
Comprehensive Identification of Main, Environment Interaction and Epistasis Quantitative Trait Nucleotides for 100-Seed Weight in Soybean (Glycine max (L.) Merr.)


 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Phenotypic Data
2.3. Genotypic Data
2.4. Population Structure Analysis
2.5. Phenotypic Data Analysis and Heritability Estimation
2.6. Genome Wide Association Analysis of Soybean HSW
- (i)
- Three kinds of association analyses were included: single environment association analysis for significant main-effect QTNs; multi-environment dataset joint analysis [19] for stable main effect QTNs and QEIs (QTN-by-environment interactions); and epistatic analysis can obtain main effect QTNs and QQIs (QTN-QTN interaction).
- (ii)
- Phenotypic data used in Single environment association analysis: E1, E2 and E3 was three single environments. Mean-phenotype was mean of 3 replications of each single environment. BLUP-phenotype was BLUP (best linear unbiased value) of 3 replications of each single environment.
- (iii)
- Phenotypic data used in multi-environment dataset joint analysis: Mean-phenotype of three environment E1, E2 and E3 were combined together for association analysis. BLUP-phenotype were processed in the same way as the Mean-phenotype.
- (iv)
- Phenotypic data used in epistatic analysis: The mean of the mean value of three environments were used as phenotypic data, namely, Mean-phenotype. BLUP-phenotype was BLUP from 3 replications of three environments.
- (v)
- stable locus: In this study, QTNs/QEIs/QQIs that appeared in at least twice in single-environment association analysis, multi-environment dataset joint analysis, and epistatic association analysis were considered as the stable locus [20] in Mean-phenotype. For BLUP-phenotype, the criterion for screening locus was the same.
2.7. Functional Annotation of Arabidopsis Homologous Genes
2.8. Differential Expression Analysis of Potential Candidate Genes in 14 Tissues
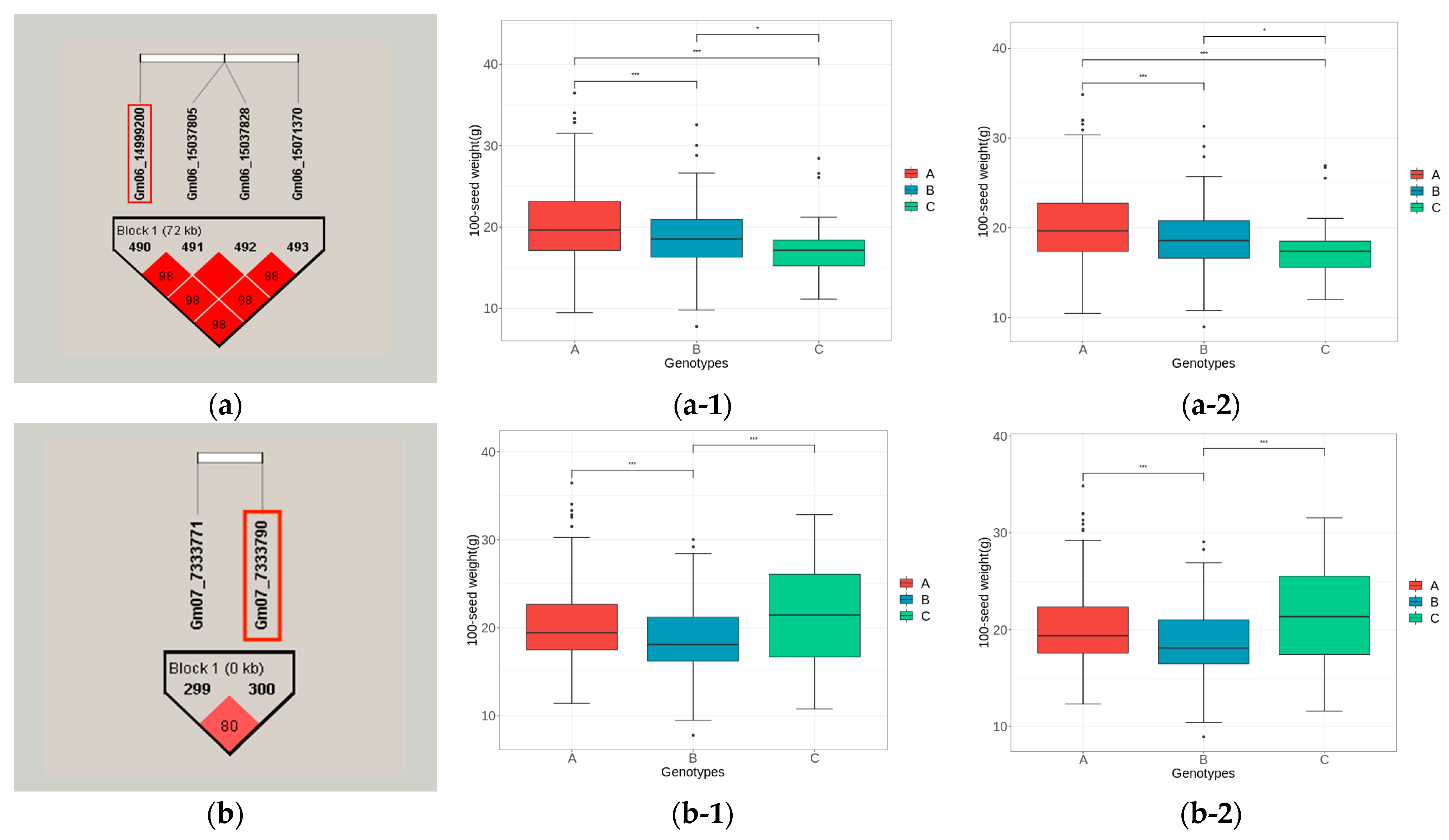
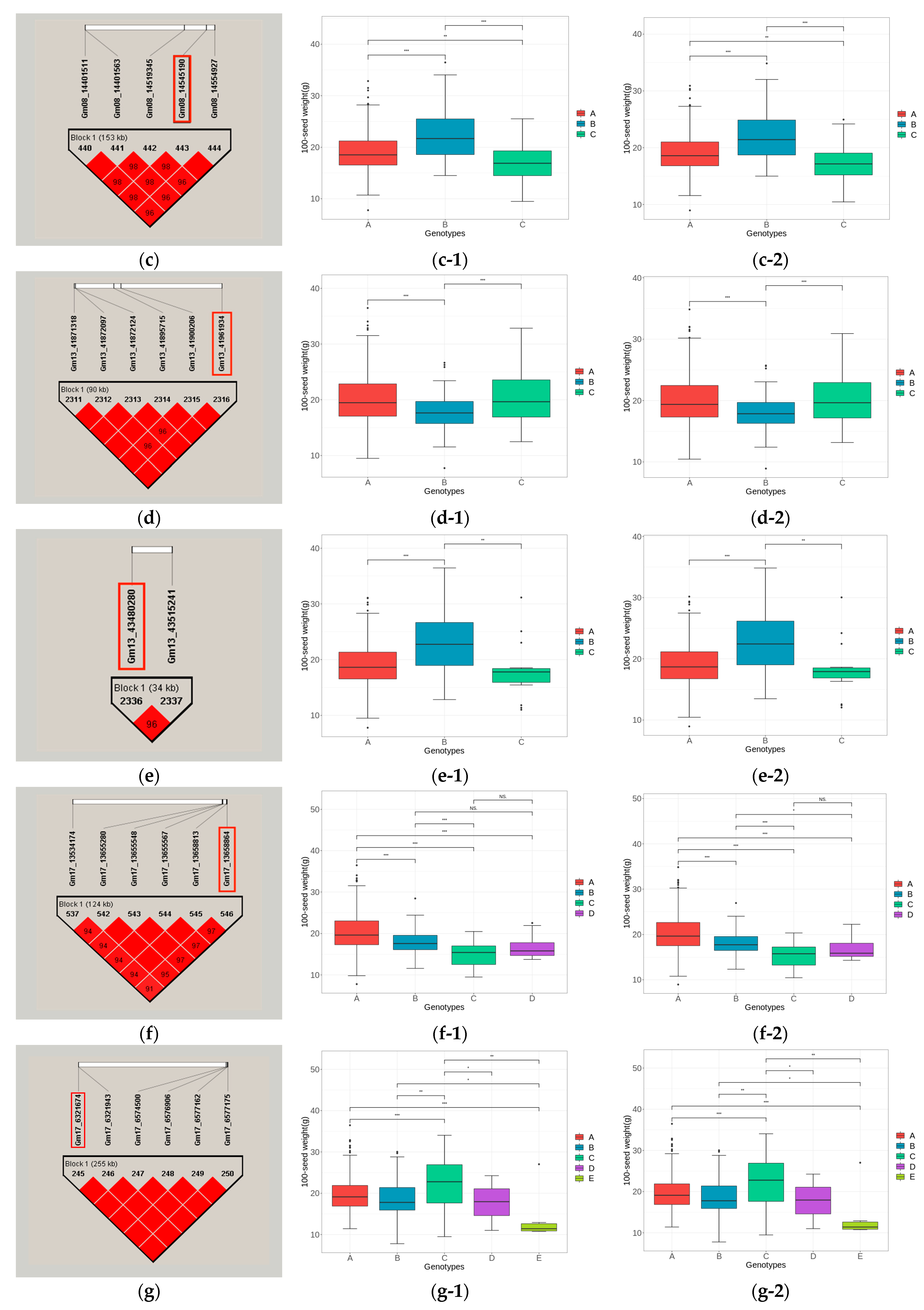
2.9. Haplotype Block Analysis and Phenotypic Difference Analysis of Potential Candidate Gene Loci
3. Results
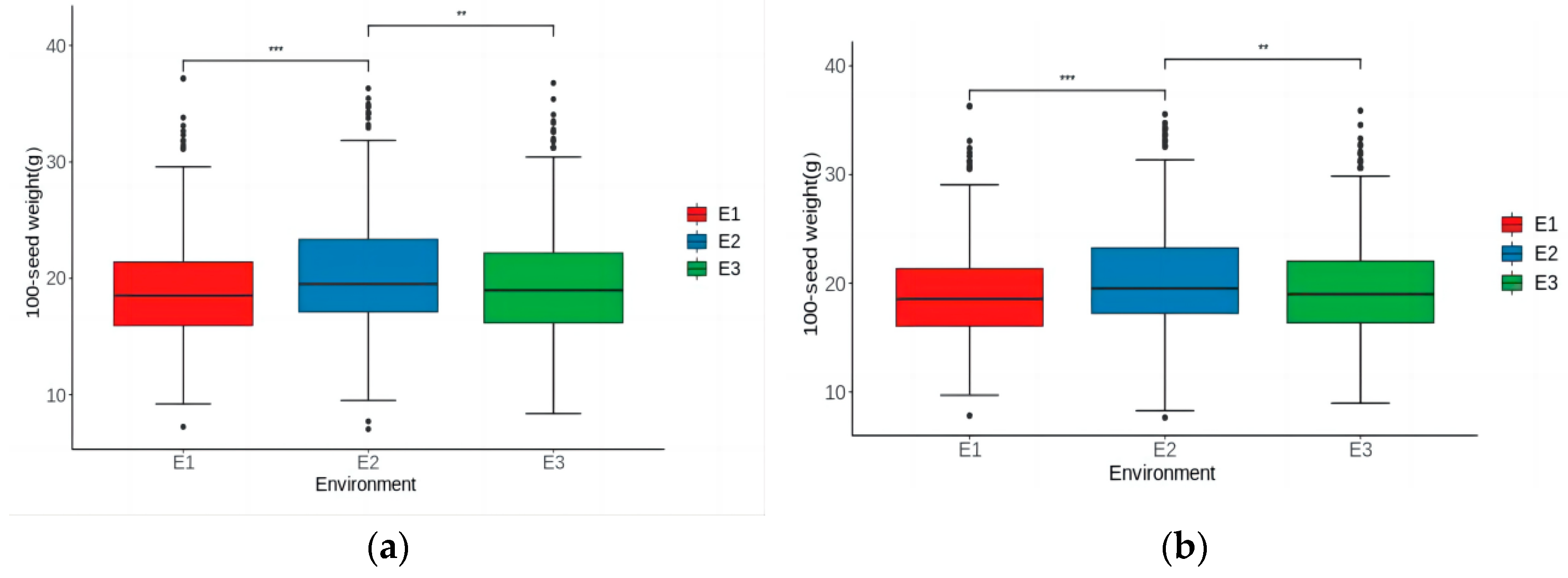
3.1. Phenotypic Analysis of 100-Seed Weight
3.2. Association Analysis of Mean-Phenotype for 100-Seed Weight
3.2.1. QTNs of Single Environment Analysis
3.2.2. QTNs and QEIs of Multi-Environment Dataset Joint Analysis
3.2.3. QTNs and QQIs of Epistasis Analysis
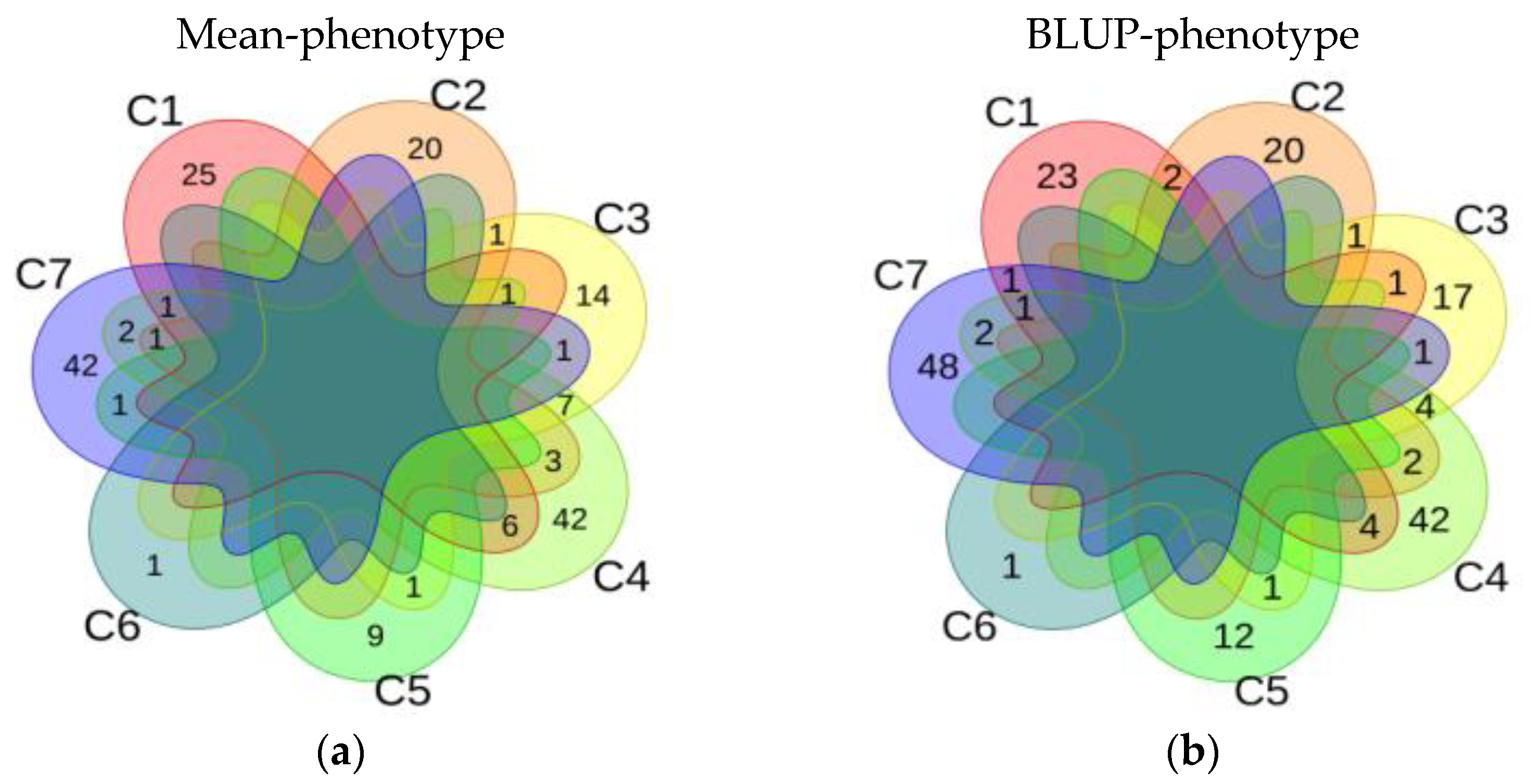
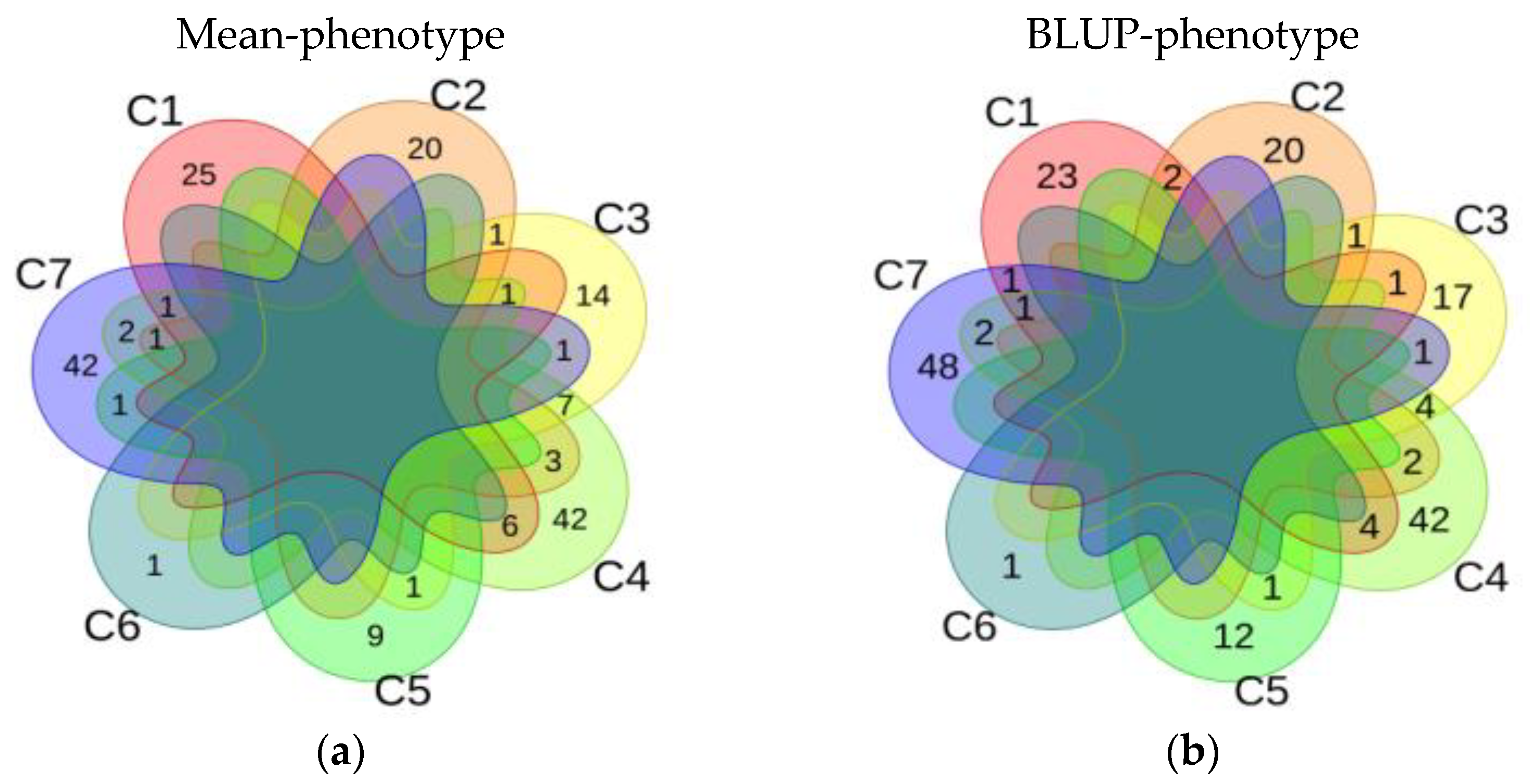
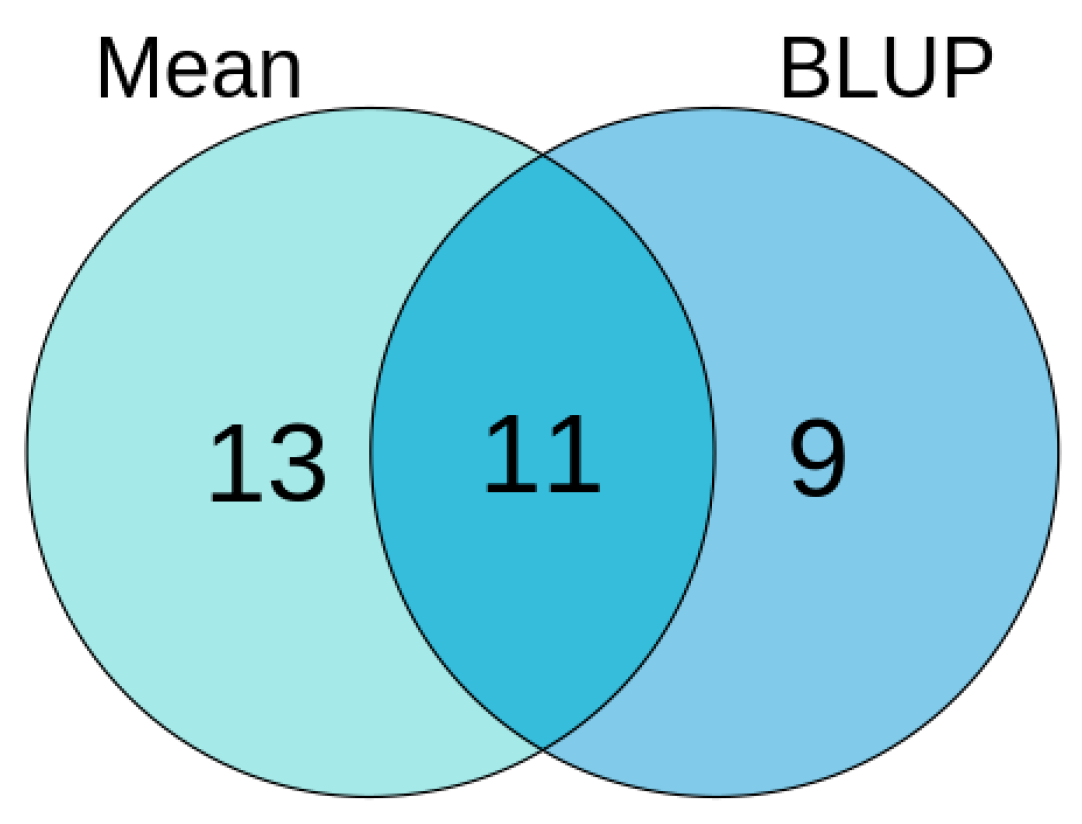
3.2.4. Stable QTNs of Single Environment, Multi-Environment and Epistasis Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| QTNs a | Chr b | LOD c | Effect d | R2 (%) e | Types of QTNs Detected f |
|---|---|---|---|---|---|
| Gm05_37024496 | 5 | 8.65, 12.41 | 0.70, 0.88 | 0.75, 1.13 | E2, E3 |
| Gm04_36981242 | 4 | 38.24, 104.26 | 1.23, 1.17 | 1.78, 1.41 | E1, Multi-environment QTN |
| Gm07_7333790 | 7 | 15.33, 52.04 | −0.74, −0.80 | 0.60, 0.60 | E1, Multi-environment QTN |
| Gm08_4692303 | 8 | 30.47, 57.58 | 1.08, 0.84 | 2.71, 1.45 | E1, Multi-environment QTN |
| Gm13_10282573 | 13 | 12.95, 22.97 | −0.68, −0.51 | 0.54, 0.27 | E1, Multi-environment QTN |
| Gm13_26750464 | 13 | 10.68, 7.65 | 0.61, 0.30 | 1.49, 0.31 | E1, Multi-environment QTN |
| Gm17_13658864 | 17 | 20.12, 7.96 | 0.86, 0.30 | 2.37, 0.26 | E1, Multi-environment QTN |
| Gm03_3724705 | 3 | 9.29–30.06 | 0.73, 0.60 | 1.17, 0.83 | E2, Multi-environment QTN |
| Gm07_22276117 | 7 | 11.28, 61.19 | 0.80, 0.87 | 0.72, 0.89 | E2, Multi-environment QTN |
