
Whole-Genome Resequencing Reveals the Genetic Diversity and Selection Signatures of the Brassica juncea from the Yunnan-Guizhou Plateau


 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Phenotyping
2.2. Resequencing and Discovery of Genomic Variations
2.3. Population Structure and Phylogenetic Analyses
2.4. Genome-Wide Selective Sweep Analysis and Selection of Candidate Genes
2.5. GWAS and Identification of Candidate Genes
3. Results
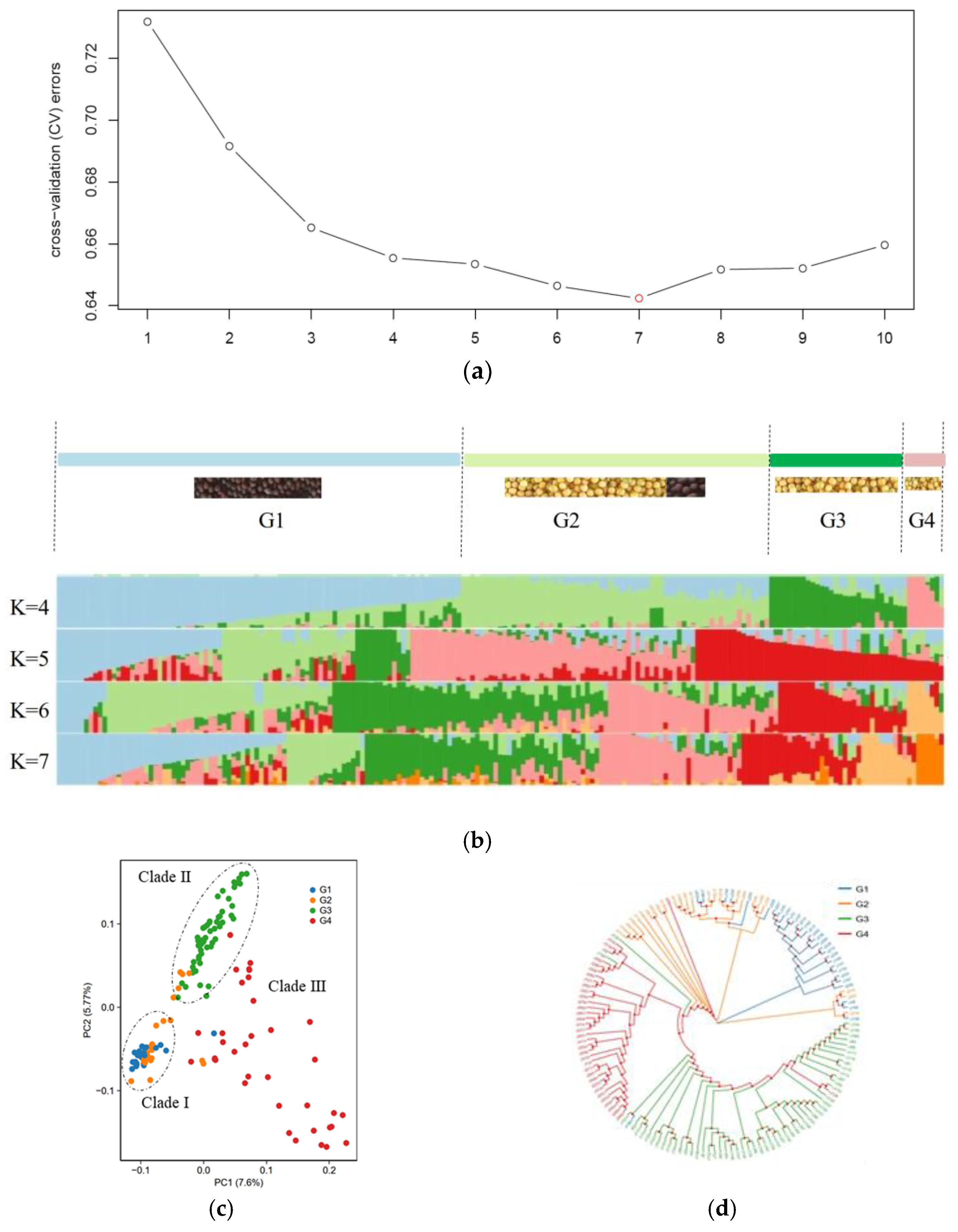
3.1. Genotype of B. juncea
3.2. Population Structure of B. juncea
3.3. Genetic Diversity of B. juncea
3.4. Kinship of B. juncea
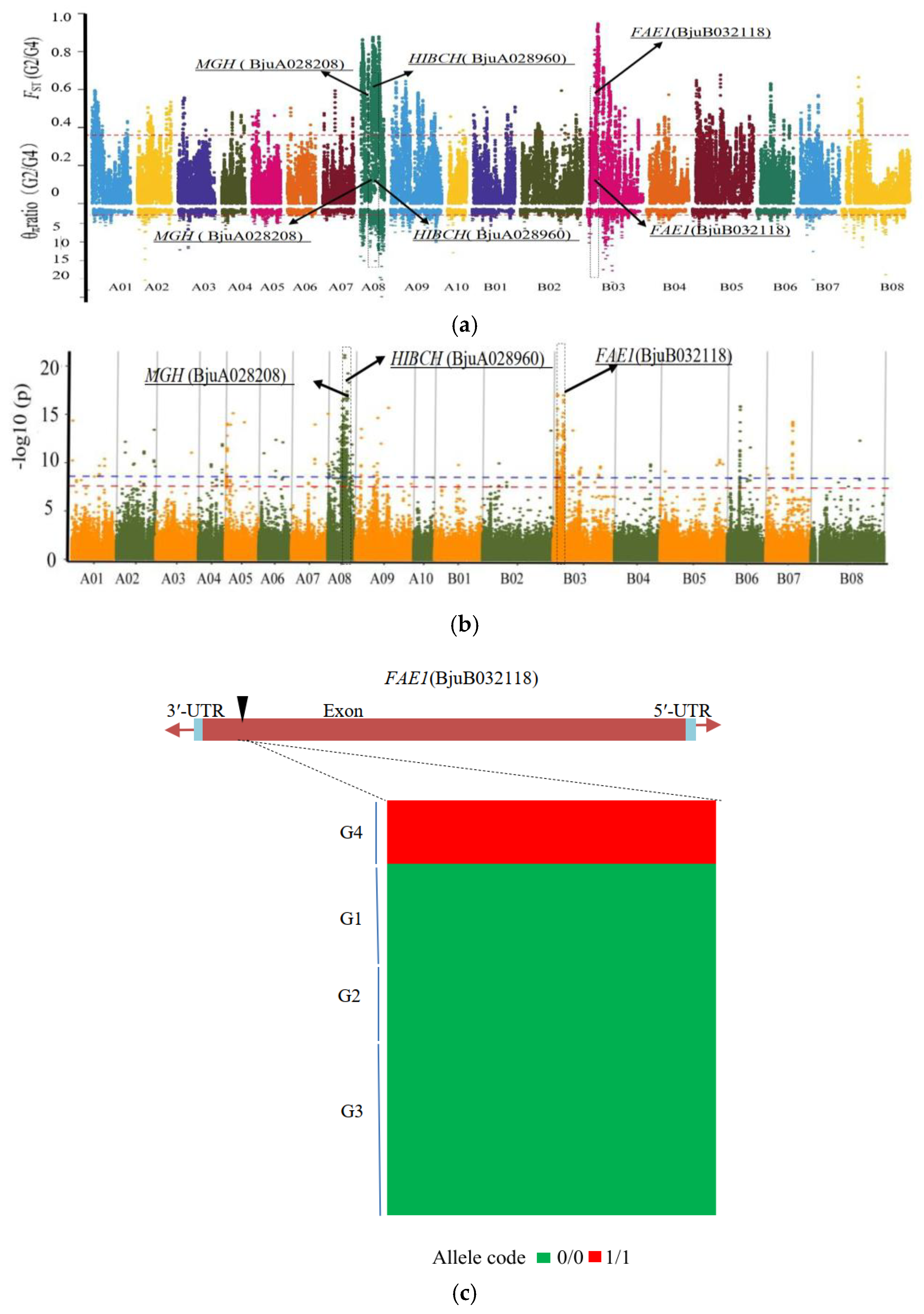
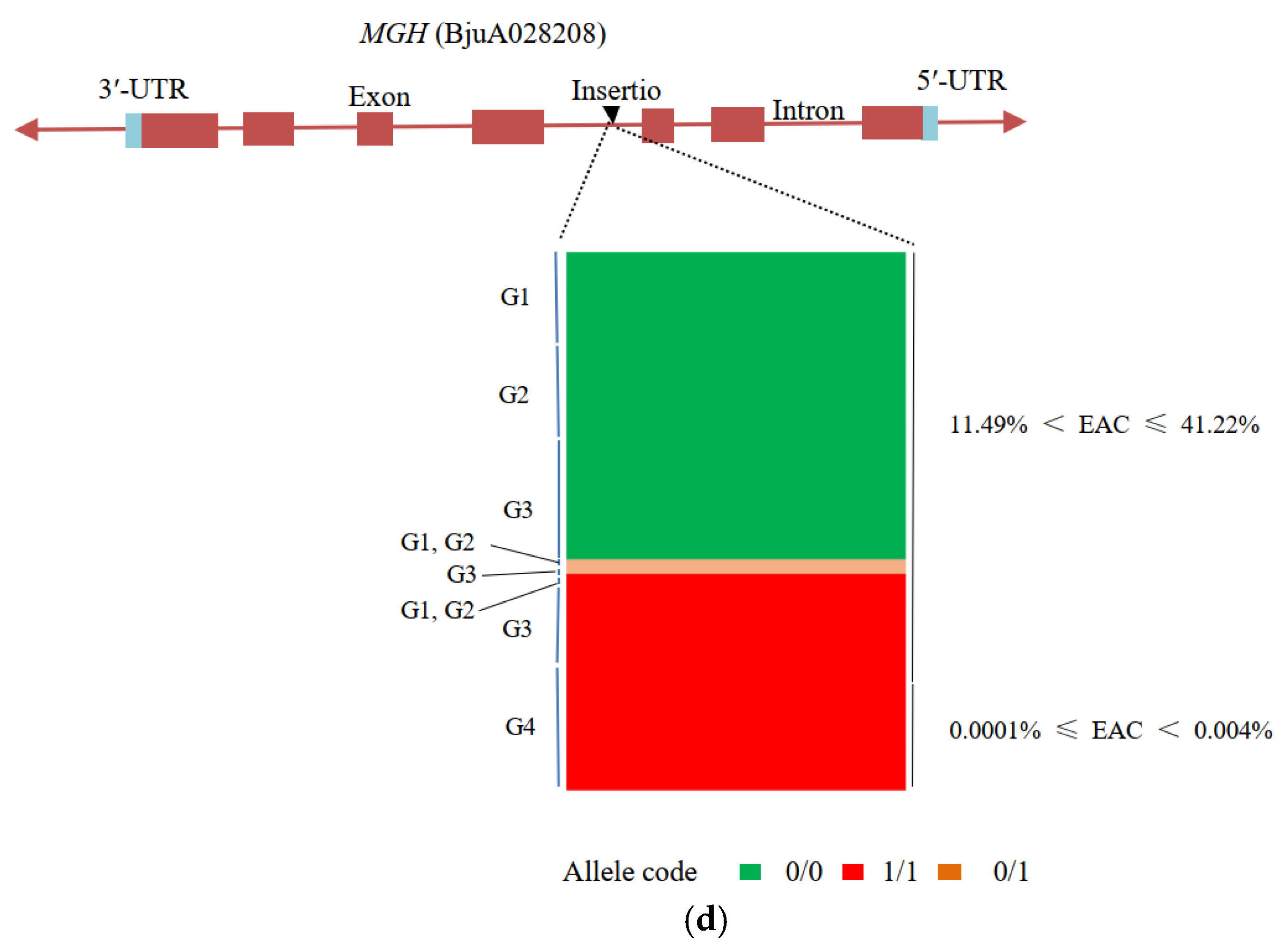
3.5. Candidate Genes for Seed Coat Color
3.6. Major Genes for Fatty Acid Composition
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nagaharu, U.; Nagaharu, N. Genome analysis in Brassica with special reference to the experimental formation of B. napus and peculiar mode of fertilization. Jpn. J. Bot. 1935, 7, 389–452. [Google Scholar]
- Yang, J.; Liu, D.; Wang, X.; Ji, C.; Cheng, F.; Liu, B.; Hu, Z.; Chen, S.; Pental, D.; Ju, Y. The genome sequence of allopolyploid Brassica juncea and analysis of differential homoeolog gene expression influencing selection. Nat. Genet. 2016, 48, 1225–1232. [Google Scholar] [CrossRef]
- Hemingway, J. The mustard species: Condiment and food ingredient use and potential as oilseed crops. In Brassica Oilseeds: Production and Utilization; Komber, D.S., McGregor, D.I., Eds.; CAB International: Wallingford, UK, 1995; pp. 373–383. [Google Scholar]
- Vavilov, N.I. Phytogeographic basis of plant breeding. The origin, variation, immunity and breeding of cultivated plants. Soil Sci. 1951, 72, 482. [Google Scholar] [CrossRef]
- Shigesaburo, T. A plant exploration in Brassica and allied genera. Tohoku J. Agric. Res. 1967, 17, 249–277. [Google Scholar]
- Hinata, K.; Prakash, S. Ethnobotany and evolutionary origin of Indian Oleiferous brassicae. Indian J. Genet. Plant Breed. 1984, 44, 102–112. [Google Scholar]
- Yang, J.; Zhang, C.; Zhao, N.; Zhang, L.; Hu, Z.; Chen, S.; Zhang, M. Chinese root-type mustard provides phylogenomic insights into the evolution of the multi-use diversified allopolyploid Brassica juncea. Mol. Plant. 2018, 11, 512–514. [Google Scholar] [CrossRef]
- Gladis, T.; Hammer, K. The Brassica collection in Gatersleben: Brassica juncea, Brassica napus, Brassica nigra, and Brassica rapa. Feddes Rep. 1992, 103, 469–507. [Google Scholar] [CrossRef]
- Mackie, S.J. Comparison of RF3 B. juncea to RF3 B. napus. Food Nutr. Sci. 2021, 12, 1041–1061. [Google Scholar]
- Woods, D.; Capcara, J.; Downey, R. The potential of mustard (Brassica juncea (L.) Coss) as an edible oil crop on the Canadian Prairies. Can. J. Plant Sci. 1991, 71, 195–198. [Google Scholar] [CrossRef]
- Liu, Z.; Guan, C.; Chen, S.; Liu, S.; Yang, L. Transfer of superior traits from Brassica juncea into Brassica napus. Agric. Sci. Technol. Hunan 2010, 11, 49–52. [Google Scholar]
- Rahman, M. Production of yellow-seeded Brassica napus through interspecific crosses. Plant Breed. 2001, 120, 463–472. [Google Scholar] [CrossRef]
