
Assessment of Genetic Diversity and Protein Content of Scandinavian Peas (Pisum sativum)



Abstract
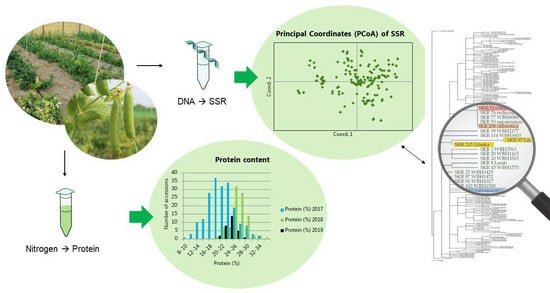
1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. Microsatellites
2.3. Thousand Seed Weight and Protein Content
2.4. Data Analysis
2.4.1. Polymorphism Information Content
2.4.2. Neighbor Joining Tree
2.4.3. Principal Coordinate Analysis of Microsatellite Data
2.4.4. ANOVA of Crude Protein across Years
3. Results
3.1. Plant Material
3.2. Microsatellites
3.3. Thousand Seed Weight and Protein Content
3.3.1. Polymorphism Information Content
3.3.2. Neighbor Joining Tree
3.3.3. Principal Coordinates of Microsatellite Data
3.3.4. Nitrogen Content and Thousand Seed Weight in Relation to Genetic Diversity
4. Discussion
4.1. Microsatellites
4.2. Polymorphism Information Content
4.3. Neighbor Joining Tree
4.4. Thousand Seed Weight and Protein Content
4.5. Future Perspectives
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Davies, D.R. Peas, Pisum sativum. In Evolution of Crop Plants, 2nd ed.; Smartt, J., Simmonds, N.W., Eds.; Longman Scientific & Technical: London, UK, 1995; pp. 294–296. ISBN 0-582-08643-4. [Google Scholar]
- FAOstat. 2022. Available online: https://www.fao.org/faostat/en/#data (accessed on 15 November 2022).
- Kreplak, J.; Madoui, M.-A.; Cápal, P.; Novak, P.; Labadie, K.; Aubert, G.; Bayer, P.E.; Gali, K.; Syme, R.A.; Main, D.; et al. A reference genome for pea provides insight into legume genome evolution. Nat. Genet. 2019, 51, 1411–1422. [Google Scholar] [CrossRef]
- Mendel, G. Versuche über Plflanzenhybriden. Verhandlungen des Naturforschenden Vereines in Brünn, Bd. IV für das Jahr 1865, Abhandlungen. 1866, pp. 3–47. Available online: http://www.esp.org/foundations/genetics/classical/gm-65.pdf (accessed on 17 July 2023).
- Bastianelli, D.; Grosjean, F.; Peyronnet, C.; Duparque, M.; Regnier, J.M. Feeding value of pea (Pisum sativum, L.). Chemical composition of different categories of pea. Anim. Sci. 1998, 67, 609–619. [Google Scholar] [CrossRef]
- Nikolopoulou, D.; Grigorakis, K.; Stasini, M.; Alexis, M.; Iliadis, K. Differences in chemical composition of field pea (Pisum sativum) cultivars: Effects of cultivation area and year. Food Chem. 2007, 103, 847–852. [Google Scholar] [CrossRef]
- Holl, F.B.; Vose, J.R. Carbohydrate and protein accumulation in the developing field pea seed. Can. J. Plant Sci. 1980, 60, 1109–1114. [Google Scholar] [CrossRef]
- McLean, L.A.; Sosulski, F.W.; Youngs, C.G. Effects of nitrogen and moisture on yield and protein in field peas. Can. J. Plant Sci. 1974, 54, 301–305. [Google Scholar] [CrossRef]
- Robertson, R.N.; Highkin, H.R.; Smydzuk, J.; Went, F.W. The Effect of Environmental Conditions on the Development of Pea Seeds. Aust. J. Biol. Sci. 1962, 15, 1–15. [Google Scholar] [CrossRef]
- Wang, N.; Daun, J.K. Effect of variety and crude protein content on nutrients and certain antinutrients in field peas (Pisum sativum). J. Sci. Food Agric. 2004, 84, 1021–1029. [Google Scholar] [CrossRef]
- Reichert, R.D.; MacKenzie, S.L. Composition of peas (Pisum sativum) varying widely in protein content. J. Agric. Food Chem. 1982, 30, 312–317. [Google Scholar] [CrossRef]
- Smykal, P.; Aubert, G.; Burstin, J.; Coyne, C.J. Pea (Pisum sativum L.) in the Genomic Era. Agronomy 2012, 2, 74–115. [Google Scholar] [CrossRef]