| Gm16_15333418 | 16 | 18.57, 16.90 | −1.05, −0.44 | 0.89, 0.17 | E2, Multi-environment QTN |
| Gm02_5698517 | 2 | 4.28, 4.49 | 0.51, 0.23 | 1.09, 0.23 | E3, Multi-environment QTN |
| Gm08_14545190 | 8 | 6.49, 51.24 | −0.63, −0.79 | 1.25, 2.07 | E3, Multi-environment QTN |
| Gm09_4376323 | 9 | 8.04, 15.31 | 0.70, 0.42 | 2.05, 0.77 | E3, Multi-environment QTN |
| Gm10_3962423 | 10 | 12.92, 6.16 | 0.90, 0.27 | 1.80, 0.17 | E3, Multi-environment QTN |
| Gm14_40721910 | 14 | 11.56, 11.27 | 0.85, 0.36 | 1.62, 0.31 | E3, Multi-environment QTN |
| Gm18_496658 | 18 | 17, 10.96 | −1.04, −0.36 | 1.26, 0.16 | E3, Multi-environment QTN |
| Gm19_34854234 | 19 | 7.86, 7.99 | −0.69, −0.30 | 1.10, 0.21 | E3, Multi-environment QTN |
| Gm18_55491235 | 18 | 10.11, 6.11 | 0.79, 0.60 | 2.33, 0.35 | E3, epistasis QQI |
| Gm15_32270601 | 15 | 15.60, 19.91 | 1.0, 0.67 | 1.59, 1.05 | E3, Multi-environment QEI |
| Gm13_41961934 | 13 | 7.97, 5.07, 16.37 | 0.52, 0.56, 0.44 | 0.80, 0.73, 0.48 | E1, E3, Multi-environment QTN |
| Gm06_1271502 | 6 | 10.97, 15.80, 3.43 | 0.62, 0.42, 0.18 | 1.62, 0.68, 0.16 | E1, Multi-environment QTN, epistasis QQI |
| Gm11_10506624 | 11 | 15.32, 12.14, 4.58 | 0.94, 0.38, 0.41 | 2.93, 0.49, 0.87 | E2, Multi-environment QTN, epistasis QQI |
| Gm01_47969266 | 1 | 18.36, 4.33 | 0.46, 0.94 | 0.46, 3.69 | Multi-environment QTN, epistasis QQI |
| Gm02_4453462 | 2 | 5.34, 4.33 | 0.24, 0.94 | 0.18, 3.70 | Multi-environment QTN, epistasis QQI |
3.3. Association Analysis of BLUP-Phenotype of 100-Seed Weight
3.3.1. QTNs of Single Environment Analysis
3.3.2. QTNs and QEIs of Multi-Environment Dataset Joint Analysis
3.3.3. QTNs and QQIs of Epistasis Analysis
3.3.4. Stable QTNs of Single Environment, Multi-Environment and Epistasis Analysis
| QTNs a | Chr b | LOD c | Effect d | R2 (%) e | Test Results f |
|---|---|---|---|---|---|
| Gm04_5395873 | 4 | 18.74, 6.99 | −0.82, −0.60 | 1.87, 0.81 | E1, E2 |
| Gm03_3724705 | 3 | 10.10, 9.30 | 0.60, 0.70 | 1.03, 1.17 | E1, E2 |
| Gm05_37024496 | 5 | 8.43, 11.85 | 0.66, 0.81 | 0.73, 1.07 | E2, E3 |
| Gm14_9347269 | 14 | 24.28, 35.47 | −0.94, −1.48 | 1.18, 2.37 | E1, E3 |
| Gm06_48581982 | 6 | 10.77, 16.44 | −0.61, −0.43 | 1.99, 0.85 | E1, Multi-environment QTN |
| Gm07_7333790 | 7 | 17.86, 68.04 | −0.79, −0.90 | 0.78, 0.86 | E1, Multi-environment QTN |
| Gm13_43480280 | 13 | 12.02, 21.88 | −0.64, −0.49 | 1.30, 0.67 | E1, Multi-environment QTN |
| Gm20_34264812 | 20 | 6.39, 8.58 | 0.47, 0.31 | 1.04, 0.39 | E1, Multi-environment QTN |
| Gm19_8351766 | 19 | 13.69, 74.54 | 0.86, 0.96 | 0.66, 0.89 | E2, Multi-environment QTN |
| Gm06_13909376 | 6 | 8.35, 14.90 | −0.65, −0.41 | 1.97, 0.82 | E2, Multi-environment QTN |
| Gm08_14545190 | 8 | 7.04, 31.76 | −0.61, −0.60 | 1.35, 1.34 | E3, Multi-environment QTN |
| Gm09_4376323 | 9 | 8.07, 9.09 | 0.65, 0.32 | 2.05, 0.48 | E3, Multi-environment QTN |
| Gm10_3962423 | 10 | 12.86, 14.01 | 0.84, 0.39 | 1.79, 0.41 | E3, Multi-environment QTN |
| Gm19_34854234 | 19 | 7.88, 13.34 | −0.65, −0.38 | 1.10, 0.34 | E3, Multi-environment QTN |
| Gm15_32270601 | 15 | 15.34, 19.58 | 0.93, 0.66 | 1.56, 1.11 | E3, Multi-environment QEI |
| Gm14_1830770 | 14 | 10.2, 3.61 | −0.59, −0.51 | 1.20, 1.20 | E1, epistasis QQI |
| Gm18_55491235 | 18 | 10.72, 4.85 | 0.77, 0.22 | 2.47, 0.33 | E3, epistasis QQI |
| Gm02_4453462 | 2 | 4.93, 13.93, 4.47 | 0.40, 0.40, 0.59 | 0.62, 0.50, 1.86 | E1, Multi-environment QTN, epistasis QQI |
| Gm06_1271502 | 6 | 44.82, 5.09 | 0.72, 0.4 | 2.16, 0.99 | Multi-environment QTN, epistasis QQI |
| Gm15_47035163 | 15 | 26.52, 5.12 | 0.55, 0.53 | 1.41, 1.87 | Multi-environment QTN, epistasis QQI |
