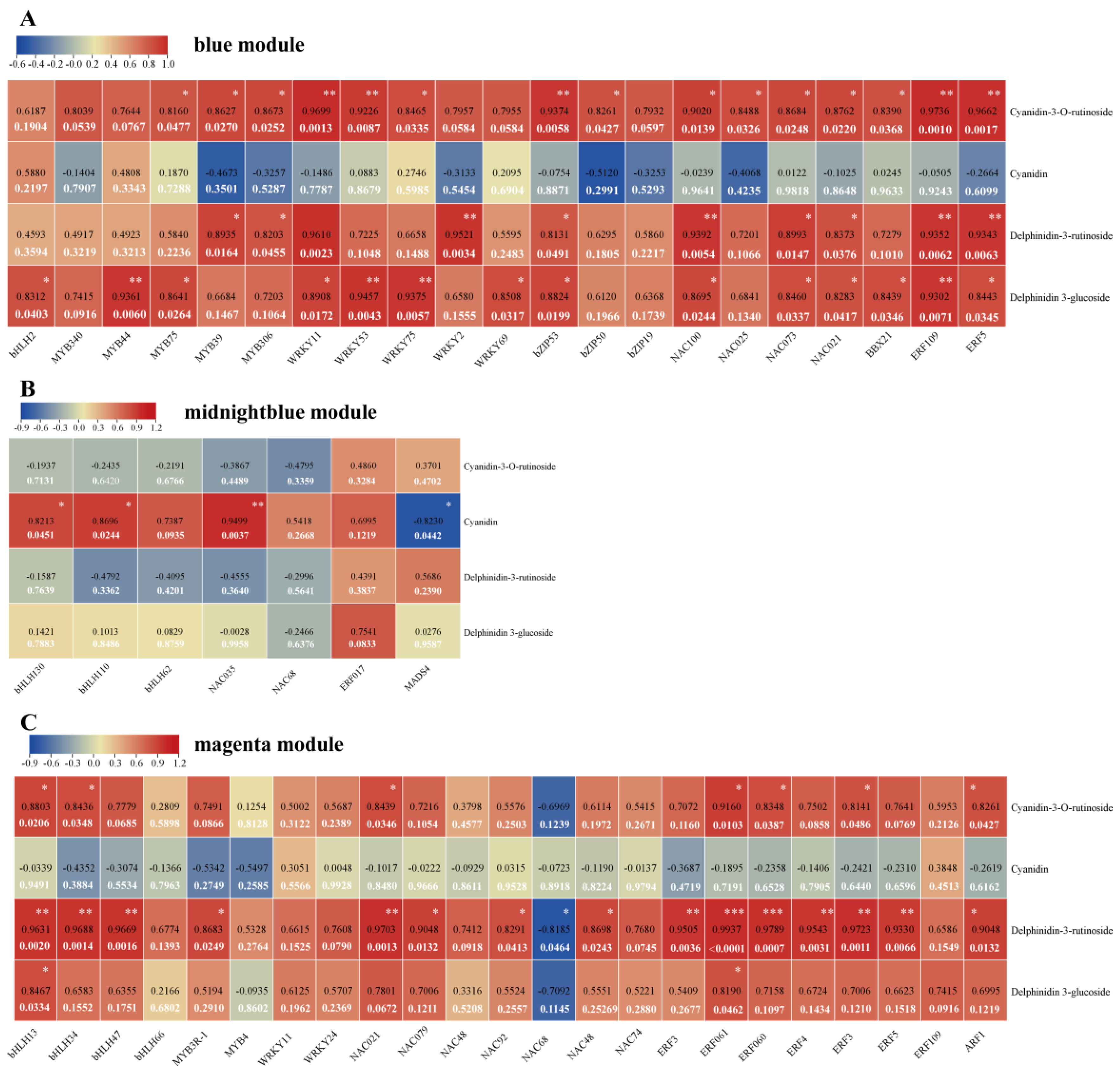
3.6. Targeting of Transcription Factors Involved in Biosynthesis of Flavonoids and Anthocyanins
3.7. Identification of Potential Component of MBW Complex
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Read, V.M. Hippeastrum: The Gardener’s Amaryllis; Timber Press: Portland, OR, USA, 2004. [Google Scholar]
- van der Kooi, C.J.; Wilts, B.D.; Leertouwer, H.L.; Staal, M.; Elzenga, J.T.M.; Stavenga, D.G. Iridescent flowers? Contribution of surface structures to optical signaling. New Phytol. 2014, 203, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Kay, Q.; Daoud, H.; Stirton, C. Pigment distribution, light reflection and cell structure in petals. Bot. J. Linn. Soc. 1981, 83, 57–83. [Google Scholar] [CrossRef]
- Delgado-Vargas, F.; Jiménez, A.R.; Paredes-López, O. Natural pigments: Carotenoids, anthocyanins, and betalains–characteristics, biosynthesis, processing, and stability. Crit. Rev. Food Sci. Nutr. 2000, 40, 173–289. [Google Scholar] [CrossRef]
- Yan, H.; Pei, X.; Zhang, H.; Li, X.; Zhang, X.; Zhao, M.; Chiang, V.L.; Sederoff, R.R.; Zhao, X. MYB-mediated regulation of anthocyanin biosynthesis. Int. J. Mol. Sci. 2021, 22, 3103. [Google Scholar] [CrossRef]
- Alexandersson, R.; Johnson, S.D. Pollinator-mediated selection on flower-tube length in a hawkmoth-pollinated Gladiolus (Iridaceae). Proc. Biol. Sci. 2002, 269, 631–636. [Google Scholar] [CrossRef]
- Sunil, L.; Shetty, N.P. Biosynthesis and regulation of anthocyanin pathway genes. Appl. Microbiol. Biotechnol. 2022, 106, 1783–1798. [Google Scholar] [CrossRef]
- Hong, J.; Yang, L.; Zhang, D.; Shi, J. Plant metabolomics: An indispensable system biology tool for plant science. Int. J. Mol. Sci. 2016, 17, 767. [Google Scholar] [CrossRef]
- Li-Beisson, Y.; Hirai, M.Y.; Nakamura, Y. Plant metabolomics. J. Exp. Bot. 2024, 75, 1651–1653. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, N.; Chen, W.; Zhang, W.; Zhang, H.; Yu, H.; Yi, Y. Integrated metabolomics and transcriptomics reveal metabolites difference between wild and cultivated Ophiocordyceps sinensis. Food Res. Int. 2023, 163, 112275. [Google Scholar] [CrossRef]
- Xu, C.; Gui, Z.; Huang, Y.; Yang, H.; Luo, J.; Zeng, X. Integrated transcriptomics and metabolomics analyses provide insights into Qingke in response to cold stress. J. Agric. Food Chem. 2023, 71, 18345–18358. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Gou, N.; Chen, C.; Huang, M.; Zhang, Y.; Bai, H.; Li, H.; Wang, L.; Wuyun, T. Transcriptome and metabolome analyses reveal sugar and acid accumulation during Apricot fruit development. Int. J. Mol. Sci. 2023, 24, 16992. [Google Scholar] [CrossRef]