- Burstin, J.; Salloignon, P.; Chabert-Martinello, M.; Magnin-Robert, J.-B.; Siol, M.; Jacquin, F.; Chauveau, A.; Pont, C.; Aubert, G.; Delaitre, C.; et al. Genetic diversity and trait genomic prediction in a pea diversity panel. BMC Genom. 2015, 16, 105. [Google Scholar] [CrossRef]
- Cieslarova, J.; Hybl, M.; Griga, M.; Smykal, P. Molecular analysis of temporal genetic structuring in pea (Pisum sativum L.) cultivars bred in the Czech Republic and in former Czechoslovakia since the mid-20th century. Czech J. Genet. Plant Breed. 2012, 48, 61–73. [Google Scholar] [CrossRef]
- Hanci, F. Genetic variability in peas (Pisum sativum L.) from Turkey asssessed with molecular and morphological markers. Folia Hortic. 2019, 31, 101–116. [Google Scholar] [CrossRef]
- Sharma, A.; Sharma, S.; Kumar, N.; Rana, R.S.; Sharma, P.; Kumar, P.; Rani, M. Morpho-molecular genetic diversity and population structure analysis in garden pea genotypes using simple sequence repeat markers. PLoS ONE 2022, 17, e0273499. [Google Scholar] [CrossRef]
- Haliloglu, K.; Turkoglu, A.; Tan, M.; Poczai, P. SSR-Based Molecular Identification and Population Structure Analysis for Forage Pea (Pisum sativum var arvense L.) Landraces. Genes 2022, 13, 1086. [Google Scholar] [CrossRef]
- Mohamed, A.; García-Martínez, S.; Loumerem, M.; Carbonell, P.; Ruiz, J.J.; Boubaker, M. Assessment of genetic diversity among local pea (Pisum sativum L.) accessions cultivated in the arid regions of Southern Tunisia using agro-morphological and SSR molecular markers. Genet. Resour. Crop Evol. 2019, 66, 1189–1203. [Google Scholar] [CrossRef]
- Hagenblad, J.; Boström, E.; Nygårds, L.; Leino, M.W. Genetic diversity in local cultivars of garden pea (Pisum sativum L.) conserved ‘on farm’ and in historical collections. Genet. Resour. Crop Evol. 2014, 61, 413–422. [Google Scholar] [CrossRef][Green Version]
- Singh, J.; Dhall, R.K.; Vikal, Y. Genetic diversity studies in Indian germplasm of pea (Pisum sativum L.) using morphological and microsatellite markers. Genetika 2021, 53, 473–491. [Google Scholar] [CrossRef]
- Dribnokhodova, O.P.; Gostimsky, S.A. Allele polymorphism of microsatellite loci in pea Pisum sativum L. lines, varieties, and mutants. Russ. J. Genet. 2009, 45, 788–793. [Google Scholar] [CrossRef]
- Gainullina, K.P.; Kuluev, B.R.; Davletov, F.A. Genetic diversity assessment in pea cultivars and lines using the SSR analysis. Proc. Appl. Bot. Genet. Breed. 2020, 181, 70–80. [Google Scholar] [CrossRef]
- Ram, H.; Hedau, N.K.; Chaudhari, G.V.; Choudhary, M.; Kant, L. Genetic Diversity Assessment in Pea (Pisum sativum L.) using Microsatellite Markers. Int. J. Bio Resour. Stress Manag. 2021, 12, 402–408. [Google Scholar] [CrossRef]
- Wu, X.; Li, N.; Hao, J.; Hu, J.; Zhang, X.; Blair, M.W. Genetic Diversity of Chinese and Global Pea (Pisum sativum L.) Collections. Crop Sci. 2017, 57, 1574–1584. [Google Scholar] [CrossRef]
- Zong, X.-X.; Guan, J.-P.; Wang, S.-M.; Liu, Q.-C.; Redden, R.R.; Ford, R. Genetic Diversity and Core Collection of Alien Pisum sativum L. Germplasm. Acta Agron. Sin. 2008, 34, 1518–1528. [Google Scholar] [CrossRef]
- Teshome, A.; Bryngelsson, T.; Dagne, K.; Geleta, M. Assessment of genetic diversity in Ethiopian field pea (Pisum sativum L.) accessions with newly developed EST-SSR markers. BMC Genet. 2015, 16, 102. [Google Scholar] [CrossRef]