- Roy, N. Interspecific transfer of Brassica juncea-type high blackleg resistance to Brassica napus. Euphytica 1984, 33, 295–303. [Google Scholar] [CrossRef]
- Raney, P.; Rakow, G.; Olson, T. Development of zero erucic, low linolenic Brassica juncea utilizing interspecific crossing. In Proceedings of the 9th International Rapeseed Congress, Cambridge, UK, 4–7 July 1995; Volume 2, pp. 413–415. [Google Scholar]
- Potts, D.; Rakow, G.; Males, D.; Woods, D. The development of canola-quality Brassica juncea. Can. J. Plant Sci. 2003, 83, 117–118. [Google Scholar]
- Rashid, A.; Rakow, G.; Downey, R. Development of yellow seeded Brassica napus through interspecific crosses. Plant Breed. 1994, 112, 127–134. [Google Scholar] [CrossRef]
- Marles, M.S.; Gruber, M.Y. Histochemical characterisation of unextractable seed coat pigments and quantification of extractable lignin in the Brassicaceae. J. Sci. Food Agric. 2004, 84, 251–262. [Google Scholar] [CrossRef]
- Li, X.; Westcott, N.; Links, M.; Gruber, M.Y. Seed coat phenolics and the developing silique transcriptome of Brassica carinata. J. Agric. Food Chem. 2010, 58, 10918–10928. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lu, Y.; Yuan, Y.; Zhang, X.; Geng, J.; Chen, Y.; Cloutier, S.; McVetty, P.B.; Li, G. Map-based cloning and characterization of a gene controlling hairiness and seed coat color traits in Brassica rapa. Plant Mol. Biol. 2009, 69, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chen, L.; Hong, M.; Zhang, Y.; Zu, F.; Wen, J.; Yi, B.; Ma, C.; Shen, J.; Tu, J. A large insertion in bHLH transcription factor BrTT8 resulting in yellow seed coat in Brassica rapa. PLoS ONE 2012, 7, e44145. [Google Scholar] [CrossRef]
- Padmaja, L.K.; Agarwal, P.; Gupta, V.; Mukhopadhyay, A.; Sodhi, Y.S.; Pental, D.; Pradhan, A.K. Natural mutations in two homoeologous TT8 genes control yellow seed coat trait in allotetraploid Brassica juncea (AABB). Theor. Appl. Genet. 2014, 127, 339–347. [Google Scholar] [CrossRef]
- Zhai, Y.; Yu, K.; Cai, S.; Hu, L.; Amoo, O.; Xu, L.; Yang, Y.; Ma, B.; Jiao, Y.; Zhang, C. Targeted mutagenesis of BnTT8 homologs controls yellow seed coat development for effective oil production in Brassica napus L. Plant Biotechnol. J. 2020, 18, 1153–1168. [Google Scholar] [CrossRef] [PubMed]
- Lian, J.; Lu, X.; Yin, N.; Ma, L.; Lu, J.; Liu, X.; Li, J.; Lu, J.; Lei, B.; Wang, R. Silencing of BnTT1 family genes affects seed flavonoid biosynthesis and alters seed fatty acid composition in Brassica napus. Plant Sci. 2017, 254, 32–47. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Rivas, J.M.; Sperling, P.; Lühs, W.; Heinz, E. Spatial and temporal regulation of three different microsomal oleate desaturase genes (FAD2) from normal-type and high-oleic varieties of sunflower (Helianthus annuus L.). Mol. Breed. 2001, 8, 159–168. [Google Scholar] [CrossRef]
- Stefansson, B.; Hougen, F.W.; Downey, R. Note on the isolation of rape plants with seed oil free from erucic acid. Can. J. Plant Sci. 1961, 41, 218–219. [Google Scholar] [CrossRef]
- Li, X.; van Loo, E.N.; Gruber, J.; Fan, J.; Guan, R.; Frentzen, M.; Stymne, S.; Zhu, L.H. Development of ultra-high erucic acid oil in the industrial oil crop Crambe abyssinica. Plant Biotechnol. J. 2012, 10, 862–870. [Google Scholar] [CrossRef]