- Liang, J.; Zhang, G.; Song, Y.; He, C.; Zhang, J. Targeted metabolome and transcriptome analyses reveal the pigmentation mechanism of Hippophae (Sea Buckthorn) Fruit. Foods 2022, 11, 3278. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.; Lin, M.; Ali, M.M.; Zheng, Y.; Yi, X.; Wang, S.; Chen, F.; Lin, Z. LhANS-rr1, LhDFR, and LhMYB114 regulate anthocyanin biosynthesis in flower buds of Lilium ‘Siberia’. Genes 2023, 14, 559. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wu, K.; Li, L.; Ma, G.; Fang, L.; Zeng, S. Identification of HpMYB1 inducing anthocyanin accumulation in Hippeastrum hybridum tepals by RNA-seq. BMC Plant Biol. 2023, 23, 594. [Google Scholar] [CrossRef] [PubMed]
- Anwar, M.; Yu, W.; Yao, H.; Zhou, P.; Allan, A.C.; Zeng, L. NtMYB3, an R2R3-MYB from Narcissus, regulates flavonoid biosynthesis. Int. J. Mol. Sci. 2019, 20, 5456. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, J.; Tian, S.; Hao, W.; Du, L. Two B-Box proteins, MaBBX20 and MaBBX51, coordinate light-induced anthocyanin biosynthesis in grape Hyacinth. Int. J. Mol. Sci. 2022, 23, 5678. [Google Scholar] [CrossRef]
- Koyama, T.; Sato, F. The function of ETHYLENE RESPONSE FACTOR genes in the light-induced anthocyanin production of Arabidopsis thaliana leaves. Plant Biotechnol. 2018, 35, 87–91. [Google Scholar] [CrossRef]
- Salas, E.; Dueñas, M.; Schwarz, M.; Winterhalter, P.; Cheynier, V.; Fulcrand, H. Characterization of pigments from different high speed countercurrent chromatography wine fractions. J. Agric. Food Chem. 2005, 53, 4536–4546. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Lim, S.H.; Kim, D.H.; Lee, J.Y. RsTTG1, a WD40 protein, interacts with the bHLH transcription factor RsTT8 to regulate anthocyanin and proanthocyanidin biosynthesis in Raphanus sativus. Int. J. Mol. Sci. 2022, 23, 11973. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wu, Y.; Li, J.; Wang, X.; Zeng, Z.; Xu, J.; Liu, Y.; Feng, J.; Chen, H.; He, Y.; et al. TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol. Plant 2023, 16, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Parrish, S.B.; Paudel, D.; Deng, Z. Transcriptome analysis of Lantana camara flower petals reveals candidate anthocyanin biosynthesis genes mediating red flower color development. G3 Genes Genomes Genet. 2023, 14, jkad259. [Google Scholar] [CrossRef]
- Ohmiya, A.; Hirashima, M.; Yagi, M.; Tanase, K.; Yamamizo, C. Identification of genes associated with chlorophyll accumulation in flower petals. PLoS ONE 2014, 9, e113738. [Google Scholar] [CrossRef]
- Iglesias-Sanchez, A.; Navarro-Carcelen, J.; Morelli, L.; Rodriguez-Concepcion, M. Arabidopsis FIBRILLIN6 influences carotenoid biosynthesis by directly promoting phytoene synthase activity. Plant Physiol. 2024, 194, 1662–1673. [Google Scholar] [CrossRef]
- van der Kooi, C.J.; Stavenga, D.G. Vividly coloured poppy flowers due to dense pigmentation and strong scattering in thin petals. J. Comp. Physiol. A Neuroethol. Sens. Neural Behav. Physiol. 2019, 205, 363–372. [Google Scholar] [CrossRef]