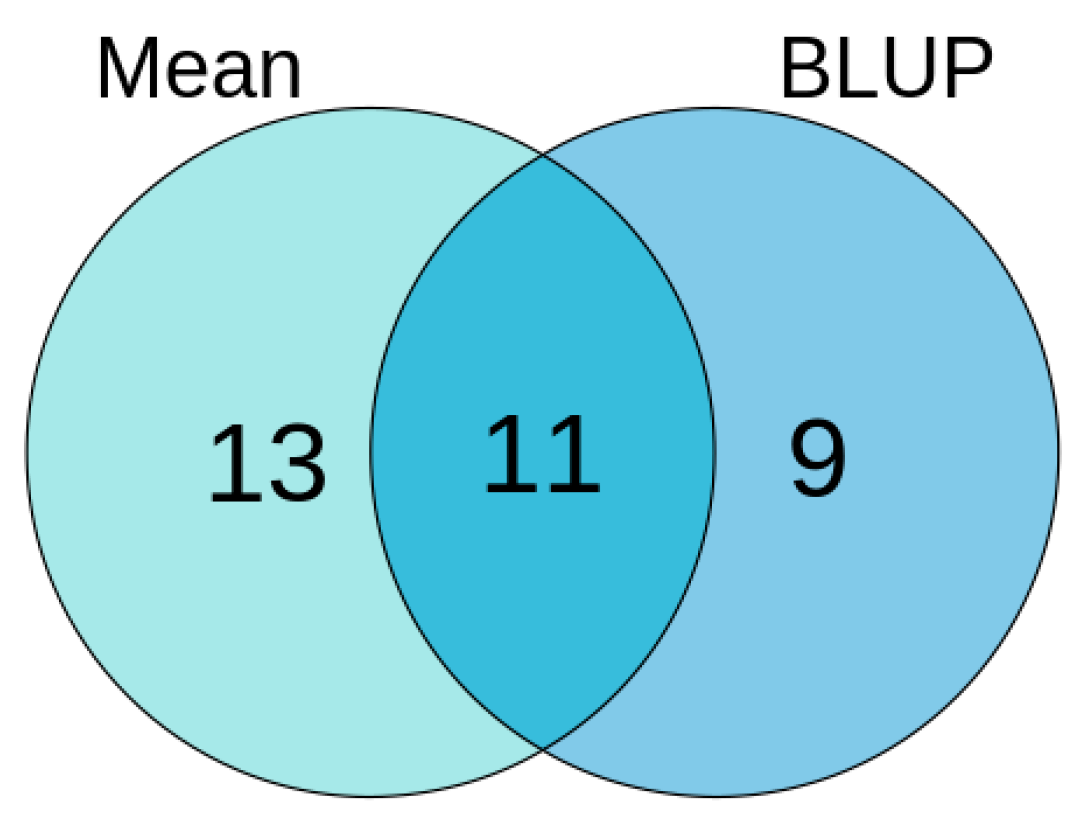
3.4. Comprehensive Results of Association Analysis of Mean-Phenotype and BLUP-Phenotype in Soybean HSW
3.5. Exploration and Analysis of Candidate Genes for Soybean 100-Seed Weight
3.5.1. Identification of Potential Candidate Genes through Functional Annotation of Homologous Genes in Arabidopsis
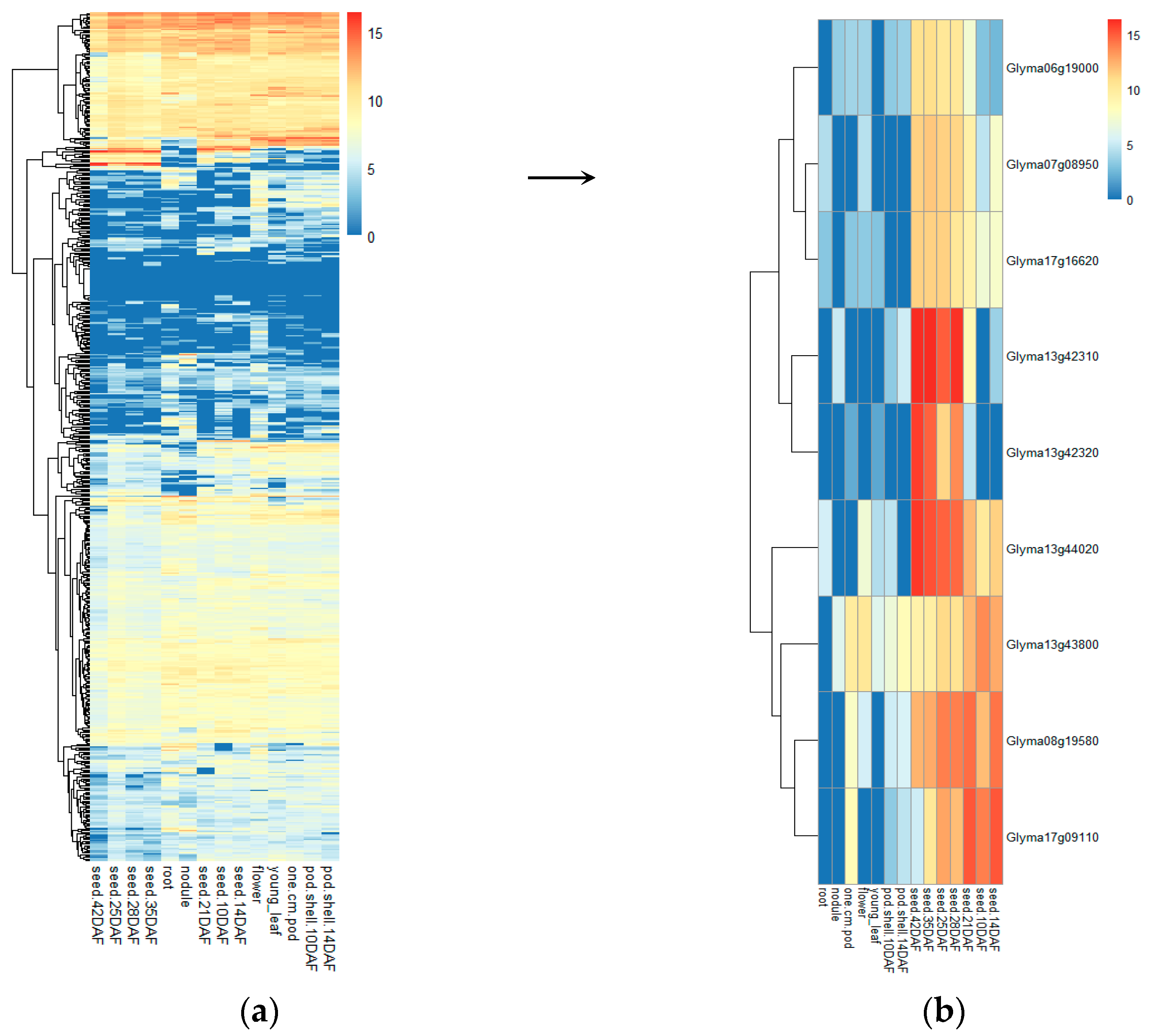
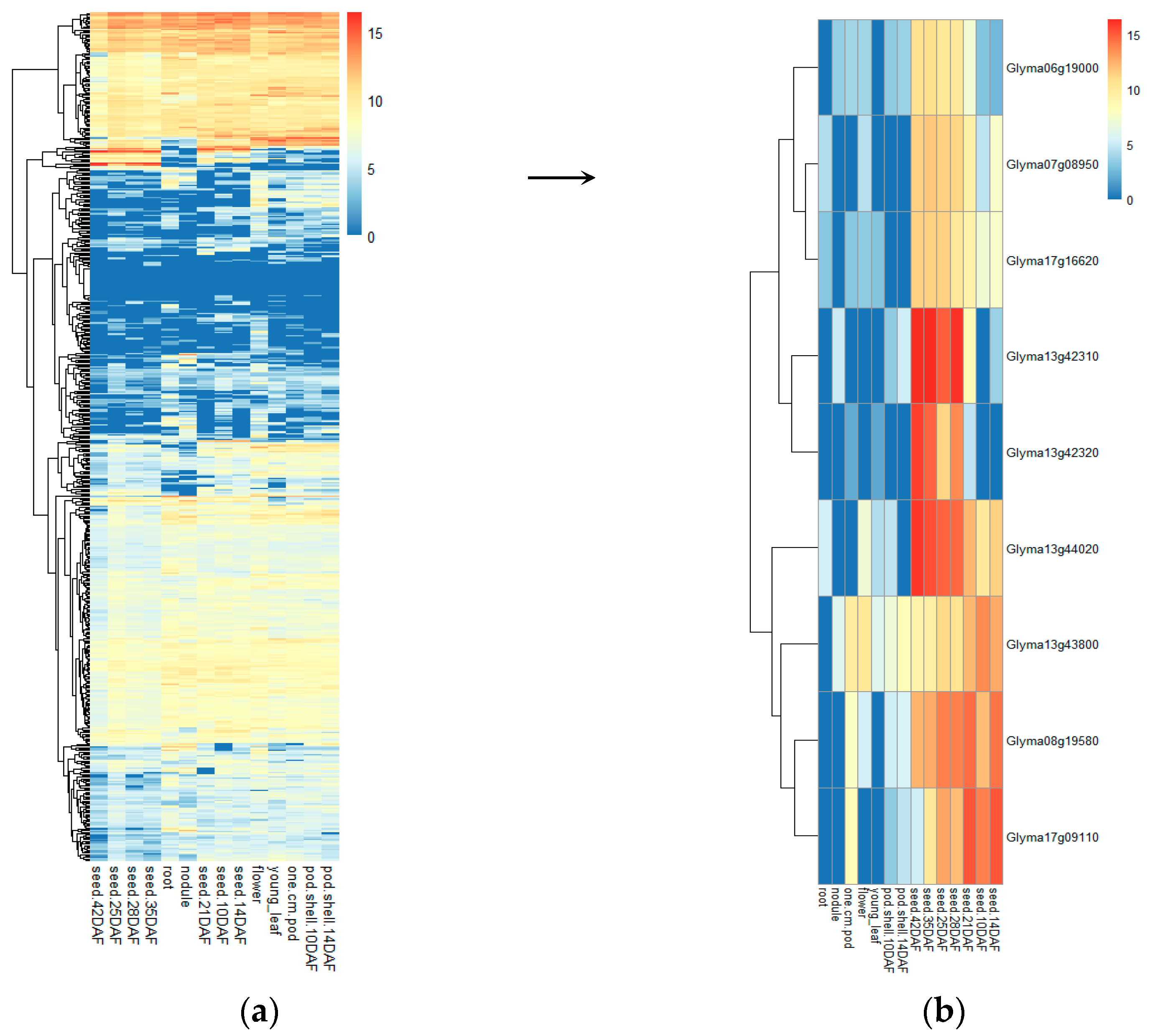
3.5.2. Identification of Candidate Genes through Differential Expression Analysis
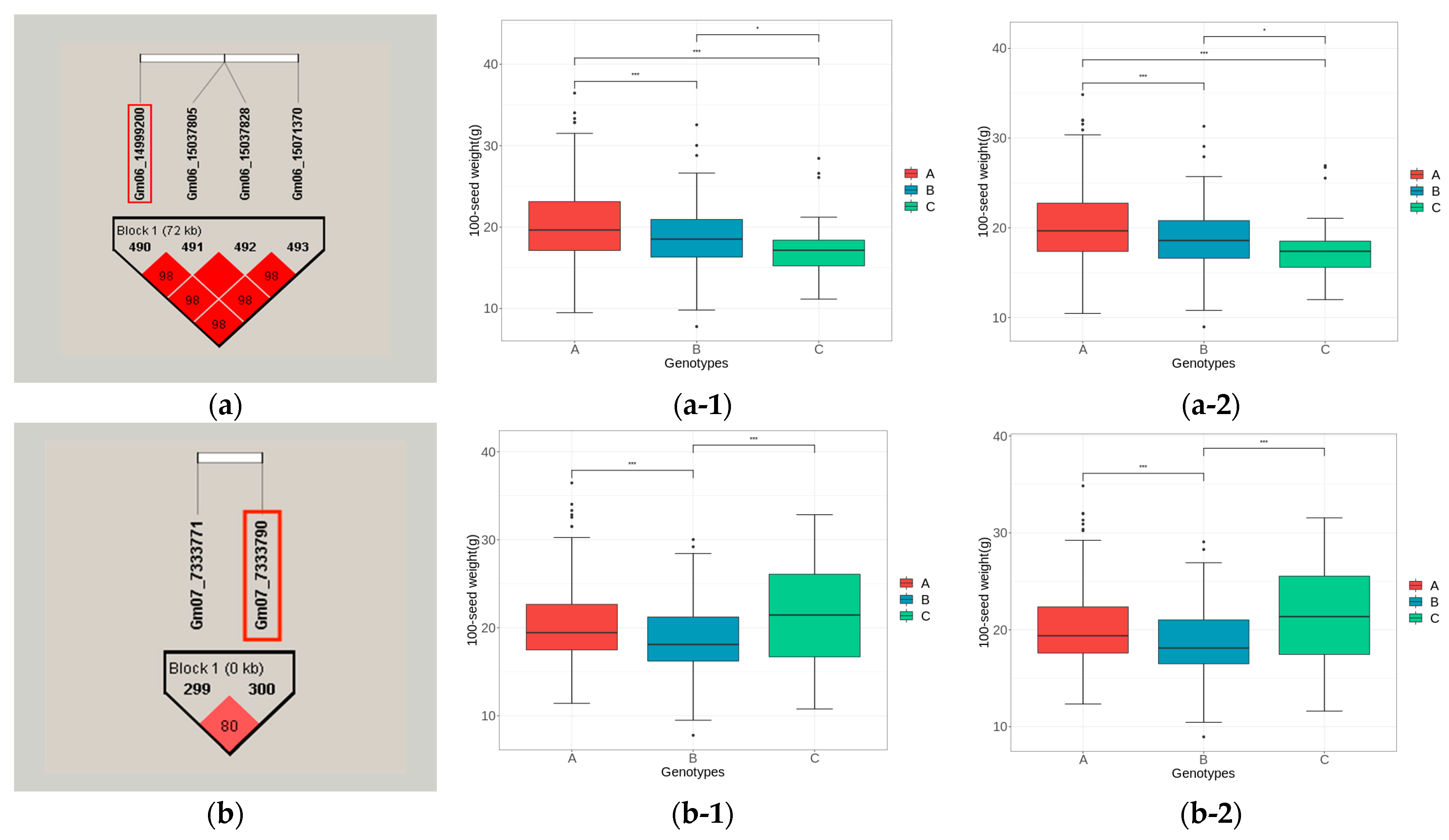
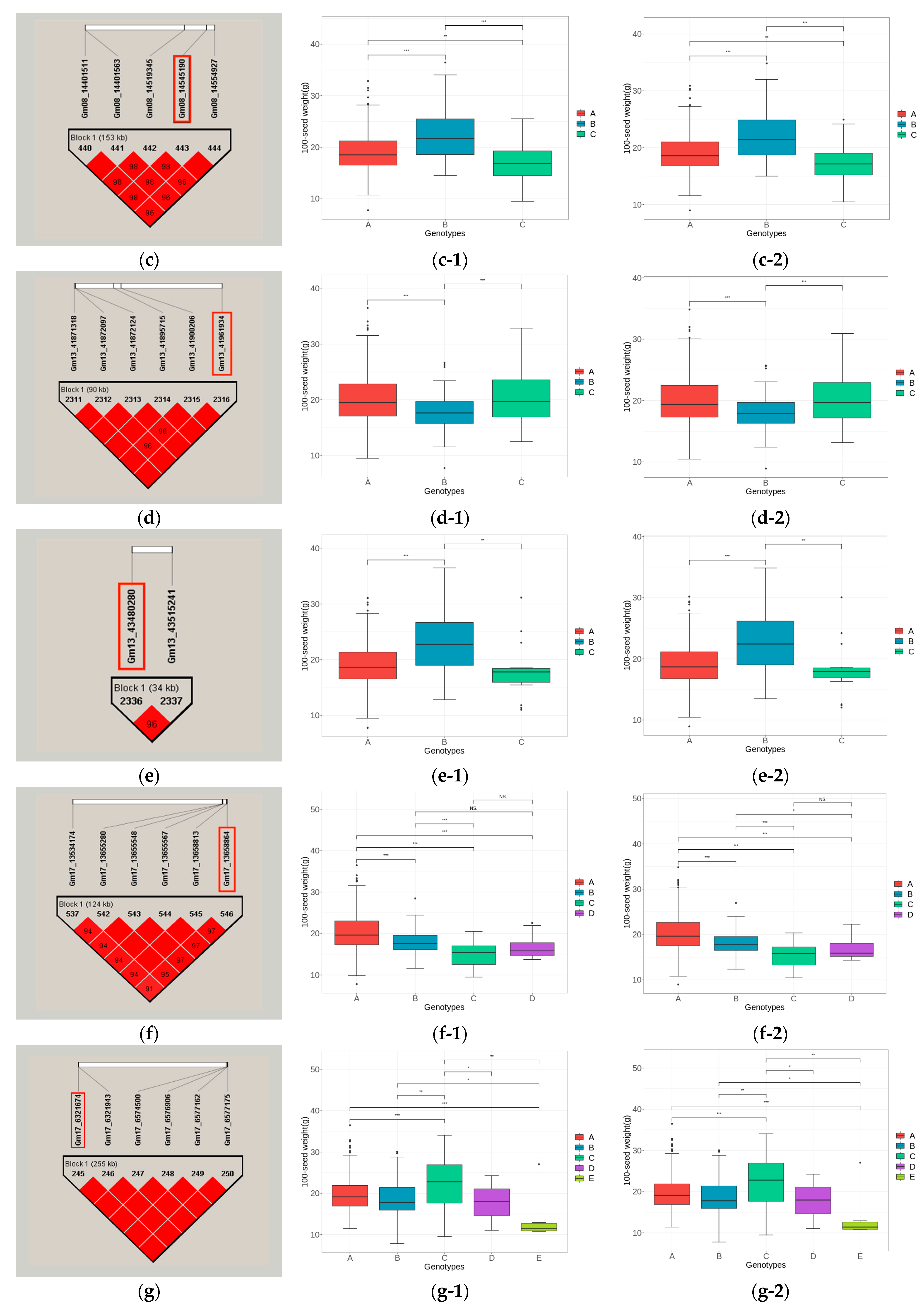
3.5.3. Haplotype Block Analysis of Candidate Genes
4. Discussion
4.1. Comparison between Stable QTNs Identified in This Study and Reported QTL
4.2. Detection Capability of the 3VmrMLM Method
4.3. Significance of QTN-Environment Interaction and QTN-QTN Interaction
4.4. Potential Candidate Genes Discovered by the Complementary Mapping Approaches
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Liu, S.L.; Zhang, M.; Feng, F.; Tian, Z.X. Toward a “Green Revolution” for Soybean. Mol. Plant 2020, 5, 688–697. [Google Scholar] [CrossRef]
- Vogel, J.T.; Liu, W.D.; Olhoft, P.; Crafts-Brandner, S.J.; Pennycooke, J.C.; Christiansen, N. Soybean Yield Formation Physiology—A Foundation for Precision Breeding Based Improvement. Front. Plant Sci. 2021, 12, 719706. [Google Scholar] [CrossRef]
- Orsi, C.H.; Tanksley, S.D. Natural Variation in an ABC Transporter Gene Associated with Seed Size Evolution in Tomato Species. PLoS Genet. 2009, 5, e1000347. [Google Scholar] [CrossRef]
- Keim, P.; Diers, B.W.; Olson, T.C.; Shoemaker, R.C. RFLP Mapping in Soybean: Association between Marker Loci and Variation in Quantitative Traits. Genetics 1990, 126, 735–742. [Google Scholar] [CrossRef]
- Qi, Z.Y.; Song, J.; Zhang, K.X.; Liu, S.L.; Tian, X.C.; Wang, Y.; Fang, Y.L.; Li, X.Y.; Wang, J.J.; Yang, C.; et al. Identification of QTNs Controlling 100-Seed Weight in Soybean Using Multilocus Genome-Wide Association Studies. Front. Gene 2020, 11, 689. [Google Scholar] [CrossRef]
- Zhang, W.; Xu, W.J.; Zhang, H.M.; Liu, X.Q.; Cui, X.Y.; Li, S.S.; Song, L.; Zhu, Y.L.; Chen, X.; Chen, H.T. Comparative Selective Signature Analysis and High-Resolution GWAS Reveal a New Candidate Gene Controlling Seed Weight in Soybean. Theor. Appl. Genet. 2021, 134, 1329–1341. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Xiong, Q.; Cheng, T.; Li, Q.T.; Liu, X.L.; Bi, Y.D.; Li, W.; Zhang, W.K.; Ma, B.; Lai, Y.C.; et al. A PP2C-1 Allele Underlying a Quantitative Trait Locus Enhances Soybean 100-Seed Weight. Mol. Plant 2017, 10, 670–684. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.Z.; Li, W.; Jiang, H.W.; Wang, Y.; Gao, H.H.; Liu, M.; Chen, Q.S.; Lai, Y.C.; He, C.Y. Differential Expression of a WRKY Gene between Wild and Cultivated Soybeans Correlates to Seed Size. J. Exp. Bot. 2017, 68, 2717–2729. [Google Scholar] [CrossRef]
- Lu, X.; Li, Q.T.; Xiong, Q.; Li, W.; Bi, Y.D.; Lai, Y.C.; Liu, X.L.; Man, W.Q.; Zhang, W.K.; Ma, B.; et al. The Transcriptomic Signature of Developing Soybean Seeds Reveals the Genetic Basis of Seed Trait Adaptation during Domestication. Plant J. 2016, 86, 530–544. [Google Scholar] [CrossRef]
- Yu, J.M.; Pressoir, G.; Briggs, W.H.; Bi, I.V.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S.; Nielsen, D.M.; Holland, J.B.; et al. A Unified Mixed-Model Method for Association Mapping That Accounts for Multiple Levels of Relatedness. Nat. Genet. 2006, 38, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of Population Structure Using Multilocus Genotype Data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Zhang, Z.W.; Ersoz, E.; Lai, C.Q.; Todhunter, R.J.; Tiwari, H.K.; Gore, M.A.; Bradbury, P.J.; Yu, J.M.; Arnett, D.K.; Ordovas, J.M.; et al. Mixed Linear Model Approach Adapted for Genome-Wide Association Studies. Nat. Genet. 2010, 42, 355–360. [Google Scholar] [CrossRef]