- Han, J.; Lühs, W.; Sonntag, K.; Zähringer, U.; Borchardt, D.S.; Wolter, F.P.; Heinz, E.; Frentzen, M. Functional characterization of [beta]-ketoacyl-CoA synthase genes from Brassica napus L. Plant Mol. Biol. 2001, 46, 229. [Google Scholar] [CrossRef]
- Bach, L.; Faure, J.-D. Role of very-long-chain fatty acids in plant development, when chain length does matter. Comptes Rendus Biol. 2010, 333, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Batsale, M.; Bahammou, D.; Fouillen, L.; Mongrand, S.; Joubès, J.; Domergue, F. Biosynthesis and functions of very-long-chain fatty acids in the responses of plants to abiotic and biotic stresses. Cells 2021, 10, 1284. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Mukhopadhyay, A.; Arumugam, N.; Sodhi, Y.; Pental, D.; Pradhan, A. Molecular tagging of erucic acid trait in oilseed mustard (Brassica juncea) by QTL mapping and single nucleotide polymorphisms in FAE1 gene. Theor. Appl. Genet. 2004, 108, 743–749. [Google Scholar] [CrossRef] [PubMed]
- Kanrar, S.; Venkateswari, J.; Dureja, P.; Kirti, P.; Chopra, V. Modification of erucic acid content in Indian mustard (Brassica juncea) by up-regulation and down-regulation of the Brassica juncea FAT TY ACID ELONGATION1 (BjFAE1) gene. Plant Cell Rep. 2006, 25, 148–155. [Google Scholar] [CrossRef]
- Mietkiewska, E.; Brost, J.M.; Giblin, E.M.; Barton, D.L.; Taylor, D.C. Cloning and functional characterization of the fatty acid elongase 1 (FAE1) gene from high erucic Crambe abyssinica cv. Prophet. Plant Biotechnol. J. 2007, 5, 636–645. [Google Scholar] [CrossRef]
- Das, S.; Roscoe, T.; Delseny, M.; Srivastava, P.; Lakshmikumaran, M. Cloning and molecular characterization of the Fatty Acid Elongase 1 (FAE 1) gene from high and low erucic acid lines of Brassica campestris and Brassica oleracea. Plant Sci. 2002, 162, 245–250. [Google Scholar] [CrossRef]
- Guo, Y.; Mietkiewska, E.; Francis, T.; Katavic, V.; Brost, J.M.; Giblin, M.; Barton, D.L.; Taylor, D.C. Increase in nervonic acid content in transformed yeast and transgenic plants by introduction of a Lunaria annua L. 3-ketoacyl-CoA synthase (KCS) gene. Plant Mol. Biol. 2009, 69, 565–575. [Google Scholar] [CrossRef]
- Taylor, D.C.; Francis, T.; Guo, Y.; Brost, J.M.; Katavic, V.; Mietkiewska, E.; Michael Giblin, E.; Lozinsky, S.; Hoffman, T. Molecular cloning and characterization of a KCS gene from Cardamine graeca and its heterologous expression in Brassica oilseeds to engineer high nervonic acid oils for potential medical and industrial use. Plant Biotechnol. J. 2009, 7, 925–938. [Google Scholar] [CrossRef]
- Xiao, L.; Zhao, H.; Zhao, Z.; Du, D.; Xu, L.; Yao, Y.; Zhao, Z.; Xing, X.; Shang, G.; Zhao, H. Genetic and physical fine mapping of a multilocular gene Bjln1 in Brassica juncea to a 208-kb region. Mol. Breed. 2013, 32, 373–383. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Wright, S.B. Variability within and among Natural Populations, 1st ed.; University of Chicago Press: Chicago, IL, USA, 1978. [Google Scholar]
- Li, C.; Bai, Y.; Li, S.; Chen, H.; Han, X.; Zhao, H.; Shao, J.; Park, S.-u.; Wu, Q. Cloning, characterization, and activity analysis of a flavonol synthase gene FtFLS1 and its association with flavonoid content in tartary buckwheat. J. Agric. Food Chem. 2012, 60, 5161–5168. [Google Scholar] [CrossRef] [PubMed]