- de Moraes Pontes, J.G.; da Silva Pinheiro, M.S.; Fill, T.P. Unveiling chemical interactions between plants and fungi using metabolomics approaches. Adv. Exp. Med. Biol. 2023, 1439, 1–20. [Google Scholar]
- Hrdlickova, R.; Toloue, M.; Tian, B. RNA-Seq methods for transcriptome analysis. Wiley Interdiscip. Rev. RNA 2017, 8, e1364. [Google Scholar] [CrossRef]
- Singh, K.S.; van der Hooft, J.J.J.; van Wees, S.C.M.; Medema, M.H. Integrative omics approaches for biosynthetic pathway discovery in plants. Nat. Prod. Rep. 2022, 39, 1876–1896. [Google Scholar] [CrossRef]
- Sharma, S.; Kumar, A.; Singh, D.; Kumari, A.; Kapoor, P.; Kaur, S.; Shreon, B.; Garg, M. Integrated transcriptional and metabolomics signature pattern of pigmented wheat to insight the seed pigmentation and other associated features. Plant Physiol. Biochem. 2022, 189, 59–70. [Google Scholar] [CrossRef]
- Guo, X.; Shakeel, M.; Wang, D.; Qu, C.; Yang, S.; Ahmad, S.; Song, Z. Metabolome and transcriptome profiling unveil the mechanisms of light-induced anthocyanin synthesis in rabbiteye blueberry (vaccinium ashei: Reade). BMC Plant Biol. 2022, 22, 223. [Google Scholar] [CrossRef]
- Rodríguez-Lorenzo, M.; Mauri, N.; Royo, C.; Rambla, J.L.; Diretto, G.; Demurtas, O.; Hilbert, G.; Renaud, C.; Tobar, V.; Huete, J.; et al. The flavour of grape colour: Anthocyanin content tunes aroma precursor composition by altering the berry microenvironment. J. Exp. Bot. 2023, 74, 6369–6390. [Google Scholar] [CrossRef] [PubMed]
- Naik, J.; Rajput, R.; Pucker, B.; Stracke, R.; Pandey, A. The R2R3-MYB transcription factor MtMYB134 orchestrates flavonol biosynthesis in Medicago truncatula. Plant Mol. Biol. 2021, 106, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Jia, N.; Wang, J.J.; Liu, J.; Jiang, J.; Sun, J.; Yan, P.; Sun, Y.; Wan, P.; Ye, W.; Fan, B. DcTT8, a bHLH transcription factor, regulates anthocyanin biosynthesis in Dendrobium candidum. Plant Physiol. Biochem. 2021, 162, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Alabd, A.; Ahmad, M.; Zhang, X.; Gao, Y.; Peng, L.; Zhang, L.; Ni, J.; Bai, S.; Teng, Y. Light-responsive transcription factor PpWRKY44 induces anthocyanin accumulation by regulating PpMYB10 expression in pear. Hortic Res. 2022, 9, uhac199. [Google Scholar] [CrossRef]
- Han, H.; Dong, L.; Zhang, W.; Liao, Y.; Wang, L.; Wang, Q.; Ye, J.; Xu, F. Ginkgo biloba GbbZIP08 transcription factor is involved in the regulation of flavonoid biosynthesis. J. Plant Physiol. 2023, 287, 154054. [Google Scholar] [CrossRef]
- Plunkett, B.J.; Henry-Kirk, R.; Friend, A.; Diack, R.; Helbig, S.; Mouhu, K.; Tomes, S.; Dare, A.P.; Espley, R.V.; Putterill, J.; et al. Apple B-box factors regulate light-responsive anthocyanin biosynthesis genes. Sci. Rep. 2019, 9, 17762. [Google Scholar] [CrossRef]
