
Identifications of Seed Vigor-Related QTLs and Candidate Genes Combined Cultivated Soybean with Wild Soybean


 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Soybean Materials
2.2. Seed Vigor Testing
2.3. QTL Mapping
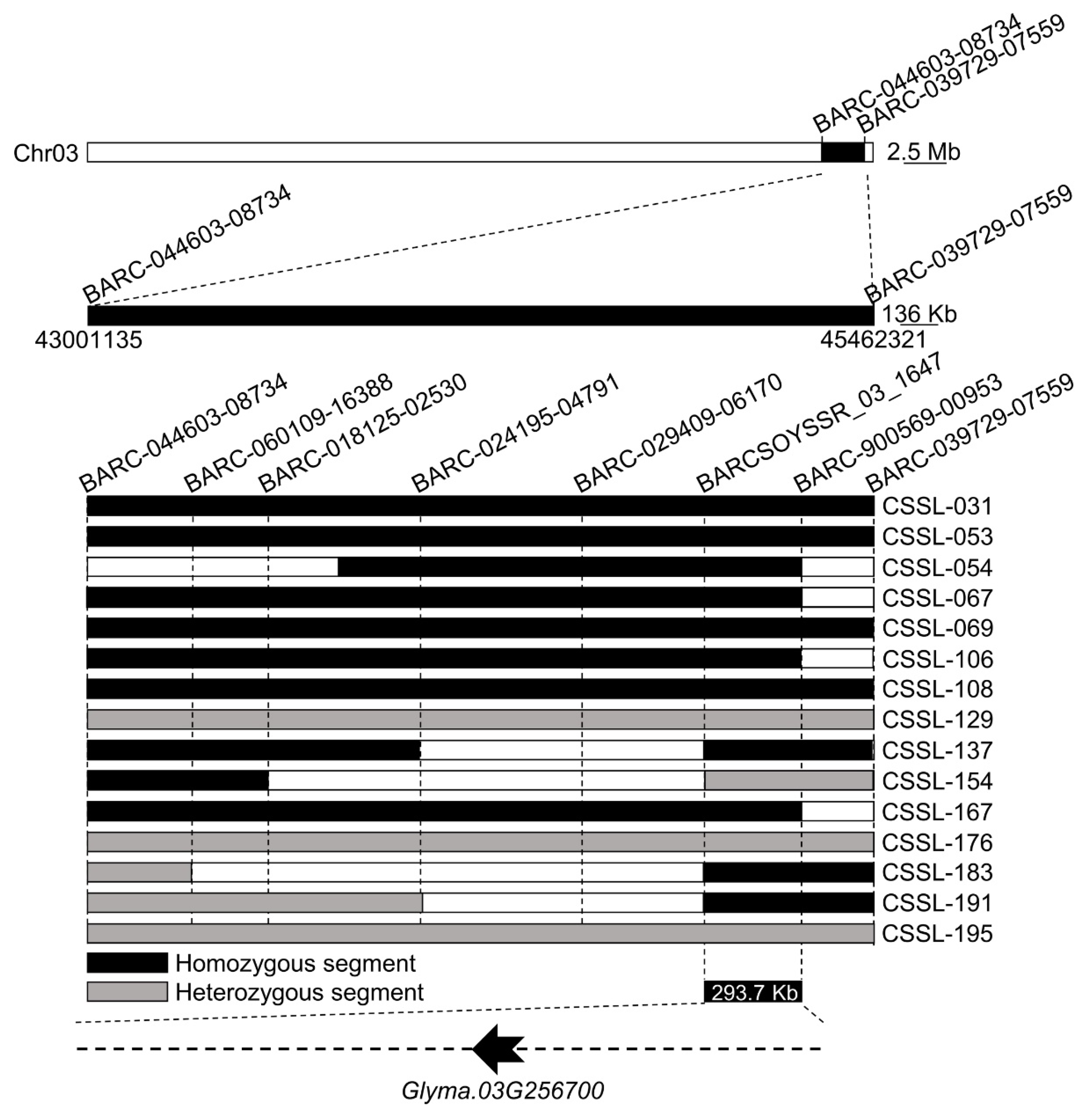
2.4. ZYD00006 Chromosome Insertion Analyses
2.5. Candidate Gene Screening
2.6. SNP Analyses of Candidate Genes
2.7. RNA-Seq
2.8. qPCR Validation
2.9. Haplotype Analyses of Glyma.03G256700
3. Results
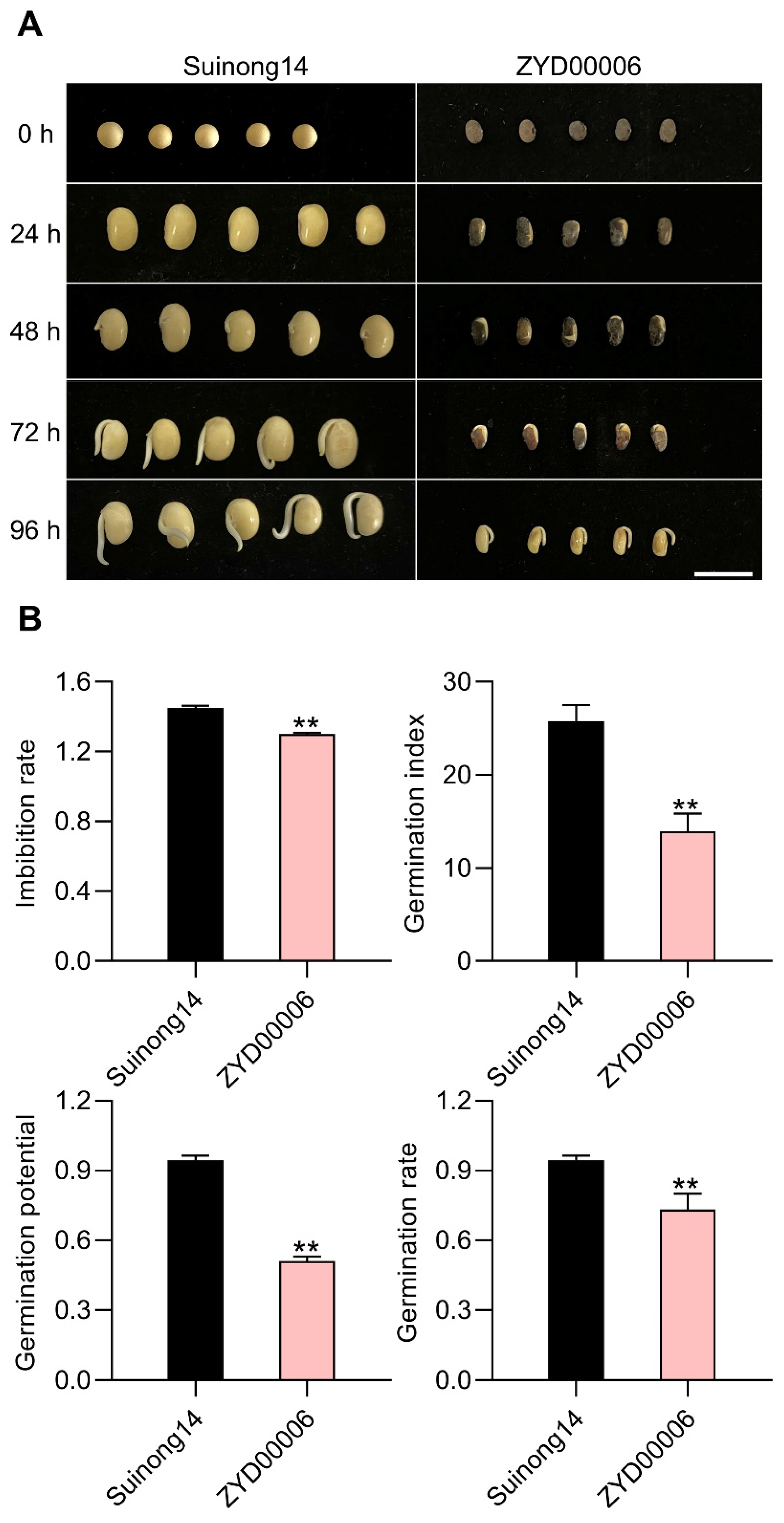
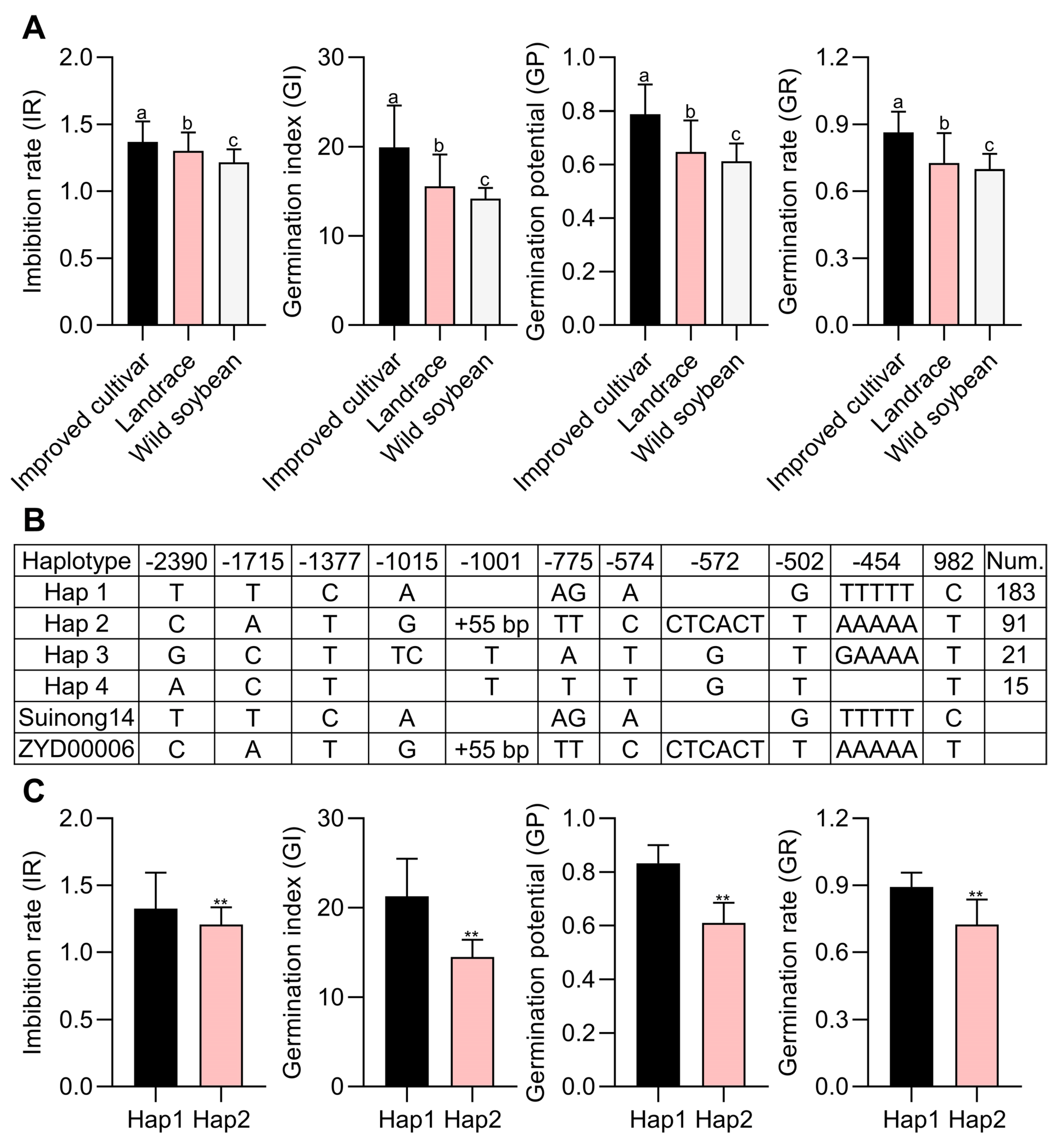
3.1. The Seed Vigor of the Improved Suinong14 Cultivar Is Superior to That of Wild ZYD00006 Soybean
3.2. Seed Vigor-Associated QTL Identification in Soybean CSSL Populations
3.3. Chromosome Fragment Insertion-Based Identification of Candidate Intervals
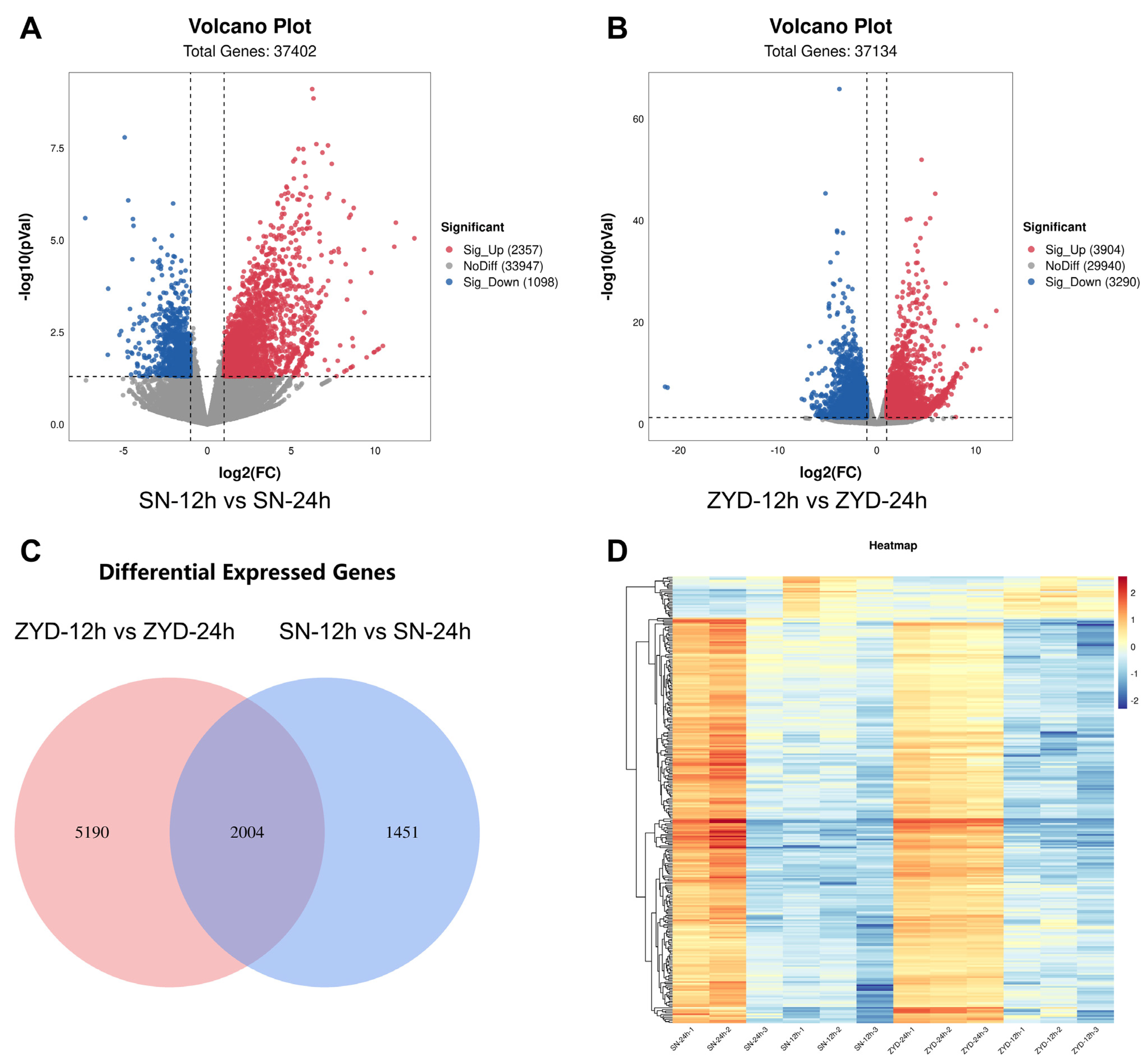
3.4. High-Throughput RNA-Seq of Germinating Suinong14 and ZYD00006 Varieties
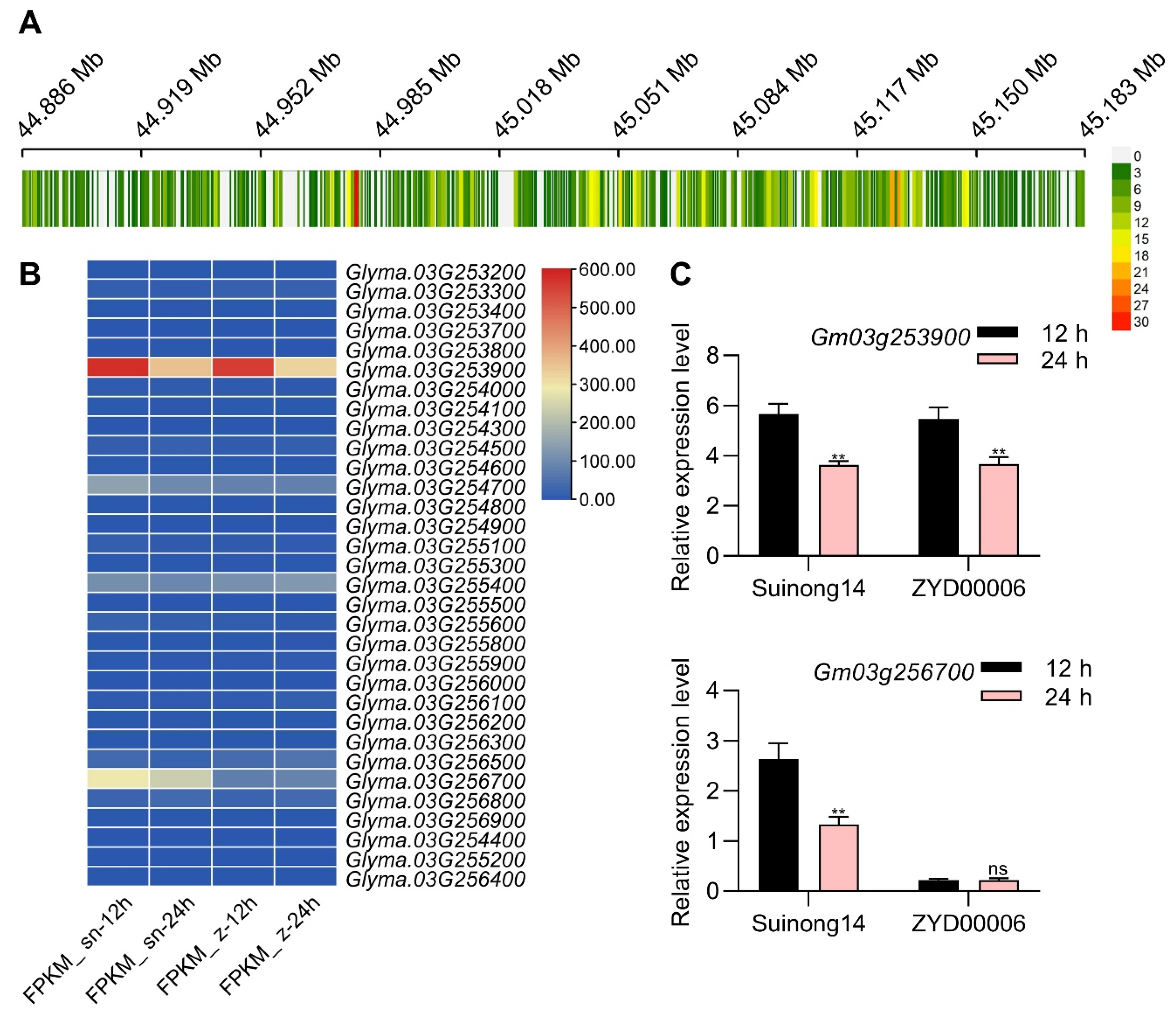
3.5. Sequencing-Based Seed Vigor-Related Candidate Gene Identification
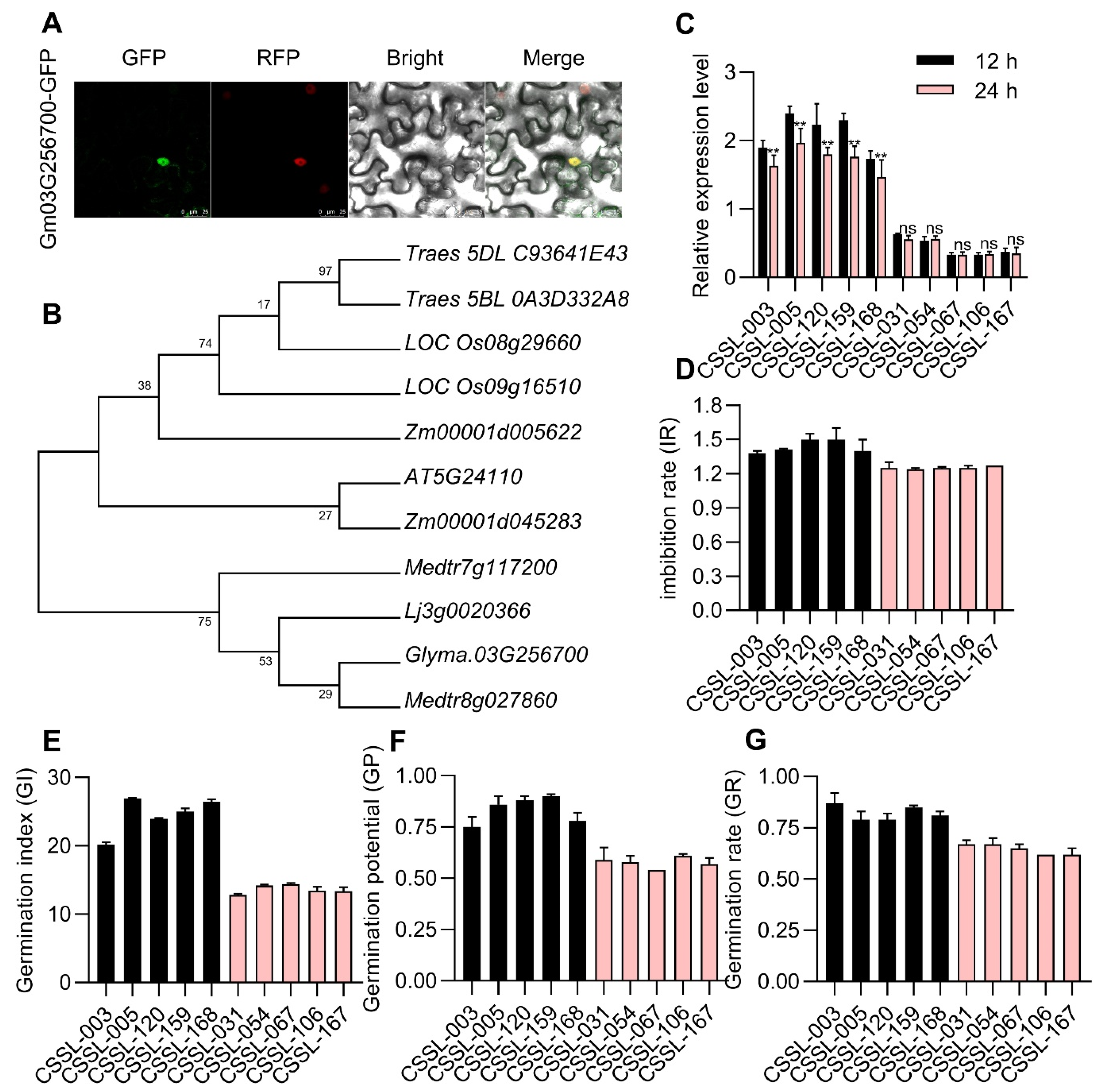
3.6. Glyma.03G256700 Encodes a WRKY53 Protein Exhibiting Differential Expression in CSSLs Exhibiting Varying Levels of Seed Vigor
3.7. Analyses of Glyma.03G256700 Haplotypes in Natural Soybean Germplasms
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, Y.H.; Zhou, G.; Ma, J.; Jiang, W.; Jin, L.G.; Zhang, Z.; Guo, Y.; Zhang, J.; Sui, Y.; Zheng, L.; et al. De novo assembly of soybean wild relatives for pan-genome analysis of diversity and agronomic traits. Nat. Biotechnol. 2014, 32, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ma, C.; Ma, S.N.; Zheng, H.Y.; Tian, H.L.; Wang, X.; Wang, Y.; Jiang, H.W.; Wang, J.X.; Zhang, Z.G.; et al. Genetic variation in GmCRP contributes to nodulation in soybean (Glycine max Merr.). Crop J. 2023, 11, 332–344. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, S.L.; Wang, Z.; Yuan, Y.Q.; Zhang, Z.F.; Liang, Q.J.; Yang, X.; Duan, Z.B.; Liu, Y.C.; Kong, F.J.; et al. Progress in soybean functional genomics over the past decade. Plant Biotechnol. J. 2022, 20, 256–282. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Guan, W.H.; Shi, Y.G.; Wang, S.G.; Fan, H.; Yang, J.W.; Chen, W.G.; Zhang, W.J.; Sun, D.Z.; Jing, R.L. QTL mapping and candidate gene analysis of seed vigor-related traits during artificial aging in wheat (Triticum aestivum). Sci. Rep. 2020, 10, 22060. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Wu, F.; Xie, X.; Yang, C. Quantitative Trait Locus Mapping of Seed Vigor in Soybean under −20 °C Storage and Accelerated Aging Conditions via RAD Sequencing. Curr. Issues Mol. Biol. 2021, 43, 1977–1996. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.Q.; Xu, D.Y.; Sui, Y.P.; Ding, X.H.; Song, X.J. A multiomic study uncovers a bZIP23-PER1A-mediated detoxification pathway to enhance seed vigor in rice. Proc. Natl. Acad. Sci. USA 2022, 119, e2026355119. [Google Scholar] [CrossRef]
- Reed, R.C.; Bradford, K.J.; Khanday, I.; Bradford, K.J.; Khanday, I. Seed germination and vigor: Ensuring crop sustainability in a changing climate. Heredity 2022, 128, 450–459. [Google Scholar] [CrossRef]
- Zhao, J.; He, Y.; Huang, S.; Wang, Z. Advances in the Identification of Quantitative Trait Loci and Genes Involved in Seed Vigor in Rice. Front. Plant Sci. 2021, 12, 659307. [Google Scholar] [CrossRef]
- Huang, Y.T.; Wu, W.; Zhao, T.Y.; Lu, M.; Wu, H.P.; Cao, D.D. Drying temperature regulates vigor of high moisture rice seeds via involvement in phytohormone, ROS, and relevant gene expression. J. Sci. Food Agric. 2021, 101, 2143–2155. [Google Scholar] [CrossRef]
- Li, T.; Zhang, Y.; Wang, D.; Liu, Y.; Dirk, L.M.A.; Goodman, J.; Downie, A.B.; Wang, J.; Wang, G.; Zhao, T. Regulation of Seed Vigor by Manipulation of Raffinose Family Oligosaccharides in Maize and Arabidopsis thaliana. Mol. Plant 2017, 10, 1540–1555. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Fei, Y.; Wang, Y.; Zhao, W.; Hou, L.; Cao, Y.; Wu, M.; Wu, H. Identification of a Seed Vigor-Related QTL Cluster Associated with Weed Competitive Ability in Direct-Seeded Rice (Oryza sativa L.). Rice 2023, 16, 45. [Google Scholar] [CrossRef]