- Harborne, J.B.; Williams, C.A. Advances in flavonoid research since 1992. Phytochemistry 2000, 55, 481–504. [Google Scholar] [CrossRef] [PubMed]
- Kunst, L.; Taylor, D.C.; Underhill, E.W. Fatty acid elongation in developing seeds of Arabidopsis thaliana. Plant Physiol. Biochem. 1992, 30, 425–434. [Google Scholar]
- Puyaubert, J.; Garcia, C.; Chevalier, S.; Lessire, R. Acyl-CoA elongase, a key enzyme in the development of high-erucic acid rapeseed? Eur. J. Lipid Sci. Technol. 2005, 107, 263–267. [Google Scholar] [CrossRef]
- Zhi-Wen, L.; Ting-Dong, F.; Jin-Xing, T.; Bao-Yuan, C. Inheritance of seed colour and identification of RAPD and AFLP markers linked to the seed colour gene in rapeseed (Brassica napus L.). Theor. Appl. Genet. 2005, 110, 303–310. [Google Scholar] [CrossRef]
- Kang, L.; Qian, L.; Zheng, M.; Chen, L.; Chen, H.; Yang, L.; You, L.; Yang, B.; Yan, M.; Gu, Y. Genomic insights into the origin, domestication and diversification of Brassica juncea. Nat. Genet. 2021, 53, 1392–1402. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wan, Z.; Nelson, M.N.; Chauhan, J.S.; Redden, R.; Burton, W.A.; Lin, P.; Salisbury, P.A.; Fu, T.; Cowling, W.A. Evidence from genome-wide simple sequence repeat markers for a polyphyletic origin and secondary centers of genetic diversity of Brassica juncea in China and India. J. Hered. 2013, 104, 416–427. [Google Scholar] [CrossRef] [PubMed]
- Ecke, W.; Clemens, R.; Honsdorf, N.; Becker, H.C. Extent and structure of linkage disequilibrium in canola quality winter rapeseed (Brassica napus L.). Theor. Appl. Genet. 2010, 120, 921–931. [Google Scholar] [CrossRef]
- Kebede, B.; Cheema, K.; Greenshields, D.L.; Li, C.; Selvaraj, G.; Rahman, H. Construction of genetic linkage map and mapping of QTL for seed color in Brassica rapa. Genome 2012, 55, 813–823. [Google Scholar] [CrossRef]
- Wang, Y.; Xiao, L.; Guo, S.; An, F.; Du, D. Fine mapping and whole-genome resequencing identify the seed coat color gene in Brassica rapa. PLoS ONE 2016, 11, e0166464. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Qu, C.; Wittkop, B.; Yi, B.; Xiao, Y.; He, Y.; Snowdon, R.J.; Li, J. A high-density SNP map for accurate mapping of seed fibre QTL in Brassica napus L. PLoS ONE 2013, 8, e83052. [Google Scholar] [CrossRef]
- Gajardo, H.A.; Wittkop, B.; Soto-Cerda, B.; Higgins, E.E.; Parkin, I.A.; Snowdon, R.J.; Federico, M.L.; Iniguez-Luy, F.L. Association mapping of seed quality traits in Brassica napus L. using GWAS and candidate QTL approaches. Mol. Breed. 2015, 35, 143. [Google Scholar] [CrossRef]
- Lu, K.; Wei, L.; Li, X.; Wang, Y.; Wu, J.; Liu, M.; Zhang, C.; Chen, Z.; Xiao, Z.; Jian, H. Whole-genome resequencing reveals Brassica napus origin and genetic loci involved in its improvement. Nat. Commun. 2019, 10, 1154. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Wang, Q.; Hu, Y.; Jia, Y.; Chen, J.; Liu, B.; Zhang, Z.; Guan, X.; Chen, S.; Zhou, B. Genomic analyses in cotton identify signatures of selection and loci associated with fiber quality and yield traits. Nat. Genet. 2017, 49, 1089–1098. [Google Scholar] [CrossRef]