- Jaakola, L.; Poole, M.; Jones, M.O.; Kämäräinen-Karppinen, T.; Koskimäki, J.J.; Hohtola, A.; Häggman, H.; Fraser, P.D.; Manning, K.; King, G.J.; et al. A SQUAMOSA MADS box gene involved in the regulation of anthocyanin accumulation in bilberry fruits. Plant Physiol. 2010, 153, 1619–1629. [Google Scholar] [CrossRef]
- Jiang, W.; Xia, Y.; Su, X.; Pang, Y. ARF2 positively regulates flavonols and proanthocyanidins biosynthesis in Arabidopsis thaliana. Planta 2022, 256, 44. [Google Scholar] [CrossRef]
- Wan, H.; Liu, Y.; Wang, T.; Jiang, P.; Wen, W.; Nie, J. Combined transcriptomic and metabolomic analyses identifies CsERF003, a citrus ERF transcription factor, as flavonoid activator. Plant Sci. 2023, 334, 111762. [Google Scholar] [CrossRef]
- Mahmood, K.; Xu, Z.; El-Kereamy, A.; Casaretto, J.A.; Rothstein, S.J. The Arabidopsis transcription factor ANAC032 represses anthocyanin biosynthesis in response to high sucrose and oxidative and abiotic stresses. Front. Plant Sci. 2016, 7, 1548. [Google Scholar] [CrossRef]
- Wei, Z.Z.; Hu, K.D.; Zhao, D.L.; Tang, J.; Huang, Z.Q.; Jin, P.; Li, Y.H.; Han, Z.; Hu, L.Y.; Yao, G.F.; et al. MYB44 competitively inhibits the formation of the MYB340-bHLH2-NAC56 complex to regulate anthocyanin biosynthesis in purple-fleshed sweet potato. BMC Plant Biol. 2020, 20, 258. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, W.; Dou, Y.; Zhang, J.; Jiang, G.; Miao, L.; Han, G.; Liu, Y.; Li, H.; Zhang, Z. Transcript quantification by RNA-Seq reveals differentially expressed genes in the red and yellow fruits of Fragaria vesca. PLoS ONE 2015, 10, e0144356. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Jeon, S.J.; Yanders, S.; Park, S.C.; Kim, H.S.; Kim, S. MYB3 plays an important role in lignin and anthocyanin biosynthesis under salt stress condition in Arabidopsis. Plant Cell Rep. 2022, 41, 1549–1560. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Qu, Y.; Sha, G.; Zhang, S.; Ma, Y.; Chen, M.; Zhai, R.; Yang, C.; Xu, L.; Wang, Z. PbWRKY75 promotes anthocyanin synthesis by activating PbDFR, PbUFGT, and PbMYB10b in pear. Physiol. Plant 2021, 173, 1841–1849. [Google Scholar] [CrossRef]
- Zuluaga, D.L.; Gonzali, S.; Loreti, E.; Pucciariello, C.; Degl’Innocenti, E.; Guidi, L.; Alpi, A.; Perata, P. Arabidopsis thaliana MYB75/PAP1 transcription factor induces anthocyanin production in transgenic tomato plants. Funct. Plant Biol. 2008, 35, 606–618. [Google Scholar] [CrossRef]
- Zhang, B.; Zhu, Z.Z.; Qu, D.; Wang, B.C.; Hao, N.N.; Yang, Y.Z.; Yang, H.J.; Zhao, Z.Y. MdBBX21, a B-Box protein, positively regulates light-induced anthocyanin accumulation in apple peel. Front. Plant Sci. 2021, 12, 774446. [Google Scholar] [CrossRef]