- Botstein, D.; White, R.L.; Skolnick, M.; Davis, R.W. Construction of a genetic linkage map in man using restriction length polymorphisms. Am. J. Hum. Genet. 1980, 32, 314–331. [Google Scholar] [PubMed]
- Loridon, K.; McPhee, K.; Morin, J.; Dubreuil, P.; Pilet-Nayel, M.L.; Aubert, G.; Rameau, C.; Baranger, A.; Coyne, C.; Lejeune-Hénaut, I.; et al. Microsatellite marker polymorphism and mapping in pea (Pisum sativum L.). Theor. Appl. Genet. 2005, 111, 1022–1031. [Google Scholar] [CrossRef] [PubMed]
- Tar’an, B.; Zhang, C.; Warkentin, T.; Tullu, A.; Vandenberg, A. Genetic diversity among varieties and wild species accessions of pea (Pisum sativum L.) based on molecular markers, and morphological and physiological characters. Genome 2005, 48, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Bouhadida, M.; Srarfi, F.; Saadi, I.; Kharrat, M.M. Molecular characterization of pea (Pisum sativum L.) using microsatellite markers. IOSR J. Appl. Chem. 2013, 5, 57–61. [Google Scholar] [CrossRef]
- Ahmad, S.; Singh, M.; Lamb-Palmer, N.D.; Lefsrud, M.; Singh, J. Assessment of genetic diversity in 35 Pisum sativum accessions using microsatellite markers. Can. J. Plant Sci. 2012, 92, 1075–1081. [Google Scholar] [CrossRef]
- Jain, S.; Kumar, A.; Mamidi, S.; McPhee, K. Genetic diversity and population structure among pea (Pisum sativum L.) cultivars as revealed by simple sequence repeat and novel genic markers. Mol. Biotechnol. 2014, 56, 925–938. [Google Scholar] [CrossRef]
- Nasiri, J.; Haghnazari, A.; Saba, J. Genetic diversity among varieties and wild species accessions of pea (Pisum sativum L.) based on SSR markers. Afr. J. Biotechnol. 2009, 8, 3405–3417. [Google Scholar] [CrossRef]
- Kumari, P.; Basal, N.; Singh, A.K.; Rai, V.P.; Srivastava, C.P.; Singh, P.K. Genetic diversity studies in pea (Pisum sativum L.) using simple sequence repeat markers. Genet. Mol. Res. 2013, 12, 3540–3550. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Saint-Denis, T.; Goupy, J. Optimization of a nitrogen analyses based on the Dumas method. Anal. Chim. Acta 2004, 515, 191–198. [Google Scholar] [CrossRef]
- Mosse, J. Nitrogen to Protein Conversion Factor for Ten Cereals and Six Legumes or Oilseeds. A Reappraisal of Its Definition and Determination. Variation According to Species and to Seed Protein Content. J. Agric. Food Chem. 1990, 38, 18–24. [Google Scholar] [CrossRef]
- Coyne, C.; Grusak, M.; Razai, L.; Baik, B. Variation for pea seed protein concentration in the USDA core collection. Pisum Genet. 2005, 37, 5–9. [Google Scholar]
- Swofford, D.L. PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods); Version 4; Sinauer Associates: Sunderland, MA, USA, 2003. [Google Scholar]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bionformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GENALEX6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Kalinowski, S. How well do evolutionary trees describe genetic relationships among populations? Heredity 2009, 102, 506–513. [Google Scholar] [CrossRef] [PubMed]
- Soltis, P.S.; Soltis, D.E. Applying the Bootstrap in Phylogeny Reconstruction. Stat. Sci. 2003, 18, 256–267. [Google Scholar] [CrossRef]
- Wiens, J.J. Missing data and the design of phylogenetic analyses. J. Biomed. Inform. 2006, 39, 34–42. [Google Scholar] [CrossRef]