- Wang, S.B.; Feng, J.Y.; Ren, W.L.; Huang, B.; Zhou, L.; Wen, Y.J.; Zhang, J.; Dunwell, J.M.; Xu, S.Z.; Zhang, Y.M. Improving Power and Accuracy of Genome-Wide Association Studies via a Multi-Locus Mixed Linear Model Methodology. Sci. Rep. 2016, 6, 19444. [Google Scholar] [CrossRef]
- Ni, G.; Van Der Werf, J.; Zhou, X.; Hypponen, E.; Wray, N.R.; Lee, S.H. Genotype-Covariate Correlation and Interaction Disentangled by a Whole-Genome Multivariate Reaction Norm Model. Nat. Commun. 2019, 10, 2239. [Google Scholar] [CrossRef]
- Carlborg, Ö.; Haley, C.S. Epistasis: Too Often Neglected in Complex Trait Studies? Nat. Rev. Genet. 2004, 5, 618–625. [Google Scholar] [CrossRef]
- Li, X.; Guo, T.; Wang, J.; Bekele, W.A.; Sukumaran, S.; Vanous, A.E.; McNellie, J.P.; Cortes, L.T.; Lopes, M.S.; Lamkey, K.R.; et al. An Integrated Framework Reinstating the Environmental Dimension for GWAS and Genomic Selection in Crops. Mol. Plant 2021, 14, 874–887. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Y.; Lin, Y.; Chen, J.B.; Yan, Q.; Xue, C.C.; Wu, R.R.; Chen, X.; Yuan, X.X. Genome-Wide Association Studies Provide Genetic Insights into Natural Variation of Seed-Size-Related Traits in Mungbean. Front. Plant Sci. 2022, 13, 997988. [Google Scholar] [CrossRef] [PubMed]
- Kerin, M.; Marchini, J. Inferring Gene-by-Environment Interactions with a Bayesian Whole-Genome Regression Model. Am. J. Hum. Genet. 2020, 107, 698–713. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhang, Y.W.; Zhang, Z.C.; Xiang, Y.; Liu, M.H.; Zhou, Y.H.; Zuo, J.F.; Zhang, H.Q.; Chen, Y.; Zhang, Y.-M. A Compressed Variance Component Mixed Model for Detecting QTNs and QTN-by-Environment and QTN-by-QTN Interactions in Genome-Wide Association Studies. Mol. Plant 2022, 15, 630–650. [Google Scholar] [CrossRef] [PubMed]
- Karikari, B.; Wang, Z.L.; Zhou, Y.L.; Yan, W.L.; Feng, J.Y.; Zhao, T.J. Identification of Quantitative Trait Nucleotides and Candidate Genes for Soybean Seed Weight by Multiple Models of Genome-Wide Association Study. BMC Plant Biol. 2020, 20, 404. [Google Scholar] [CrossRef] [PubMed]
- Bates, D.M.; Maechler, M.; Bolker, B.M.; Walker, S. Lme4: Linear Mixed-Effects Models Using Eigen and S4. 2015. R Package. Available online: http://CRAN.R-Project.Org/Package=lme4 (accessed on 20 March 2023).
- Yi, X.; Liang, Y.; Huerta-Sanchez, E.; Jin, X.; Cuo, Z.X.P.; Pool, J.E.; Xu, X.; Jiang, H.; Vinckenbosch, N.; Korneliussen, T.S.; et al. Sequencing of 50 Human Exomes Reveals Adaptation to High Altitude. Science 2010, 329, 75–78. [Google Scholar] [CrossRef]
- Hubisz, M.J.; Falush, D.; Stephens, M.; Pritchard, J.K. Inferring Weak Population Structure with the Assistance of Sample Group Information. Mol. Ecol. Resour. 2009, 9, 1322–1332. [Google Scholar] [CrossRef] [PubMed]
- Pattee, H.E.; Giesbrecht, F.G.; Mozingo, R.W. A Note on Broad-Sense Heritability of Selected Sensory Descriptors in Virginia-Type Arachis hypogaea L. Peanut Sci. 1993, 20, 24–26. [Google Scholar] [CrossRef]
- Severin, A.J.; Woody, J.L.; Bolon, Y.; Joseph, B.; Diers, B.W.; Farmer, A.D.; Muehlbauer, G.J.; Nelson, R.T.; Grant, D.; Specht, J.E.; et al. RNA-Seq Atlas of Glycine Max: A Guide to the Soybean Transcriptome. BMC Plant Biol. 2010, 10, 160. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: Analysis and Visualization of LD and Haplotype Maps. Bioinformatics 2005, 21, 263–265. [Google Scholar] [CrossRef]