- Zeng, Z.; Guo, C.; Yan, X.; Song, J.; Wang, C.; Xu, X.; Hao, Y. QTL mapping and KASP marker development for seed vigor related traits in common wheat. Front. Plant Sci. 2022, 13, 994973. [Google Scholar] [CrossRef]
- Nguyen, D.T.; Hayes, J.E.; Harris, J.; Sutton, T. Fine Mapping of a Vigor QTL in Chickpea (Cicer arietinum L.) Reveals a Potential Role for Ca4_TIFY4B in Regulating Leaf and Seed Size. Front. Plant Sci. 2022, 13, 829566. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Zhang, F.; Zafar, S.; Wang, J.; Lu, H.; Naveed, S.; Lou, J.; Xu, J. Genetic dissection of heterosis of indica-japonica by introgression line, recombinant inbred line and their testcross populations. Sci. Rep. 2021, 11, 10265. [Google Scholar] [CrossRef]
- Dang, X.; Thi, T.G.; Dong, G.; Wang, H.; Edzesi, W.M.; Hong, D. Genetic diversity and association mapping of seed vigor in rice (Oryza sativa L.). Planta 2014, 239, 1309–1319. [Google Scholar] [CrossRef]
- Gu, J.; Hou, D.; Li, Y.; Chao, H.; Zhang, K.; Wang, H.; Xiang, J.; Raboanatahiry, N.; Wang, B.; Li, M. Integration of proteomic and genomic approaches to dissect seed germination vigor in Brassica napus seeds differing in oil content. BMC Plant Biol. 2019, 19, 21. [Google Scholar] [CrossRef] [PubMed]
- Luo, T.; Zhang, Y.; Zhang, C.; Nelson, M.N.; Yuan, J.; Guo, L.; Xu, Z. Genome-Wide Association Mapping Unravels the Genetic Control of Seed Vigor under Low-Temperature Conditions in Rapeseed (Brassica napus L.). Plants 2021, 10, 426. [Google Scholar] [CrossRef]
- Zhou, Z.; Jiang, Y.; Wang, Z.; Gou, Z.; Lyu, J.; Li, W.; Yu, Y.; Shu, L.; Zhao, Y.; Ma, Y.; et al. Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean. Nat. Biotechnol. 2015, 33, 408–414. [Google Scholar] [CrossRef]
- Liu, Y.; Du, H.; Li, P.; Shen, Y.; Peng, H.; Liu, S.; Zhou, G.A.; Zhang, H.; Liu, Z.; Shi, M.; et al. Pan-Genome of Wild and Cultivated Soybeans. Cell 2020, 182, 162–176.e13. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Hou, L.; Xie, J.; Cao, F.; Wei, R.; Yang, M.; Qi, Z.; Zhu, R.; Zhang, Z.; Xin, D.; et al. Construction of Chromosome Segment Substitution Lines and Inheritance of Seed-Pod Characteristics in Wild Soybean. Front. Plant Sci. 2022, 13, 869455. [Google Scholar] [CrossRef]