- Lu, Y.; Liu, X.; Liu, S.; Yue, Y.; Guan, C.; Liu, Z. A simple and rapid procedure for identification of seed coat colour at the early developmental stage of Brassica juncea and Brassica napus seeds. Plant Breed. 2012, 131, 176–179. [Google Scholar] [CrossRef]
- Auger, B.; Marnet, N.; Gautier, V.; Maia-Grondard, A.; Leprince, F.; Renard, M.; Guyot, S.; Nesi, N.; Routaboul, J.-M. A detailed survey of seed coat flavonoids in developing seeds of Brassica napus L. J. Agric. Food Chem. 2010, 58, 6246–6256. [Google Scholar] [CrossRef]
- Debeaujon, I.; Nesi, N.; Perez, P.; Devic, M.; Grandjean, O.; Caboche, M.; Lepiniec, L. Proanthocyanidin-accumulating cells in Arabidopsis testa: Regulation of differentiation and role in seed development. Plant Cell 2003, 15, 2514–2531. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, S.; Shikazono, N.; Tanaka, A. TRANSPARENT TESTA 19 is involved in the accumulation of both anthocyanins and proanthocyanidins in Arabidopsis. Plant J. 2004, 37, 104–114. [Google Scholar] [CrossRef]
- Xie, D.-Y.; Sharma, S.B.; Paiva, N.L.; Ferreira, D.; Dixon, R.A. Role of anthocyanidin reductase, encoded by BANYULS in plant flavonoid biosynthesis. Science 2003, 299, 396–399. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Dubos, C.; Lepiniec, L. Transcriptional control of flavonoid biosynthesis by MYB–bHLH–WDR complexes. Trends Plant Sci. 2015, 20, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Marles, M.S.; Ray, H.; Gruber, M.Y. New perspectives on proanthocyanidin biochemistry and molecular regulation. Phytochemistry 2003, 64, 367–383. [Google Scholar] [CrossRef]
- Park, K.I.; Ishikawa, N.; Morita, Y.; Choi, J.D.; Hoshino, A.; Iida, S. A bHLH regulatory gene in the common morning glory, Ipomoea purpurea, controls anthocyanin biosynthesis in flowers, proanthocyanidin and phytomelanin pigmentation in seeds, and seed trichome formation. Plant J. 2007, 49, 641–654. [Google Scholar] [CrossRef]
- Lepiniec, L.; Debeaujon, I.; Routaboul, J.-M.; Baudry, A.; Pourcel, L.; Nesi, N.; Caboche, M. Genetics and biochemistry of seed flavonoids. Annu. Rev. Plant Biol. 2006, 57, 405–430. [Google Scholar] [CrossRef] [PubMed]
- von Wettstein, D. From analysis of mutants to genetic engineering. Annu. Rev. Plant Biol. 2007, 58, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Han, Z.-y.; Zhang, J.; Hu, Y.; Song, T.; Yao, Y. The balance of expression of dihydroflavonol 4-reductase and flavonol synthase regulates flavonoid biosynthesis and red foliage coloration in crabapples. Sci. Rep. 2015, 5, 12228. [Google Scholar] [CrossRef]
- Luo, P.; Ning, G.; Wang, Z.; Shen, Y.; Jin, H.; Li, P.; Huang, S.; Zhao, J.; Bao, M. Disequilibrium of flavonol synthase and dihydroflavonol-4-reductase expression associated tightly to white vs. red color flower formation in plants. Front. Plant Sci. 2016, 6, 1257. [Google Scholar] [CrossRef]
- Yao, P.; Zhao, H.; Luo, X.; Gao, F.; Li, C.; Yao, H.; Chen, H.; Park, S.-U.; Wu, Q. Fagopyrum tataricum FtWD40 functions as a positive regulator of anthocyanin biosynthesis in transgenic tobacco. J. Plant Growth Regul. 2017, 36, 755–765. [Google Scholar] [CrossRef]