- Bertero, H.D.; Vega, A.J.D.L.; Correa, G.; Jacobsen, S.E.; Mujica, A. Genotype and Genotype-by-Environment Interaction Effects for Grain Yield and Grain Size of Quinoa (Chenopodium quinoa Willd.) as Revealed by Pattern Analysis of International Multi-Environment Trials. Field Crop. Res. 2004, 89, 299–318. [Google Scholar] [CrossRef]
- Hickey, L.T.; Hafeez, A.N.; Robinson, H.; Jackson, S.A.; Leal-Bertioli, S.C.M.; Tester, M.; Gao, C.X.; Godwin, I.D.; Hayes, B.J.; Wulff, B.B.H. Breeding Crops to Feed 10 Billion. Nat. Biotechnol. 2019, 37, 744–754. [Google Scholar] [CrossRef]
- Li, N.; Xu, R.; Li, Y.H. Molecular Networks of Seed Size Control in Plants. Annu. Rev. Plant Biol. 2019, 70, 435–463. [Google Scholar] [CrossRef]
- McCouch, S.R.; Chen, X.; Panaud, O.; Temnykh, S.; Xu, Y.; Yong, G.C.; Huang, N.; Ishii, T.; Blair, M. Microsatellite marker development, mapping and applications in rice genetics and breeding. Plant Mole. Biol. 1997, 35, 89–99. [Google Scholar] [CrossRef]
- Hu, Z.B.; Zhang, H.R.; Kan, G.Z.; Ma, D.Y.; Zhang, D.; Shi, G.X.; Hong, E.L.; Zhang, G.Z.; Yu, D.Y. Determination of the Genetic Architecture of Seed Size and Shape via Linkage and Association Analysis in Soybean (Glycine max L. Merr.). Genetica 2013, 141, 247–254. [Google Scholar] [CrossRef]
- Chen, Y.J.; Xiong, Y.J.; Hong, H.L.; Li, G.; Gao, J.; Guo, Q.Y.; Sun, R.J.; Ren, H.L.; Zhang, F.; Wang, J.; et al. Genetic Dissection and Genomic Selection for Seed Weight, Pod Length, and Pod Width in Soybean. Crop. J. 2023, 11, 832–841. [Google Scholar] [CrossRef]
- Elattar, M.A.; Karikari, B.; Li, S.G.; Song, S.Y.; Cao, Y.C.; Aslam, M.; Hina, A.; Abou-Elwafa, S.F.; Zhao, T.J. Identification and Validation of Major QTLs, Epistatic Interactions, and Candidate Genes for Soybean Seed Shape and Weight Using Two Related RIL Populations. Front. Genet. 2021, 12, 666440. [Google Scholar] [CrossRef]
- Mian, M.A.R.; Bailey, M.A.; Tamulonis, J.P.; Shipe, E.R.; Carter, T.E., Jr.; Parrott, W.A.; Ashley, D.A.; Hussey, R.S.; Boerma, H.R. Molecular Markers Associated with Seed Weight in Two Soybean Populations. Theor. Appl. Genet. 1996, 93, 1011–1016. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Y.W.; Jiao, Z.J.; Liu, P.P.; Chang, W. Identification of QTLs and Candidate Genes for 100-Seed Weight Trait Using PyBSASeq Algorithm in Soybean. Acta Agron. Sinica 2022, 48, 635–643. [Google Scholar] [CrossRef]
- Orf, J.H.; Chase, K.; Jarvik, T.; Mansur, L.M.; Cregan, P.B.; Adler, F.R.; Lark, K.G. Genetics of Soybean Agronomic Traits: I. Comparison of Three Related Recombinant Inbred Populations. Crop. Sci. 1999, 39, 1642–1651. [Google Scholar] [CrossRef]
- Djanaguiraman, M.; Prasad, P.V.V.; Schapaugh, W.T. High Day or Nighttime Temperature Alters Leaf Assimilation, Reproductive Success, and Phosphatidic Acid of Pollen Grain in Soybean [Glycine max (L.) Merr.]. Crop. Sci. 2013, 53, 1594–1604. [Google Scholar] [CrossRef]
- Vantoai, T.T.; Beuerlein, J.E.; Chmitthenner, A.F.; Stmartin, S.K. Genetic Variability for Flooding Tolerance in Soybeans. Crop. Sci. 1994, 34, 1112–1115. [Google Scholar] [CrossRef]
- Manavalan, L.P.; Guttikonda, S.K.; Phan Tran, L.S.; Nguyen, H.T. Physiological and Molecular Approaches to Improve Drought Resistance in Soybean. Plant Cell Physiol. 2009, 50, 1260–1276. [Google Scholar] [CrossRef] [PubMed]
- Manuck, S.B.; McCaffery, J.M. Gene-Environment Interaction. Annu. Rev. Psychol. 2014, 65, 41–70. [Google Scholar] [CrossRef]