- Xin, D.; Qi, Z.; Jiang, H.; Hu, Z.; Zhu, R.; Hu, J.; Han, H.; Hu, G.; Liu, C.; Chen, Q.; et al. QTL Location and Epistatic Effect Analysis of 100-Seed Weight Using Wild Soybean (Glycine soja Sieb. & Zucc.) Chromosome Segment Substitution Lines. PLoS ONE 2016, 11, e0149380. [Google Scholar] [CrossRef]
- Qi, Z.; Guo, C.; Li, H.; Qiu, H.; Li, H.; Jong, C.; Yu, G.; Zhang, Y.; Hu, L.; Wu, X.; et al. Natural variation in Fatty Acid 9 is a determinant of fatty acid and protein content. Plant Biotechnol. J. 2023. Version of Record online. [Google Scholar] [CrossRef]
- Zhang, W.; Liao, X.; Cui, Y.; Ma, W.; Zhang, X.; Du, H.; Ma, Y.; Ning, L.; Wang, H.; Huang, F.; et al. A cation diffusion facilitator, GmCDF1, negatively regulates salt tolerance in soybean. PLoS Genet. 2019, 15, e1007798. [Google Scholar] [CrossRef]
- Törönen, P.; Medlar, A.; Holm, L. PANNZER2: A rapid functional annotation web server. Nucleic Acids Res. 2018, 46, W84–W88. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, J.; Ma, C.; Zhou, Z.; Yang, D.; Zheng, J.; Wang, Q.; Li, H.; Zhou, H.; Sun, Z.; et al. QTL Mapping and Data Mining to Identify Genes Associated With the Sinorhizobium fredii HH103 T3SS Effector NopD in Soybean. Front. Plant Sci. 2020, 11, 453. [Google Scholar] [CrossRef] [PubMed]
- Bobby, R.; Bazzelle, R.; Clark, W.; Kantartzi, S.K.; Meksem, K.; Akond, M.; Kassem, M.A. Genetic Analysis of Yield Components in the PI 438489B by ‘Hamilton’ Recombinant Inbred Line (RIL) Population of Soybean [Glycine max (L.) Merr.]. J. Agric. Sci. 2012, 4, 98–105. [Google Scholar] [CrossRef]
- Zuo, J.; Liu, J.; Gao, F.; Yin, G.; Wang, Z.; Chen, F.; Li, X.; Xu, J.; Chen, T.; Li, L.; et al. Genome-Wide Linkage Mapping Reveals QTLs for Seed Vigor-Related Traits Under Artificial Aging in Common Wheat (Triticum aestivum). Front. Plant Sci. 2018, 9, 1101. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Y.; Li, X.; Hu, J.; Xu, R.; Zhang, D. Expanding the gene pool for soybean improvement with its wild relatives. aBIOTECH 2022, 3, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Fang, C.; Liu, B.; Kong, F. Natural variation and artificial selection of photoperiodic flowering genes and their applications in crop adaptation. aBIOTECH 2021, 2, 156–169. [Google Scholar] [CrossRef]