- Klein, A.; Houtin, H.; Rond-Coissieux, C.; Naudet-Huart, M.; Touratier, M.; Marget, P.; Burstin, J. Meta-analysis of QTL reveals the genetic control of yield-related traits and seed protein content in pea. Sci. Rep. 2020, 10, 15925. [Google Scholar] [CrossRef] [PubMed]
- Solberg, S.Ø.; Yndgaard, F.; Poulsen, G.; von Bothmer, R. Seed yield and protein content in the Weibullsholm Pisum collection. Genet. Resour. Crop Evol. 2017, 64, 2035–2047. [Google Scholar] [CrossRef]
- Carlson-Nilsson, U.; Aloisi, K.; Vågen, I.M.; Rajala, A.; Mølmann, J.B.; Rasmussen, S.K.; Niemi, M.; Wojciechowska, E.; Pärssinen, P.; Poulsen, G.; et al. Trait Expression and Environmental Responses of Pea (Pisum sativum L.) Genetic Resources Targeting Cultivation in the Arctic. Front. Plant Sci. 2021, 12, 688067. [Google Scholar] [CrossRef] [PubMed]
- Slinkard, A.E. Percent Protein in the USDA World Collection of Peas Grown at Saskatoon in 1971; Crop Science Department, University of Saskatchewan: Saskatoon, SK, Canada, 1972. [Google Scholar]
- Wittenberg, A.H.J.; van der Lee, T.A.J.; Visser, R.G.F.; Schouten, H.J.; Kilian, A. Genome-Wide Profiling Using Diversity Arrays Technology (DArT); Theoretical Considerations and Practical Protocols. In Genetic Mapping Using the Diversity Arrays Technology (DArT)—Application and Validation Using the Whole-Genome Sequences of Arabidopsis Thaliana and the Fungal Wheat Pathogen Mycosphaerella Graminicola; Wittenberg, A.H.J., Ed.; Wageningen University: Wageningen, The Netherlands, 2007; Chapter 2; pp. 25–59. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SKR. No. | Accession | Name | AA5 | AC58 | AA9 | TSW 2017 (g) | Protein 2017 (%) | Protein 2018 (%) | Protein 2019 (%) |
|---|---|---|---|---|---|---|---|---|---|
| SKR1 | NGB103441 | WBH 3441 | x | x | x | 178.1 | 23.4 | 26.9 | |
| SKR2 | NGB101418 | WBH 1418 | x | x | x | 107.3 | 20.3 | 20.8 | |
| SKR3 | NGB103563 | WBH 3563 | x | x | x | 16.0 | |||
| SKR4 | NGB101515 | WBH 1515 | x | x | x | ||||
| SKR5 | NGB100756 | WBH 756 | x | x | x | 185.0 | 25.0 | 29.9 | 25.0 |
| SKR6 | NGB101687 | WBH 1687 | x | x | x | 19.7 | |||
| SKR7 | NGB105795 | multimicrodentatus | x | x | x | 15.6 | |||
| SKR8 | NGB103423 | Latah | x | x | 14.3 | ||||
| SKR9 | NGB100463 | WBH 463 | x | x | x | 16.0 | |||
| SKR10 | NGB103565 | WBH 3565 | x | x | x | 12.0 | |||
| SKR11 | NGB101735 | Improved Harbinger | x | x | x | 244 | 21.0 | 25.6 | |
| SKR12 | NGB101736 | rouge C15 | x | x | x | 17.7 | |||
| SKR13 | NGB101339 | Bonneville | x | x | x | 180.2 | 26.4 | 22.0 | |
| SKR14 | NGB101741 | New Season | x | x | x | 19.5 | |||
| SKR15 | NGB101603 | WBH 1603 | x | x | x | ||||
| SKR16 | NGB103546 | W.S.U.-28 | x | x | x | 22.3 | 25.9 | ||
| SKR17 | NGB101772 | Wellensiek’s tester | x | x | x | 18.6 | |||
| SKR18 | NGB101325 | WBH 1325 | x | x | x | ||||
| SKR19 | NGB101330 | WBH 1330 | x | x | x | 20.0 | |||
| SKR20 | NGB101165 | WBH 1165 | x | x | x | 142.2 | 23.0 | 24.1 | |
| SKR21 | NGB103436 | WBH 3436 | x | x | x | 18.3 | |||
| SKR22 | NGB102963 | Wilt Resistant Thomas Laxton | 172.8 | 24.7 | 25.8 | ||||
| SKR23 | NGB101889 | x | x | x | |||||
| SKR24 | NGB103439 | WBH 3439 | x | x | x | 13.9 | |||
| SKR25 | NGB103429 | WBH 3429 | x | x | x | 20.6 | 24.1 | ||
| SKR26 | NGB103583 | WBH 3583 | x | x | x | ||||