- Holzinger, E.R.; Szymczak, S.; Dasgupta, A.; Malley, J.; Li, Q.; Bailey-Wilson, J.E. Variable Selection Method for the Identification of Epistatic Models. In Proceedings of the Pacific Symposium, Kohala Coast, HI, USA, 4–8 January 2015; pp. 195–206. [Google Scholar] [CrossRef]
- Sakai, T.; Abe, A.; Shimizu, M.; Terauchi, R. Ril-Step: Epistasis Analysis of Rice Recombinant Inbred Lines Reveals Candidate Interacting Genes That Control Seed Hull Color and Leaf Chlorophyll Content. G3-Genes Genom. Genet. 2021, 11, jkab130. [Google Scholar] [CrossRef]
- Kim, K.H.; Kim, J.Y.; Lim, W.J.; Jeong, S.; Lee, H.Y.; Cho, Y.; Moon, J.K.; Kim, N. Genome-Wide Association and Epistatic Interactions of Flowering Time in Soybean Cultivar. PLoS ONE 2020, 15, e0228114. [Google Scholar] [CrossRef] [PubMed]
- Medic, J.; Atkinson, C.; Hurburgh, C.R. Current Knowledge in Soybean Composition. J. Am. Oil Chem. Soc. 2014, 91, 363–384. [Google Scholar] [CrossRef]
- Zhuang, W.J.; Chen, H.; Yang, M.; Wang, J.P.; Pandey, M.K.; Zhang, C.; Chang, W.C.; Zhang, L.S.; Zhang, X.T.; Tang, R.H.; et al. The Genome of Cultivated Peanut Provides Insight into Legume Karyotypes, Polyploid Evolution and Crop Domestication. Nat. Genet. 2019, 51, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Liu, S.L.; Wang, Z.; Yuan, Y.Q.; Zhang, Z.F.; Liang, Q.J.; Yang, X.; Duan, Z.B.; Liu, Y.C.; Kong, F.J.; et al. Progress in Soybean Functional Genomics over the Past Decade. Plant Biotechnol. J. 2022, 20, 256–282. [Google Scholar] [CrossRef]
- Li, S.X.; Cong, Y.H.; Liu, Y.P.; Wang, T.T.; Shuai, Q.; Chen, N.N.; Gai, J.Y.; Li, Y. Optimization of Agrobacterium-Mediated Transformation in Soybean. Front. Plant Sci. 2017, 24, 246. [Google Scholar] [CrossRef]
- Liu, J.; Gunapati, S.; Mihelich, N.T.; Stec, A.O.; Michno, J.M.; Stupar, R.M. Genome Editing in Soybean with CRISPR/Cas9: Methods and Protocols. Method. Mol. Biol. 2019, 1917, 217–234. [Google Scholar] [CrossRef]
- Xiao, Y.T.; Karikari, B.; Wang, L.; Chang, F.G.; Zhao, T.J. Structure Characterization and Potential Role of Soybean Phospholipases A Multigene Family in Response to Multiple Abiotic Stress Uncovered by CRISPR/Cas9 Technology. Environ. Exp. Bot. 2021, 188, 104521. [Google Scholar] [CrossRef]
- Patil, G.; Valliyodan, B.; Deshmukh, R.; Prince, S.; Nicander, B.; Zhao, M.Z.; Sonah, H.; Song, L.; Lin, L.; Chaudhary, J. Soybean (Glycine Max) SWEET Gene Family: Insights through Comparative Genomics, Transcriptome Profiling and Whole Genome Re-Sequence Analysis. BMC Genom. 2015, 16, 520. [Google Scholar] [CrossRef]
- Timilsina, S.; Potnis, N.; Newberry, E.A.; Liyanapathiranage, P.; Iruegas-Bocardo, F.; White, F.F.; Goss, E.M.; Jones, J.B. Xanthomonas Diversity, Virulence and Plant-Pathogen Interactions. Nat. Rev. Microbiol. 2020, 18, 415–427. [Google Scholar] [CrossRef]
- Langner, T.; Kamoun, S.; Belhaj, K. CRISPR Crops: Plant Genome Editing toward Disease Resistance. Annu. Rev. Phytopathol. 2018, 56, 479–512. [Google Scholar] [CrossRef]
- Saijo, Y.; Loo, E.P. Plant Immunity in Signal Integration between Biotic and Abiotic Stress Responses. New Phytol. 2020, 225, 87–104. [Google Scholar] [CrossRef]
- Weschke, W.; Panitz, R.; Gubatz, S.; Wang, Q.; Radchuk, R.; Weber, H.; Wobus, U. The Role of Invertases and Hexose Transporters in Controlling Sugar Ratios in Maternal and Filial Tissues of Barley Caryopses during Early Development. Plant J. 2003, 33, 395–411. [Google Scholar] [CrossRef]
- Hernández-Sánchez, I.E.; Maruri-López, I.; Martinez-Martinez, C.; Janis, B.; Jiménez-Bremont, J.F.; Covarrubias, A.A.; Menze, M.A.; Graether, S.P.; Thalhammer, A. LEAfing through Literature: Late Embryogenesis Abundant Proteins Coming of Age-Achievements and Perspectives. J. Exp. Bot. 2022, 73, 6525–6546. [Google Scholar] [CrossRef] [PubMed]