- Cai, X.; Jia, B.; Sun, M.; Sun, X. Insights into the regulation of wild soybean tolerance to salt-alkaline stress. Front. Plant Sci. 2022, 13, 1002302. [Google Scholar] [CrossRef]
- Du, H.; Fang, C.; Li, Y.; Kong, F.; Liu, B. Understandings and future challenges in soybean functional genomics and molecular breeding. J. Integr. Plant Biol. 2023, 65, 468–495. [Google Scholar] [CrossRef]
- Singh, G.; Gudi, S.; Amandeep Upadhyay, P.; Shekhawat, P.K.; Nayak, G.; Goyal, L.; Kumar, D.; Kumar, P.; Kamboj, A.; Thada, A.; et al. Unlocking the hidden variation from wild repository for accelerating genetic gain in legumes. Front. Plant Sci. 2022, 13, 1035878. [Google Scholar] [CrossRef]
- Lu, S.; Fang, C.; Abe, J.; Kong, F.; Liu, B. Current overview on the genetic basis of key genes involved in soybean domestication. aBIOTECH 2022, 3, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Wani, S.H.; Anand, S.; Singh, B.; Bohra, A.; Joshi, R. WRKY transcription factors and plant defense responses: Latest discoveries and future prospects. Plant Cell Rep. 2021, 40, 1071–1085. [Google Scholar] [CrossRef] [PubMed]
- Javed, T.; Gao, S.J. WRKY transcription factors in plant defense. Trends Genet. 2023, 39, 787–801. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Yu, D. Activated expression of AtWRKY53 negatively regulates drought tolerance by mediating stomatal movement. Plant Cell Rep. 2015, 34, 1295–1306. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Huhn, K.; Brandt, R.; Potschin, M.; Bieker, S.; Straub, D.; Doll, J.; Drechsler, T.; Zentgraf, U.; Wenkel, S. REVOLUTA and WRKY53 connect early and late leaf development in Arabidopsis. Development 2014, 141, 4772–4783. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Ge, J.; Bao, C.; Chang, W.; Liu, J.; Shao, J.; Liu, X.; Su, L.; Pan, L.; Zhou, D.X. Histone Deacetylase HDA9 and WRKY53 Transcription Factor Are Mutual Antagonists in Regulation of Plant Stress Response. Mol. Plant 2020, 13, 598–611. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Lu, L.; Mayer, K.S.; Scalf, M.; Qian, S.; Lomax, A.; Smith, L.M.; Zhong, X. POWERDRESS interacts with HISTONE DEACETYLASE 9 to promote aging in Arabidopsis. eLife 2016, 5, e17214. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Liu, C.; Li, S.; Zhang, Y.; Zhang, Y.; Wang, X.; Xiang, W. The Transcription Factors WRKY41 and WRKY53 Mediate Early Flowering Induced by the Novel Plant Growth Regulator Guvermectin in Arabidopsis thaliana. Int. J. Mol. Sci. 2023, 24, 8424. [Google Scholar] [CrossRef]
- Jiao, C.; Li, K.; Zuo, Y.; Gong, J.; Guo, Z.; Shen, Y. CALMODULIN1 and WRKY53 Function in Plant Defense by Negatively Regulating the Jasmonic Acid Biosynthesis Pathway in Arabidopsis. Int. J. Mol. Sci. 2022, 23, 7718. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, M.; Zhou, J.; Gao, X.; Zhu, S.; Yuan, L.; Hou, X.; Liu, T.; Chen, G.; Tang, X. Transcriptome analysis and differential gene expression profiling of wucai (Brassica campestris L.) in response to cold stress. BMC Genom. 2022, 23, 137. [Google Scholar] [CrossRef] [PubMed]
- Freeborough, W.; Gentle, N.; Rey, M.E.C. WRKY Transcription Factors in Cassava Contribute to Regulation of Tolerance and Susceptibility to Cassava Mosaic Disease through Stress Responses. Viruses 2021, 13, 1820. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Huang, L.; Dai, Y.; Liu, S.; Hong, Y.; Tian, L.; Huang, L.; Cao, Z.; Li, D.; Song, F. Arabidopsis AtERF15 positively regulates immunity against Pseudomonas syringae pv. tomato DC3000 and Botrytis cinerea. Front. Plant Sci. 2015, 6, 686. [Google Scholar] [CrossRef]
- Tian, X.; He, M.; Mei, E.; Zhang, B.; Tang, J.; Xu, M.; Liu, J.; Li, X.; Wang, Z.; Tang, W. WRKY53 integrates classic brassinosteroid signaling and the mitogen-activated protein kinase pathway to regulate rice architecture and seed size. Plant Cell 2021, 33, 2753–2775. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Tian, X.; Mei, E.; He, M.; Gao, J.; Yu, J.; Xu, M.; Liu, J.; Song, L.; Li, X.; et al. WRKY53 negatively regulates rice cold tolerance at the booting stage by fine-tuning anther gibberellin levels. Plant Cell 2022, 34, 4495–4515. [Google Scholar] [CrossRef]