| SKR27 | NGB100592 | WBH 592 | x | x | |||||
| SKR28 | NGB103452 | WBH 3452 | x | x | x | ||||
| SKR29 | NGB102158 | WBH 2158 | x | x | x | 135.5 | 18.6 | 21.5 | |
| SKR30 | NGB103422 | Alaska | x | x | x | 18.2 | |||
| SKR31 | NGB103449 | Feltham First | x | x | x | 178.9 | 21.5 | 24.4 | |
| SKR32 | NGB103426 | Juneau | x | x | x | 17.8 | |||
| SKR33 | NGB103442 | WBH 3442 | x | x | x | 14.4 | |||
| SKR34 | NGB103576 | WBH 3576 | x | x | x | 187 | 31.7 | ||
| SKR35 | NGB103578 | WBH 3578 | x | x | x | 193 | 25.6 | 26.1 | 20.5 |
| SKR36 | NGB100851 | procumbens | x | x | x | 165 | 23.4 | 28.5 | |
| SKR37 | NGB101338 | Salzmunder Edelperle | x | x | x | 17.1 | |||
| SKR38 | NGB103433 | WBH 3433 | x | x | x | 16.6 | |||
| SKR39 | NGB102177 | WBH 2177 | x | x | x | 132 | 23.7 | 27.3 | |
| SKR40 | NGB103438 | WBH 3438 | x | x | x | 20.1 | |||
| SKR41 | NGB101608 | WBH 1608 | x | x | x | ||||
| SKR42 | NGB103435 | WBH 3435 | x | x | 12.1 | ||||
| SKR43 | NGB102058 | WBH 2058 | x | x | x | 20.0 | |||
| SKR44 | NGB101524 | patelliformis | x | x | x | ||||
| SKR45 | NGB101570 | WBH 1570 | x | x | x | 13.1 | |||
| SKR46 | NGB103568 | WBH 3568 | x | x | x | 64.4 | 21.7 | 22.2 | |
| SKR47 | NGB103581 | WBH 3581 | x | x | x | ||||
| SKR48 | NGB105136 | chlorotica | x | x | x | 76.4 | 30.0 | 22.9 | 24.4 |
| SKR49 | NGB102184 | New Era | x | x | x | 16.6 | |||
| SKR50 | NGB102160 | WBH 2160 | x | x | x | 144.4 | 22.4 | 28.5 | |
| SKR51 | NGB103580 | WBH 3580 | x | x | x | 156 | 23.7 | 21.3 | |
| SKR52 | NGB102022 | WBH 2022 | x | x | x | 104 | 30.2 | 23.8 | |
| SKR53 | NGB102663 | WBH 2663 | x | x | x | ||||
| SKR54 | NGB102203 | WBH 2203 | x | x | x | 15.6 | |||
| SKR55 | NGB102217 | chlorotica | x | x | x | 19.9 | |||
| SKR56 | NGB105350 | /chlorina | 157.2 | 27.3 | 24.8 | ||||
| SKR57 | NGB102431 | Laxtonian | 193 | 23.8 | 24.8 | 24.1 | |||
| SKR58 | NGB105124 | ageotropum | x | x | x | 17.0 | |||
| SKR59 | NGB102496 | x | x | x | 151 | 32.1 | |||
| SKR60 | NGB102214 | chlorotica | x | x | x | 123 | 23.3 | 28.9 | |
| SKR61 | NGB102210 | chlorotica | x | x | x | 145 | 23.8 | 24.3 | |
| SKR62 | NGB102136 | WBH 2136 | x | x | x | 162 | 25.4 | 26.3 | |
| SKR63 | NGB102216 | chlorotica | x | x | x | 104.8 | 29.4 | 30.6 | 24.3 |
| SKR64 | NGB105862 | densinodosum | 155 | 20.9 | 34.1 | ||||
| SKR65 | NGB105428 | chlorotica | x | x | x | 15.6 | |||
| SKR66 | NGB102574 | Beta | x | x | x | 150.1 | 22.4 | 24.6 | |
| SKR67 | NGB102622 | x | x | x | 164 | 22.4 | 24.9 | 21.1 | |
| SKR68 | NGB102988 | WBH 2988 | x | x | x | 20.2 | |||
| SKR69 | NGB106051 | reduced in wax | x | x | x | 166.6 | 21.4 | 29.0 | |
| SKR70 | NGB102432 | Hundredfold | x | x | x | 18.2 | |||
| SKR71 | NGB105789 | /compactum | x | x | x | 19.0 | |||
| SKR72 | NGB106060 | supaeromaculata | 16.3 | ||||||
| SKR73 | NGB102239 | x | x | x | 20.3 | ||||
| SKR74 | NGB102369 | WBH 2369 | x | x | x | 129.2 | 21.9 | 25.2 | |
| SKR75 | NGB105051 | /compactum | x | x | x | ||||
| SKR76 | NGB105432 | reductus | x | x | x | 84 | 26.2 | 25.1 | |
| SKR77 | NGB106080 | WBH 6080 | x | x | x | 17.4 | |||
| SKR78 | NGB102579 | x | x | 231 | 27.0 | 23.1 | |||
| SKR79 | NGB102581 | x | x | x | 24.5 | ||||
| SKR80 | NGB105765 | chlorotica | 132 | 20.9 | 26.1 | ||||
| SKR81 | NGB103431 | WBH 3431 | x | x | x | 18.1 | |||