- Magwanga, R.O.; Lu, P.; Kirungu, J.N.; Lu, H.J.; Wang, X.X.; Cai, X.Y.; Zhou, Z.L.; Zhang, Z.M.; Salih, H.R.; Wang, K.B.; et al. Characterization of the Late Embryogenesis Abundant (LEA) Proteins Family and Their Role in Drought Stress Tolerance in Upland Cotton. BMC Genet. 2018, 19, 6. [Google Scholar] [CrossRef] [PubMed]
- Assefa, T.; Otyama, P.I.; Brown, A.V.; Brown, A.V.; Kalberer, S.R.; Kulkarni, R.S.; Cannon, S.B. Genome-wide associations and epistatic interactions for internode number, plant height, seed weight and seed yield in soybean. BMC Genom. 2019, 20, 257. [Google Scholar] [CrossRef]
- Zhang, X.L.; Ding, W.T.; Xue, D.; Li, X.N.; Zhou, Y.; Shen, J.C.; Feng, J.Y.; Guo, N.; Qiu, L.J.; Xing, H.; et al. Genome-wide association studies of plant architecture-related traits and 100-seed weight in soybean landraces. BMC Genom. Data 2021, 22, 10–24. [Google Scholar] [CrossRef]
- Csanádi, G.Y.; Vollmann, J.; Stift, G.; Lelley, T. Seed quality QTLs identified in a molecular map of early maturing soybean. Theor. Appl. Genet. 2001, 103, 912–919. [Google Scholar] [CrossRef]
- Hoeck, J.A.; Fehr, W.R.; Shoemaker, R.C.; Shoemaker, R.C.; Welke, G.A.; Johnson, S.L.; Cianzio, S.R. Molecular marker analysis of seed size in soybean. Crop Sci. 2003, 43, 68–74. [Google Scholar] [CrossRef]
- Kato, S.; Sayama, T.; Fujii, K.; Yumoto, S.; Kono, Y.; Hwang, T.; Kikuchi, A.; Takada, Y.; Tanaka, Y.; Shiraiwa, T.; et al. A major and stable QTL associated with seed weight in soybean across multiple environments and genetic backgrounds. Theor. Appl. Genet. 2014, 127, 1365–1374. [Google Scholar] [CrossRef]
- Kulkarni, K.P.; Asekova, S.; Lee, D.H.; Bilyeu, K.; Song, J.T.; Lee, J. Mapping QTLs for 100-seed weight in an interspecific soybean cross of Williams 82 (Glycine max) and PI 366121 (Glycine soja). Crop Pasture Sci. 2017, 68, 148–155. [Google Scholar] [CrossRef]
- Kuroda, Y.; Kaga, A.; Tomooka, N.; Yano, H.; Takada, T.; Kato, S.; Vaughan, D. QTL affecting fitness of hybrids between wild and cultivated soybeans in experimental fields. Ecol. Evol. 2013, 3, 2150–2168. [Google Scholar] [CrossRef]
- Liang, Q.; Cheng, X.H.; Mei, M.T.; Yan, X.L.; Liao, H. QTL analysis of root traits as related to phosphorus efficiency in soybean. Ann. Bot. 2010, 106, 223–234. [Google Scholar] [CrossRef]
- Teng, W.; Han, Y.; Du, Y.; Sun, D.; Zhang, Z.; Qiu, L.; Sun, G.; Li, W. QTL analyses of seed weight during the development of soybean (Glycine max L. Merr.). Heredity 2009, 102, 372–380. [Google Scholar] [CrossRef]
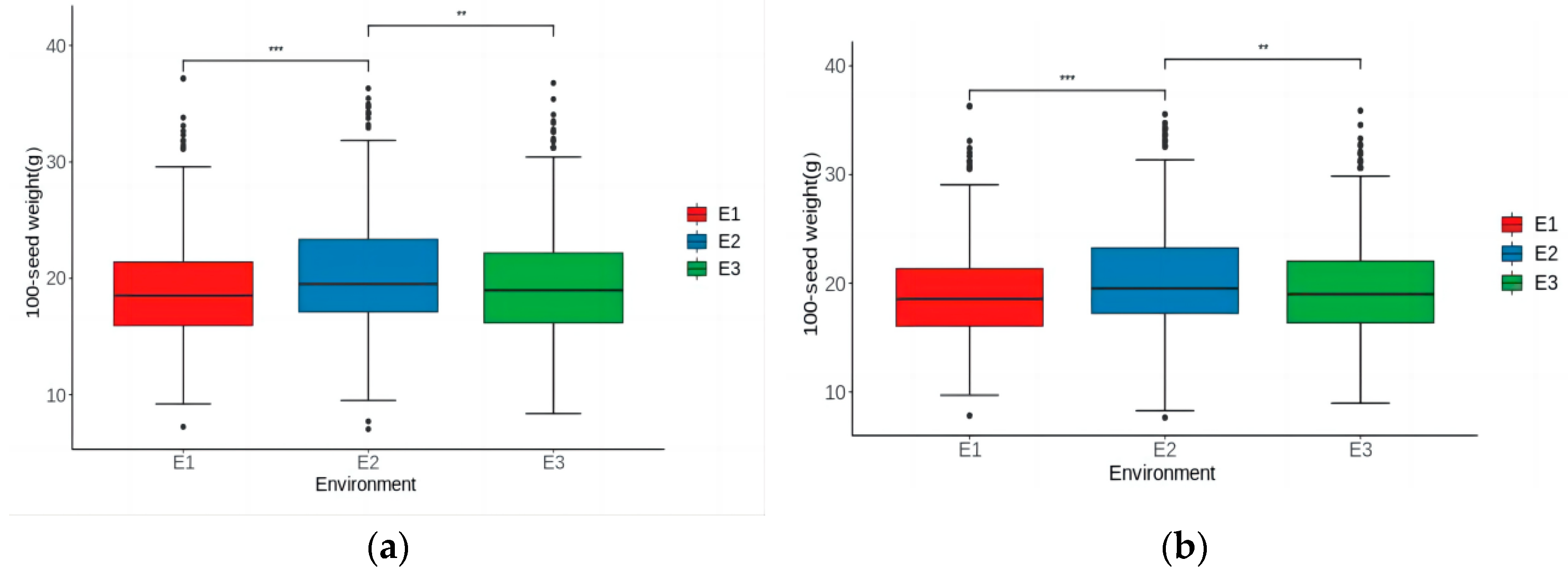
| Phenotype a | Environment b | Mean | Min | Max | SD | Skew | Kurt | CV (%) | h2 (%) |
|---|---|---|---|---|---|---|---|---|---|
| Mean-phenotype | E1 | 18.99 | 7.24 | 37.19 | 4.46 | 0.78 | 1.01 | 23 | 95.10 |
| E2 | 20.29 | 7.04 | 36.32 | 4.88 | 0.54 | 0.44 | 24 | 96.40 | |
| E3 | 19.47 | 8.38 | 36.78 | 4.87 | 0.64 | 0.46 | 25 | 94.60 | |
| BLUP-phenotype | E1 | 18.99 | 7.81 | 36.3 | 4.22 | 0.71 | 1.02 | 22 | 95.13 |
| E2 | 20.28 | 7.63 | 35.55 | 4.69 | 0.53 | 0.41 | 23 | 96.87 | |
| E3 | 19.49 | 8.96 | 35.88 | 4.60 | 0.66 | 0.45 | 24 | 94.80 |
| QQIs a | Mean-Phenotype | BLUP-Phenotype | |||||||
|---|---|---|---|---|---|---|---|---|---|
| QTN1 | QTN2 | LOD b | Effect c | Variance d | R2 (%) e | LOD b | Effect c | Variance d | R2 (%) e |
| Gm01_47969266 | Gm02_4453462 | 4.33 | 0.94 | 0.71 | 3.70 | 4.47 | 0.60 | 0.28 | 1.86 |
| Gm02_5698503 | Gm08_5258168 | 10.20 | 1.05 | 0.82 | 4.29 | 5.19 | 0.58 | 0.25 | 1.64 |