- Gao, Y.; Xue, C.Y.; Liu, J.M.; He, Y.; Mei, Q.; Wei, S.; Xuan, Y.H. Sheath blight resistance in rice is negatively regulated by WRKY53 via SWEET2a activation. Biochem. Biophys. Res. Commun. 2021, 585, 117–123. [Google Scholar] [CrossRef]
- Yang, Y.; Zhou, Y.; Chi, Y.; Fan, B.; Chen, Z. Characterization of Soybean WRKY Gene Family and Identification of Soybean WRKY Genes that Promote Resistance to Soybean Cyst Nematode. Sci. Rep. 2017, 7, 17804. [Google Scholar] [CrossRef]
- Yu, Y.; Wang, N.; Hu, R.; Xiang, F. Genome-wide identification of soybean WRKY transcription factors in response to salt stress. Springerplus 2016, 5, 920. [Google Scholar] [CrossRef]
- Yang, Y.; Chi, Y.; Wang, Z.; Zhou, Y.; Fan, B.; Chen, Z. Functional analysis of structurally related soybean GmWRKY58 and GmWRKY76 in plant growth and development. J. Exp. Bot. 2016, 67, 4727–4742. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Chen, H.W.; Li, Q.T.; Wei, W.; Li, W.; Zhang, W.K.; Ma, B.; Bi, Y.D.; Lai, Y.C.; Liu, X.L.; et al. GmWRKY27 interacts with GmMYB174 to reduce expression of GmNAC29 for stress tolerance in soybean plants. Plant J. 2015, 83, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Baillo, E.H.; Kimotho, R.N.; Zhang, Z.; Xu, P. Transcription Factors Associated with Abiotic and Biotic Stress Tolerance and Their Potential for Crops Improvement. Genes 2019, 10, 771. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Parents | CSSL Population (n = 207) | ||||
|---|---|---|---|---|---|---|
| ZYD00006 | Suinong14 | Mean | SD a | Kurtosis b | Skewness c | |
| IR | 1.30 ** | 1.45 | 1.38 | 0.15 | −0.75 | −0.32 |
| GI | 13.94 ** | 25.78 | 19.82 | 7.60 | −0.75 | −0.08 |
| GP | 0.51 ** | 0.94 | 0.76 | 0.24 | −0.83 | −0.19 |
| GR | 0.73 ** | 0.94 | 0.79 | 0.20 | −1.00 | −0.06 |
| Trait | Chr | QTL | Position (Mb) | LOD | R² (%) | ADD | Putative Causal Genes or QTLs Identified in Previous Studies |
|---|---|---|---|---|---|---|---|
| IR | Chr02 | qIR-02 | 1.3 | 3.62 | 5 | −0.28 | |
| Chr03 | qIR-03 | 45.2 | 4.54 | 2 | 0.70 | ||