| SKR82 | NGB102823 | Austrian Winter | x | x | x | 13.1 | |||
| SKR83 | NGB103571 | WBH 3571 | x | x | x | 14.4 | |||
| SKR84 | NGB103572 | WBH 3572 | x | x | x | ||||
| SKR85 | NGB103573 | WBH 3573 | x | x | x | 17.3 | |||
| SKR86 | NGB103585 | WBH 3585 | x | x | x | 13.5 | |||
| SKR87 | NGB101452 | WBH 1452 | x | x | x | 159 | 22.3 | 22.0 | |
| SKR88 | NGB101192 | WBH 1192 | 152 | 28.3 | 24.6 | 24.8 | |||
| SKR89 | NGB101391 | WBH 1391 | x | x | x | ||||
| SKR90 | NGB101689 | WBH 1689 | x | x | x | 25.1 | |||
| SKR91 | NGB101017 | WBH 1017 | x | x | x | 151.4 | 24.9 | 28.9 | |
| SKR92 | NGB101351 | WBH 1351 | x | x | x | ||||
| SKR93 | NGB105534 | supaeromaculata 68 | x | x | x | 113.1 | 28.9 | 28.7 | 23.4 |
| SKR94 | NGB102188 | WBH 2188 | x | x | x | 181.87 | 22.6 | 27.7 | |
| SKR95 | NGB103420 | WA 788 | x | x | x | 14.6 | |||
| SKR96 | NGB103434 | WBH 3434 | 18.3 | ||||||
| SKR97 | NGB103421 | Lilaska | x | x | 18.9 | ||||
| SKR98 | NGB103428 | WBH 3428 | 18.5 | ||||||
| SKR99 | NGB103432 | WBH 3432 | x | x | 19.6 | ||||
| SKR100 | NGB102070 | Puke | x | x | 16.2 | ||||
| SKR101 | NGB103577 | WBH 3577 | x | x | x | 174.8 | 21.9 | 26.0 | |
| SKR102 | NGB100909 | WBH 909 | 160 | 22.0 | 24.0 | ||||
| SKR103 | NGB101500 | WBH 1500 | x | x | 99.1 | 20.5 | 26.3 | ||
| SKR104 | NGB103437 | WBH 3437 | 67 | 23.7 | 24.6 | ||||
| SKR105 | NGB102069 | Patea | x | x | x | 212.4 | 22.0 | 19.4 | |
| SKR106 | NGB103561 | WBH 3561 | x | x | 21.2 | ||||
| SKR107 | NGB103567 | WBH 3567 | x | x | x | 17.0 | |||
| SKR108 | NGB105310 | cerosa | x | x | x | 108.8 | 26.4 | 27.0 | 22.5 |
| SKR109 | NGB103574 | WBH 3574 | x | x | x | ||||
| SKR110 | NGB103575 | WBH 3575 | x | x | |||||
| SKR111 | NGB103579 | WBH 3579 | x | x | x | 17.0 | |||
| SKR112 | NGB103584 | WBH 3584 | x | x | x | ||||
| SKR113 | NGB103602 | WBH 3602 | x | x | x | 11.2 | |||
| SKR114 | NGB103603 | WBH 3603 | x | x | x | 18.7 | |||
| SKR115 | NGB102901 | x | x | x | 12.8 | ||||
| SKR116 | NGB103430 | WBH 3430 | x | x | x | 18.3 | |||
| SKR117 | NGB103427 | WBH 3427 | x | x | 17.1 | ||||
| SKR118 | NGB102159 | WBH 2159 | x | x | 18.5 | ||||
| SKR119 | NGB102844 | x | x | 19.2 | |||||
| SKR120 | NGB102185 | New Wales | x | x | x | 209 | 24.6 | 25.4 | 23.0 |
| SKR121 | NGB101132 | WBH 1132 | x | x | x | ||||
| SKR122 | NGB103570 | WBH 3570 | x | x | x | 19.5 | |||
| SKR123 | NGB103582 | WBH 3582 | x | x | 18.7 | ||||
| SKR124 | NGB103604 | WBH 3604 | x | x | x | 12.4 | |||
| SKR125 | NGB103605 | WBH 3605 | x | x | 13.3 | ||||
| SKR126 | NGB101784 | Mrkos horizontale | x | x | x | ||||
| SKR127 | NGB102063 | WBH 2063 | x | x | x | 172 | 24.4 | 26.9 | 20.7 |
| SKR128 | NGB105765 | chlorotica | 125.1 | 26.1 | 25.1 | ||||
| SKR129 | NGB102578 | x | x | 148 | 33.6 | 26.0 | 21.2 | ||
| SKR130 | NGB102537 | 19.1 | |||||||
| SKR131 | NGB103458 | x | x | 118.4 | 20.6 | 26.7 | 22.2 | ||
| SKR132 | NGB105161 | costata | x | x | x | 92 | 25.8 | 23.3 | |
| SKR133 | NGB102999 | WBH 2999 | x | x | x | 17.4 | |||
| SKR134 | NGB102687 | WBH 2687 | x | x | 178.5 | 21.2 | 26.9 | 22.7 | |
| SKR135 | NGB105267 | variomaculata | x | x | 148 | 27.3 | 29.8 | 22.0 | |
| SKR136 | NGB105814 | chlorotica | x | x | x | 118 | 21.9 | 26.8 | |
| SKR137 | NGB102037 | WBH 2037 | x | x | x | 17.2 | |||
| SKR138 | NGB102190 | WBH 2190 | x | x | 223 | 28.8 | 26.1 | 22.4 | |