- Mahajan, M.; Joshi, R.; Gulati, A.; Yadav, S. Increase in flavan-3-ols by silencing flavonol synthase mRNA affects the transcript expression and activity levels of antioxidant enzymes in tobacco. Plant Biol. 2012, 14, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Kim, D.-H.; Lee, J.-Y.; Ha, S.-H.; Lim, S.-H. Comparative analysis of two flavonol synthases from different-colored onions provides insight into flavonoid biosynthesis. J. Agric. Food Chem. 2017, 65, 5287–5298. [Google Scholar] [CrossRef]
- Millar, A.A.; Kunst, L. Very-long-chain fatty acid biosynthesis is controlled through the expression and specificity of the condensing enzyme. Plant J. 1997, 12, 121–131. [Google Scholar] [CrossRef]
- Roscoe, T.; Lessire, R.; Puyaubert, J.; Renard, M.; Delseny, M. Mutations in the fatty acid elongation 1 gene are associated with a loss of β-ketoacyl-CoA synthase activity in low erucic acid rapeseed. FEBS Lett. 2001, 492, 107–111. [Google Scholar] [CrossRef]
- Katavic, V.; Mietkiewska, E.; Barton, D.L.; Giblin, E.M.; Reed, D.W.; Taylor, D.C. Restoring enzyme activity in nonfunctional low erucic acid Brassica napus fatty acid elongase 1 by a single amino acid substitution. Eur. J. Biochem. 2002, 269, 5625–5631. [Google Scholar] [CrossRef]
- Katavic, V.; Barton, D.L.; Giblin, E.M.; Reed, D.W.; Kumar, A.; Taylor, D.C. Gaining insight into the role of serine 282 in B. napus FAE1 condensing enzyme. FEBS Lett. 2004, 562, 118–124. [Google Scholar] [CrossRef]
- Wang, N.; Shi, L.; Tian, F.; Ning, H.; Wu, X.; Long, Y.; Meng, J. Assessment of FAE1 polymorphisms in three Brassica species using EcoTILLING and their association with differences in seed erucic acid contents. BMC Plant Biol. 2010, 10, 137. [Google Scholar] [CrossRef]
- Scarth, R.; Tang, J. Modification of Brassica oil using conventional and transgenic approaches. Crop Sci. 2006, 46, 1225–1236. [Google Scholar] [CrossRef]
- Wu, Y.; Xiao, L.; Wu, G.; Lu, C. Cloning of fatty acid elongase1 gene and molecular identification of A and C genome in Brassica species. Sci. China Ser. C Life Sci. 2007, 50, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Wu, Y.; Xiao, L.; Li, X.; Lu, C. Zero erucic acid trait of rapeseed (Brassica napus L.) results from a deletion of four base pairs in the fatty acid elongase 1 gene. Theor. Appl. Genet. 2008, 116, 491–499. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Trait | Group | Mean | SD | Max. | Min. | CV (%) | Skewness | Kurtosis | G | E | h2 (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Stearic acid (%) | G1 | 3.779 | 0.412 | 5.235 | 3.178 | 10.900 | 4.139 | 1.560 | ** | ** | 93.390 |
| G2 | 3.687 | 0.576 | 4.714 | 2.669 | 15.621 | −0.661 | 0.212 | ||||
| G3 | 3.845 | 0.456 | 4.799 | 2.705 | 11.866 | 0.152 | −0.022 | ||||
| G4 | 5.187 | 1.168 | 11.531 | 4.324 | 22.517 | 26.341 | 4.818 | ||||
| Palmitic acid (%) | G1 | 0.995 | 0.152 | 1.339 | 0.714 | 15.302 | 0.359 | 0.315 | ** | ** | 87.640 |
| G2 | 1.102 | 0.151 | 1.464 | 0.763 | 13.671 | 1.141 | 0.380 | ||||
| G3 | 1.126 | 0.119 | 1.474 | 0.710 | 10.537 | 2.717 | −0.499 | ||||
| G4 | 1.375 | 0.238 | 2.455 | 1.082 | 17.284 | 11.608 | 2.734 | ||||
| Oleic acid (%) | G1 | 19.773 | 5.619 | 34.944 | 9.295 | 28.419 | 0.440 | 0.339 | ** | ** | 87.070 |