| Chr08 | qIR-08 | 24.4 | 3.89 | 3 | 0.90 | qSTIR-8 [23] | |
| Chr12 | qIR-12 | 15.2 | 3.22 | 5 | −0.48 | ||
| Chr14 | qIR-14 | 32.2 | 3.50 | 5 | −0.68 | ||
| GI | Chr03 | qGI-03 | 43.0 | 4.95 | 1 | 0.24 | |
| Chr05 | qGI-05 | 16.5 | 3.42 | 1 | 0.27 | ||
| Chr10 | qGI-10 | 33.8 | 3.06 | 7 | 1.29 | ||
| Chr12 | qGI-12 | 32.2 | 10.02 | 5 | −0.42 | ||
| Chr15 | qGI-15 | 15.9 | 3.41 | 4 | −0.47 | ||
| GP | Chr03 | qGP-03 | 45.5 | 4.40 | 8 | 0.75 | |
| Chr06 | qGP-06 | 14.9 | 3.97 | 6 | 0.48 | qGP-6 [5] | |
| Chr07 | qGP-07 | 20.3 | 3.12 | 5 | −0.38 | ||
| Chr13 | qGP-13 | 39.4 | 3.52 | 3 | −0.58 | qDG003 [27] | |
| GR | Chr03 | qGR-03 | 44.7 | 3.42 | 6 | 0.49 | |
| Chr06 | qGR-06 | 1.8 | 3.29 | 2 | 0.31 | qGR-6 [5] | |
| Chr09 | qGR-09 | 16.3 | 3.91 | 5 | −0.59 | ||
| Chr12 | qGR-12 | 35.2 | 14.42 | 4 | −0.32 | ||
| Chr16 | qGR-16 | 36.4 | 3.40 | 1 | −0.42 | ||
| Chr18 | qGR-18 | 15.3 | 3.21 | 5 | −0.42 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, S.; Feng, H.; Sun, Y.; Yu, L.; Tang, C.; Zhao, Y.; Xue, L.; Wang, J.; Liu, C.; Xin, D.; et al. Identifications of Seed Vigor-Related QTLs and Candidate Genes Combined Cultivated Soybean with Wild Soybean. Agronomy 2024, 14, 332. https://doi.org/10.3390/agronomy14020332
Ma S, Feng H, Sun Y, Yu L, Tang C, Zhao Y, Xue L, Wang J, Liu C, Xin D, et al. Identifications of Seed Vigor-Related QTLs and Candidate Genes Combined Cultivated Soybean with Wild Soybean. Agronomy. 2024; 14(2):332. https://doi.org/10.3390/agronomy14020332
Chicago/Turabian StyleMa, Shengnan, Haojie Feng, Yiran Sun, Lin Yu, Chunshuang Tang, Yanqiang Zhao, Liansong Xue, Jinhui Wang, Chunyan Liu, Dawei Xin, and et al. 2024. "Identifications of Seed Vigor-Related QTLs and Candidate Genes Combined Cultivated Soybean with Wild Soybean" Agronomy 14, no. 2: 332. https://doi.org/10.3390/agronomy14020332
APA StyleMa, S., Feng, H., Sun, Y., Yu, L., Tang, C., Zhao, Y., Xue, L., Wang, J., Liu, C., Xin, D., Chen, Q., & Yang, M. (2024). Identifications of Seed Vigor-Related QTLs and Candidate Genes Combined Cultivated Soybean with Wild Soybean. Agronomy, 14(2), 332. https://doi.org/10.3390/agronomy14020332