| SKR139 | NGB102763 | Wonder Van Amerika I2048 | x | x | x | 142 | 22.2 | 27.4 | |
| SKR140 | NGB106000 | vixcerata | 232 | 29.4 | 28.6 | 23.0 | |||
| SKR141 | NGB102582 | 19.5 | |||||||
| SKR142 | NGB105449 | variomaculata | x | x | x | 120.2 | 22.9 | 27.5 | |
| SKR144 | NGB102370 | WBH 2370 | x | x | x | 22.2 | |||
| SKR145 | NGB102621 | x | x | x | 19.2 | ||||
| SKR146 | NGB102927 | x | x | x | 17.5 | ||||
| SKR147 | NGB103459 | x | x | x | 150 | 25.5 | 25.7 | ||
| SKR148 | NGB105820 | /xantha | x | x | x | ||||
| SKR149 | NGB102521 | x | x | x | 238 | 20.5 | 27.3 | 22.4 | |
| SKR150 | NGB102617 | 16.8 | |||||||
| SKR151 | NGB103457 | x | x | x | 144 | 22.8 | 21.7 | ||
| SKR152 | NGB105410 | variomaculata | x | x | x | 130.6 | 26.2 | 26.9 | 23.3 |
| SKR153 | NGB105981 | precocious yellowing | x | x | x | 107 | 24.5 | 29.1 | 23.6 |
| SKR154 | NGB102480 | x | x | x | 124.4 | 25.5 | 29.5 | 21.4 | |
| SKR155 | NGB102588 | x | x | x | |||||
| SKR156 | NGB105961 | chlorotica | x | x | x | 156.6 | 23.2 | 29.0 | |
| SKR157 | NGB102205 | chlorotica | x | x | x | 134 | 22.2 | 25.8 | |
| SKR158 | NGB102497 | 193 | 31.6 | ||||||
| SKR159 | NGB102533 | 19.7 | |||||||
| SKR160 | NGB102831 | x | x | x | 149 | 24.2 | 25.2 | ||
| SKR161 | NGB103052 | WBH 3052 | x | x | x | 123 | 28.7 | 27.5 | 24.0 |
| SKR162 | NGB103061 | WBH 3061 | x | x | x | ||||
| SKR163 | NGB103116 | x | x | x | 19.4 | ||||
| SKR164 | NGB105261 | variomaculata | x | x | x | 150 | 24.3 | 33.1 | 20.0 |
| SKR165 | NGB105806 | chlorotica | x | x | x | 131 | 23.5 | 25.7 | 22.5 |
| SKR166 | NGB105983 | precocious yellowing | 90 | 25.6 | 26.9 | 23.2 | |||
| SKR167 | NGB102005 | Nischkes Riesengefärgs Wi. Erbse | x | x | x | 17.2 | |||
| SKR168 | NGB100993 | WBH 993 | x | x | x | 173 | 27.9 | 32.0 | 19.9 |
| SKR169 | NGB103559 | WBH 3559 | x | x | x | 12.7 | |||
| SKR170 | NGB102405 | Supergrade | x | x | x | 194 | 21.9 | 24.6 | 21.8 |
| SKR171 | NGB101742 | New Wales | x | x | x | 182 | 22.5 | 29.9 | |
| SKR172 | NGB101888 | x | x | x | |||||
| SKR173 | NGB102423 | Senator | x | x | x | 18.4 | |||
| SKR174 | NGB103560 | WBH 3560 | 10.9 | ||||||
| SKR175 | NGB102429 | Gradus | 19.7 | ||||||
| SKR176 | NGB103569 | WBH 3569 | x | x | x | 12.9 | |||
| SKR177 | NGB103545 | Puget | x | x | x | ||||
| SKR178 | NGB103566 | WBH 3566 | x | x | x | 16.2 | |||
| SKR179 | NGB103484 | Dhamar | x | x | x | ||||
| SKR180 | NGB103425 | WBH 3425 | x | x | x | 18.7 | |||
| SKR181 | NGB103547 | Grant | x | x | x | 145 | 22.4 | 25.6 | |
| SKR182 | NGB102130 | WBH 2130 | x | x | 9.3 | ||||
| SKR183 | NGB103609 | WBH 3609 | 17.2 | ||||||
| SKR184 | NGB101979 | Ambrosia | x | x | x | 144 | 24.0 | 27.4 | 21.8 |
| SKR185 | NGB100800 | Primus | x | x | x | 149 | 22.5 | 26.8 | |
| SKR186 | NGB101721 | Midfreezer | x | x | x | 19.1 | |||
| SKR187 | NGB101395 | WBH 1395 | x | x | x | 148 | 24.4 | 27.1 | 23.1 |
| SKR188 | NGB101677 | Mexique 4 | x | x | x | 16.7 | |||
| SKR189 | NGB103440 | WBH 3440 | x | x | x | 143 | 22.7 | 26.8 | |
| SKR190 | NGB101462 | WBH 1462 | x | x | x | 25.0 | 24.2 | ||
| SKR191 | NGB101362 | clavicula | x | x | x | 156 | 17.0 | ||
| SKR192 | NGB100640 | WBH 640 | x | x | x | 18.4 | |||
| SKR193 | NGB103450 | Meteor | x | x | x | 18.6 | |||
| SKR195 | NGB103562 | WBH 3562 | x | x | x | 164 | 20.9 | 26.5 | |