- Ma, H.; Yang, T.; Li, Y.; Zhang, J.; Wu, T.; Song, T.; Yao, Y.; Tian, J. The long noncoding RNA MdLNC499 bridges MdWRKY1 and MdERF109 function to regulate early-stage light-induced anthocyanin accumulation in apple fruit. Plant Cell 2021, 33, 3309–3330. [Google Scholar] [CrossRef]
- Mo, R.; Han, G.; Zhu, Z.; Essemine, J.; Dong, Z.; Li, Y.; Deng, W.; Qu, M.; Zhang, C.; Yu, C. The ethylene response factor ERF5 regulates anthocyanin biosynthesis in ‘Zijin’ mulberry fruits by interacting with MYBA and F3H genes. Int. J. Mol. Sci. 2022, 23, 7615. [Google Scholar] [CrossRef]
- Wang, Z.; Luo, Z.; Liu, Y.; Li, Z.; Liu, P.; Bai, G.; Zhou, Z.; Xie, H.; Yang, J. Molecular cloning and functional characterization of NtWRKY11b in promoting the biosynthesis of flavonols in Nicotiana tabacum. Plant Sci. 2021, 304, 110799. [Google Scholar] [CrossRef]
- Busche, M.; Pucker, B.; Weisshaar, B.; Stracke, R. Three R2R3-MYB transcription factors from banana (Musa acuminata) activate structural anthocyanin biosynthesis genes as part of an MBW complex. BMC Res. Notes 2023, 16, 103. [Google Scholar] [CrossRef]
- Moglia, A.; Florio, F.E.; Iacopino, S.; Guerrieri, A.; Milani, A.M.; Comino, C.; Barchi, L.; Marengo, A.; Cagliero, C.; Rubiolo, P.; et al. Identification of a new R3 MYB type repressor and functional characterization of the members of the MBW transcriptional complex involved in anthocyanin biosynthesis in eggplant (S. melongena L.). PLoS ONE 2020, 15, e0232986. [Google Scholar]
- Lloyd, A.; Brockman, A.; Aguirre, L.; Campbell, A.; Bean, A.; Cantero, A.; Gonzalez, A. Advances in the MYB-bHLH-WD repeat (MBW) pigment regulatory model: Addition of a WRKY factor and co-option of an anthocyanin MYB for betalain regulation. Plant Cell Physiol. 2017, 58, 1431–1441. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, P.; Xing, C.; Ye, J.; Xue, J.; Mur, L.A.J.; Di, B.; Hu, Z.; Chen, G.; Zhang, X.; Chen, X. Molecular Elucidation of Anthocyanin Accumulation Mechanisms in Hippeastrum hybridum Cultivars. Agronomy 2025, 15, 1722. https://doi.org/10.3390/agronomy15071722
Guo P, Xing C, Ye J, Xue J, Mur LAJ, Di B, Hu Z, Chen G, Zhang X, Chen X. Molecular Elucidation of Anthocyanin Accumulation Mechanisms in Hippeastrum hybridum Cultivars. Agronomy. 2025; 15(7):1722. https://doi.org/10.3390/agronomy15071722
Chicago/Turabian StyleGuo, Pengyu, Chuanji Xing, Jiacheng Ye, Jing Xue, Luis A. J. Mur, Bao Di, Zongli Hu, Guoping Chen, Xiuhai Zhang, and Xuqing Chen. 2025. "Molecular Elucidation of Anthocyanin Accumulation Mechanisms in Hippeastrum hybridum Cultivars" Agronomy 15, no. 7: 1722. https://doi.org/10.3390/agronomy15071722
APA StyleGuo, P., Xing, C., Ye, J., Xue, J., Mur, L. A. J., Di, B., Hu, Z., Chen, G., Zhang, X., & Chen, X. (2025). Molecular Elucidation of Anthocyanin Accumulation Mechanisms in Hippeastrum hybridum Cultivars. Agronomy, 15(7), 1722. https://doi.org/10.3390/agronomy15071722