| G2 | 17.328 | 6.332 | 30.210 | 2.071 | 36.541 | 0.983 | −0.085 | ||||
| G3 | 19.776 | 5.942 | 38.429 | 9.714 | 30.046 | 0.736 | 0.822 | ||||
| G4 | 52.561 | 4.766 | 72.017 | 44.181 | 9.067 | 7.206 | 1.811 | ||||
| Linoleic acid (%) | G1 | 19.768 | 1.338 | 23.051 | 17.133 | 6.770 | 0.440 | 0.273 | ** | ** | 87.830 |
| G2 | 19.834 | 1.510 | 22.432 | 16.888 | 7.614 | −0.266 | −0.174 | ||||
| G3 | 19.979 | 1.590 | 25.559 | 16.081 | 7.961 | 2.079 | 0.425 | ||||
| G4 | 29.966 | 1.811 | 33.458 | 26.466 | 6.042 | −0.893 | 0.024 | ||||
| Linolenic acid (%) | G1 | 12.480 | 1.055 | 14.572 | 10.292 | 8.455 | −0.099 | −0.365 | ** | 86.820 | |
| G2 | 12.754 | 0.893 | 14.464 | 11.212 | 7.000 | −0.848 | 0.189 | ||||
| G3 | 12.504 | 0.885 | 14.722 | 10.215 | 7.079 | 0.424 | −0.186 | ||||
| G4 | 16.885 | 1.940 | 25.399 | 13.211 | 11.489 | 10.346 | 2.099 | ||||
| Arachidonic acid (%) | G1 | 8.820 | 1.912 | 12.296 | 4.508 | 21.680 | −0.596 | −0.218 | ** | ** | 94.960 |
| G2 | 9.017 | 2.039 | 12.826 | 3.450 | 22.619 | 1.543 | −0.676 | ||||
| G3 | 8.639 | 2.011 | 12.005 | 0.001 | 23.274 | 5.050 | −1.571 | ||||
| G4 | 0.474 | 0.732 | 3.559 | 0.001 | 54.342 | 8.026 | 2.471 | ||||
| Erucic acid (%) | G1 | 31.064 | 4.590 | 39.020 | 18.781 | 14.775 | 1.297 | −0.857 | ** | ** | 87.380 |
| G2 | 30.633 | 5.565 | 42.222 | 17.130 | 18.168 | 1.088 | −0.391 | ||||
| G3 | 29.296 | 4.940 | 38.920 | 11.492 | 16.862 | 1.885 | −0.873 | ||||
| G4 | 0.002 | 0.001 | 0.004 | 0.001 | 36.464 | −0.337 | 0.791 |
| Gene ID | Chr. | Length | Start | End | Annotation | Selection Groups |
|---|---|---|---|---|---|---|
| BjuB018407 | B08 | 915 | 51,641,320 | 51,642,396 | Flavonol synthase/flavanone 3-hydroxylase | G1/G2, G1/G3 |
| BjuA034859 | A09 | 846 | 32,483,465 | 32,484,597 | Heme oxygenase 1 | G1/G2, G1/G3 |
| BjuA034858 | A09 | 351 | 32,474,088 | 32,474,438 | Biliverdin-producing, ferredoxin | G1/G2, G1/G3 |
| BjuB019589 | B03 | 1332 | 43,953,714 | 43,955,125 | Cytokinin-N-glucosyltransferase | G1/G2, G1/G3 |
| BjuA040986 | A02 | 1083 | 1,316,124 | 1,318,173 | Flavonol synthase | G1/G2 |
| BjuA025255 | A07 | 2475 | 1,472,948 | 1,477,830 | C-type lectin domain family 16, member A | G1/G2 |
| BjuA036020 | A07 | 1290 | 806,774 | 808,218 | Shikimate O-hydroxycinnamoyltransferase | G1/G2 |
| BjuA034936 | A09 | 861 | 27,359,538 | 27,361,184 | TRANSPARENT TESTA 1 | G1/G2 |
| BjuA035531 | A09 | 1896 | 42,739,146 | 42,742,220 | Chalcone isomerase | G1/G2 |
| BjuA036650 | A09 | 1491 | 49,057,032 | 49,059,418 | Isoflavone/4′-methoxyisoflavone 2′-hydroxylase | G1/G2 |
| BjuB003779 | B02 | 1008 | 48,552,117 | 48,553,791 | Flavonol synthase/flavanone 3-hydroxylase | G1/G2 |
| BjuB004510 | B02 | 1011 | 48,559,974 | 48,561,651 | Flavonol synthase/flavanone 3-hydroxylase | G1/G2 |
| BjuA034990 | A09 | 633 | 34,170,686 | 34,172,497 | Shikimate O-hydroxycinnamoyltransferase | G1/G3 |
| BjuA036409 | A09 | 756 | 47,855,094 | 47,856,199 | Fatty-acid-binding protein 1 | G1/G3 |
| BjuB029287 | B04 | 1077 | 19,998,112 | 20,000,046 | Isoflavone/4′-methoxyisoflavone 2′-hydroxylase | G1/G3 |
| Gene ID | Chr. | Length | Start | End | Annotation | Selection Groups |
|---|---|---|---|---|---|---|