| Gm02_25227246 | Gm11_16022081 | 5.72 | −0.87 | 0.67 | 3.49 | 3.75 | −0.74 | 0.48 | 3.16 |
| Gm06_13595169 | Gm06_14999200 | 4.62 | 0.72 | 0.51 | 2.68 | 4.64 | 0.80 | 0.63 | 4.17 |
| Gm07_22276137 | Gm13_7337797 | 7.30 | −0.67 | 0.24 | 1.27 | 6.74 | −0.88 | 0.42 | 2.73 |
| Gm07_36350977 | Gm18_55491235 | 6.12 | 0.61 | 0.36 | 1.85 | 4.86 | 0.23 | 0.05 | 0.33 |
| Gm10_5133417 | Gm20_41100226 | 3.01 | 0.59 | 0.29 | 1.52 | 3.26 | 0.32 | 0.09 | 0.57 |
| Gm13_2423497 | Gm17_6321674 | 4.61 | −0.29 | 0.07 | 0.35 | 5.59 | −0.24 | 0.05 | 0.31 |
| Gm13_18560842 | Gm16_30314260 | 3.45 | 0.41 | 0.16 | 0.83 | 4.67 | 0.27 | 0.07 | 0.44 |
| Gm14_43246298 | Gm18_46567218 | 5.82 | −0.10 | 0.01 | 0.05 | 4.29 | −0.13 | 0.02 | 0.11 |
| QTNs a | Position b | Candidate Gene | ||||
|---|---|---|---|---|---|---|
| Wm82.a1.v1 c | Wm82.a2.v1 c | Position (bp) d | Gene Symbol e | Functional Annotation f | ||
| qHSW-6-2 | Gm06_14999200 | Glyma06g19000 | Glyma.06g180000 | Gm06:15230602 15235616 | LOC100776762 | Cell division cycle protein 48 homolog (CDC48) |
| qHSW-7-1 | Gm07_7333790 | Glyma07g08950 | Glyma.07g081700 | Gm07:7480416- 7483393 | GA20OX | Gibberellin 20 oxidase 2; Response to gibberellin stimulation, flower development |
| qHSW-8-2 | Gm08_14545190 | Glyma08g19580 | Glyma.08g183500 | Gm08:14793461- 14795629 | GmSWEET24 | Sucrose transportation |
| qHSW-13-2 | Gm13_41961934 | Glyma13g42310 | Glyma.13g347500 | Gm13:42321510- 42325915 | LOX2 | Lipoxygenase 1; Response to abscisic acid stimulation, Response to jasmonic acid stimulation |
| Glyma13g42320 | Glyma.13g347600 | Gm13:42328964- 42333252 | LOX1.1 | Lipoxygenase 1; Response to abscisic acid stimulation, Response to jasmonic acid stimulation | ||
| qHSW-13-3 | Gm13_43480280 | Glyma13g44020 | Glyma.13g363300 | Gm13:43580634- 43581833 | GmPM30 | Rich late embryogenesis (plant) LEA related |
| Glyma13g43800 | Glyma.13g361200 | Gm13:43396690-43398891 | LOC100500488 | AUX/IAA family protein | ||
| qHSW-17-1 | Gm17_13658864 | Glyma17g16620 | Glyma.17g155000 | Gm17:13354202- 13355951 | GmPM16 | Rich late embryogenesis (plant) LEA related |
| qHSW-17-2 | Gm17_6321674 | Glyma17g09110 | Glyma.17g083600 | Gm17:6748647 6750218 | LOC100812289 | Response to jasmonic acid stimulus; protein STRICTOSIDINE SYNTHASE-LIKE 12 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Karikari, B.; Zhang, H.; Zhang, C.; Wang, Z.; Zhao, T.; Feng, J. Comprehensive Identification of Main, Environment Interaction and Epistasis Quantitative Trait Nucleotides for 100-Seed Weight in Soybean (Glycine max (L.) Merr.). Agronomy 2024, 14, 483. https://doi.org/10.3390/agronomy14030483
Wang L, Karikari B, Zhang H, Zhang C, Wang Z, Zhao T, Feng J. Comprehensive Identification of Main, Environment Interaction and Epistasis Quantitative Trait Nucleotides for 100-Seed Weight in Soybean (Glycine max (L.) Merr.). Agronomy. 2024; 14(3):483. https://doi.org/10.3390/agronomy14030483
Chicago/Turabian StyleWang, Li, Benjamin Karikari, Hu Zhang, Chunting Zhang, Zili Wang, Tuanjie Zhao, and Jianying Feng. 2024. "Comprehensive Identification of Main, Environment Interaction and Epistasis Quantitative Trait Nucleotides for 100-Seed Weight in Soybean (Glycine max (L.) Merr.)" Agronomy 14, no. 3: 483. https://doi.org/10.3390/agronomy14030483
APA StyleWang, L., Karikari, B., Zhang, H., Zhang, C., Wang, Z., Zhao, T., & Feng, J. (2024). Comprehensive Identification of Main, Environment Interaction and Epistasis Quantitative Trait Nucleotides for 100-Seed Weight in Soybean (Glycine max (L.) Merr.). Agronomy, 14(3), 483. https://doi.org/10.3390/agronomy14030483