| SKR196 | NGB101463 | Sigyn | x | x | x | 132 | 23.2 | 24.9 | |
| SKR198 | NGB101341 | Klema Vereduna | x | x | x | 20.1 | |||
| SKR199 | NGB103424 | WBH 3424 | x | x | x | 16.2 | |||
| SKR200 | NGB103544 | Ranger | x | x | 214 | 21.7 | 25.2 | ||
| SKR201 | NGB103564 | WBH 3564 | x | x | x | 16.9 | |||
| SKR202 | NGB103610 | WBH 3610 | x | x | 15.7 | ||||
| SKR203 | NGB105454 | viridis | x | x | x | 161 | 22.8 | 25.3 | |
| SKR204 | NGB105995 | vixcerata | x | x | x | 134 | 29.0 | 28.8 | 26.4 |
| SKR205 | NGB102212 | chlorotica | x | x | x | 18.7 | |||
| SKR206 | NGB102688 | WBH 2688 | x | x | x | ||||
| SKR207 | NGB105271 | costata | x | x | x | 17.2 | |||
| SKR208 | NGB105565 | chlorotica | x | x | x | 23.7 | |||
| SKR209 | NGB105848 | fasciata | x | x | x | 26.0 | |||
| SKR210 | NGB106116 | narrow leaflet base | x | x | x | 21.1 | |||
| SKR211 | NGB102178 | Thomas Laxton | x | x | x | 225 | 22.0 | 33.0 | |
| SKR212 | NGB102737 | WW 709 | x | x | x | 17.0 | |||
| SKR213 | NGB102071 | Piri | x | x | x | 18.6 | |||
| SKR214 | NGB102183 | Darkskin Perfection | x | x | x | 154 | 22.2 | 25.2 | |
| SKR215 | NGB103451 | Lilaska | x | x | x | 16.7 | |||
| SKR216 | NGB102072 | Pania | x | x | 175 | 20.8 | 27.4 | ||
| SKR217 | NGB103606 | WBH 3606 | x | x | x | ||||
| SKR218 | NGB103607 | WBH 3607 | x | x | 254.2 | 23.9 | 26.3 | ||
| SKR219 | NGB103628 | ramosus | x | x | x | 96 | 21.6 | 25.1 | |
| SKR220 | NGB101304 | WBH 1304-1 | 159 | 22.4 | 27.9 | ||||
| SKR221 | NGB101304 | WBH 1304-2 | 16.1 | ||||||
| SKR222 | NGB101304 | WBH 1304-3 | 150 | 22.4 | 27.7 | ||||
| SKR223 | NGB101304 | WBH 1304-4 | 18.3 | ||||||
| SKR224 | NGB101304 | WBH 1304-5 | 14.6 | ||||||
| SKR225 | NGB101304 | WBH 1304-6 | 19.1 | ||||||
| SKR226 | NGB101304 | WBH 1304-7 | 163 | 22.1 | 23.6 | ||||
| SKR227 | NGB101304 | WBH 1304-8 | 154 | 21.4 | 20.9 | ||||
| SKR228 | NGB101304 | WBH 1304-9 | 14.5 | ||||||
| SKR229 | NGB101304 | WBH 1304-10 | 102 | 21.3 | 26.5 | ||||
| SKR230 | NGB101836 | 1 | 123 | 20.9 | 25.1 |
| Marker | Forward Primer (5′—3′) | Reverse Primer (5′—3′) | Color |
|---|---|---|---|
| AA5 | tgccaatcctgaggtattaacacc + M13 | catttttgcagttgcaatttcgt | FAM, Blue |
| A9 | gtgcagaagcatttgttcagat + M13 | cccacatatatttggttggtca | NED, Yellow |
| AC58 | Tccgcaatttggtaacactg + M13 | cgtcaatttcttttatgctgag | VIC, Green |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Winther, L.; Rasmussen, S.K.; Poulsen, G.; Lange, C.B.A. Assessment of Genetic Diversity and Protein Content of Scandinavian Peas (Pisum sativum). Agronomy 2023, 13, 2307. https://doi.org/10.3390/agronomy13092307
Winther L, Rasmussen SK, Poulsen G, Lange CBA. Assessment of Genetic Diversity and Protein Content of Scandinavian Peas (Pisum sativum). Agronomy. 2023; 13(9):2307. https://doi.org/10.3390/agronomy13092307
Chicago/Turabian StyleWinther, Louise, Søren Kjærsgaard Rasmussen, Gert Poulsen, and Conny Bruun Asmussen Lange. 2023. "Assessment of Genetic Diversity and Protein Content of Scandinavian Peas (Pisum sativum)" Agronomy 13, no. 9: 2307. https://doi.org/10.3390/agronomy13092307
APA StyleWinther, L., Rasmussen, S. K., Poulsen, G., & Lange, C. B. A. (2023). Assessment of Genetic Diversity and Protein Content of Scandinavian Peas (Pisum sativum). Agronomy, 13(9), 2307. https://doi.org/10.3390/agronomy13092307