| BjuA046600 | A08 | 876 | 10,993,813 | 10,995,009 | SNF1-related protein kinase regulatory subunit beta-2 | G2/G4, G3/G4 |
| BjuA029874 | A08 | 1215 | 20,449,210 | 20,451,344 | Pyruvate dehydrogenase | G2/G4, G3/G4 |
| BjuA029561 | A08 | 885 | 18,709,038 | 18,709,922 | Fatty acid desaturase 4 | G2/G4, G3/G4 |
| BjuA029559 | A08 | 885 | 18,701,624 | 18,702,508 | Fatty acid desaturase 4 | G2/G4, G3/G4 |
| BjuA029009 | A08 | 1341 | 17,215,938 | 17,218,074 | Omega-6 fatty acid desaturase | G2/G4, G3/G4 |
| BjuA028960 | A08 | 1677 | 16,923,302 | 16,927,478 | 3-hydroxyisobutyryl-CoA hydrolase-like protein | G2/G4, G3/G4 |
| BjuA028763 | A08 | 774 | 14,533,575 | 14,534,960 | Short-chain dehydrogenase/reductase SDRA | G2/G4, G3/G4 |
| BjuA028614 | A08 | 798 | 13,046,485 | 13,048,555 | Palmitoyl-protein thioesterase 1 | G2/G4, G3/G4 |
| BjuA028208 | A08 | 903 | 12,546,658 | 12,548,359 | Probable enoyl-CoA hydratase 2 | G2/G4, G3/G4 |
| BjuA028013 | A08 | 1065 | 2,947,736 | 2,949,499 | Electron transfer flavoprotein subunit alpha | G2/G4, G3/G4 |
| BjuA000690 | A08 | 798 | 13,120,281 | 13,122,367 | Palmitoyl-protein thioesterase 1 | G2/G4, G3/G4 |
| BjuA000215 | A08 | 1338 | 17,360,042 | 17,362,175 | Omega-6 fatty acid desaturase | G2/G4, G3/G4 |
| BjuB047302 | B03 | 1518 | 8,750,374 | 8,751,891 | 3-ketoacyl-CoA synthase 18 | G2/G4, G3/G4 |
| BjuB047301 | B03 | 1422 | 8,747,481 | 8,748,902 | 3-ketoacyl-CoA synthase 17 | G2/G4, G3/G4 |
| BjuB032119 | B03 | 1425 | 8,975,890 | 8,977,314 | 3-ketoacyl-CoA synthase 17 | G2/G4, G3/G4 |
| BjuB032118 | B03 | 1518 | 8,978,894 | 8,980,411 | 3-ketoacyl-CoA synthase 18 | G2/G4, G3/G4 |
| BjuB032028 | B03 | 1008 | 9,742,346 | 9,743,766 | Short-chain dehydrogenase/reductase | G2/G4, G3/G4 |
| BjuB032027 | B03 | 771 | 9,744,629 | 9,745,972 | Short-chain dehydrogenase/reductase | G2/G4, G3/G4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, X.; Fu, M.; Li, G.; Qu, C.; Liu, H.; Li, X.; Zhang, Y.; Zhang, Y.; Zhao, K.; Zhang, L.; et al. Whole-Genome Resequencing Reveals the Genetic Diversity and Selection Signatures of the Brassica juncea from the Yunnan-Guizhou Plateau. Agronomy 2023, 13, 1053. https://doi.org/10.3390/agronomy13041053
Yuan X, Fu M, Li G, Qu C, Liu H, Li X, Zhang Y, Zhang Y, Zhao K, Zhang L, et al. Whole-Genome Resequencing Reveals the Genetic Diversity and Selection Signatures of the Brassica juncea from the Yunnan-Guizhou Plateau. Agronomy. 2023; 13(4):1053. https://doi.org/10.3390/agronomy13041053
Chicago/Turabian StyleYuan, Xiaoyan, Minglian Fu, Genze Li, Cunmin Qu, Hao Liu, Xuan Li, Yunyun Zhang, Yusong Zhang, Kaiqin Zhao, Lifan Zhang, and et al. 2023. "Whole-Genome Resequencing Reveals the Genetic Diversity and Selection Signatures of the Brassica juncea from the Yunnan-Guizhou Plateau" Agronomy 13, no. 4: 1053. https://doi.org/10.3390/agronomy13041053
APA StyleYuan, X., Fu, M., Li, G., Qu, C., Liu, H., Li, X., Zhang, Y., Zhang, Y., Zhao, K., Zhang, L., Luo, Y., Li, J., He, X., He, L., & Liu, F. (2023). Whole-Genome Resequencing Reveals the Genetic Diversity and Selection Signatures of the Brassica juncea from the Yunnan-Guizhou Plateau. Agronomy, 13(4), 1053. https://doi.org/10.3390/agronomy13041053