Accelerating Genetic Gain in Sugarcane Breeding Using Genomic Selection

 ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
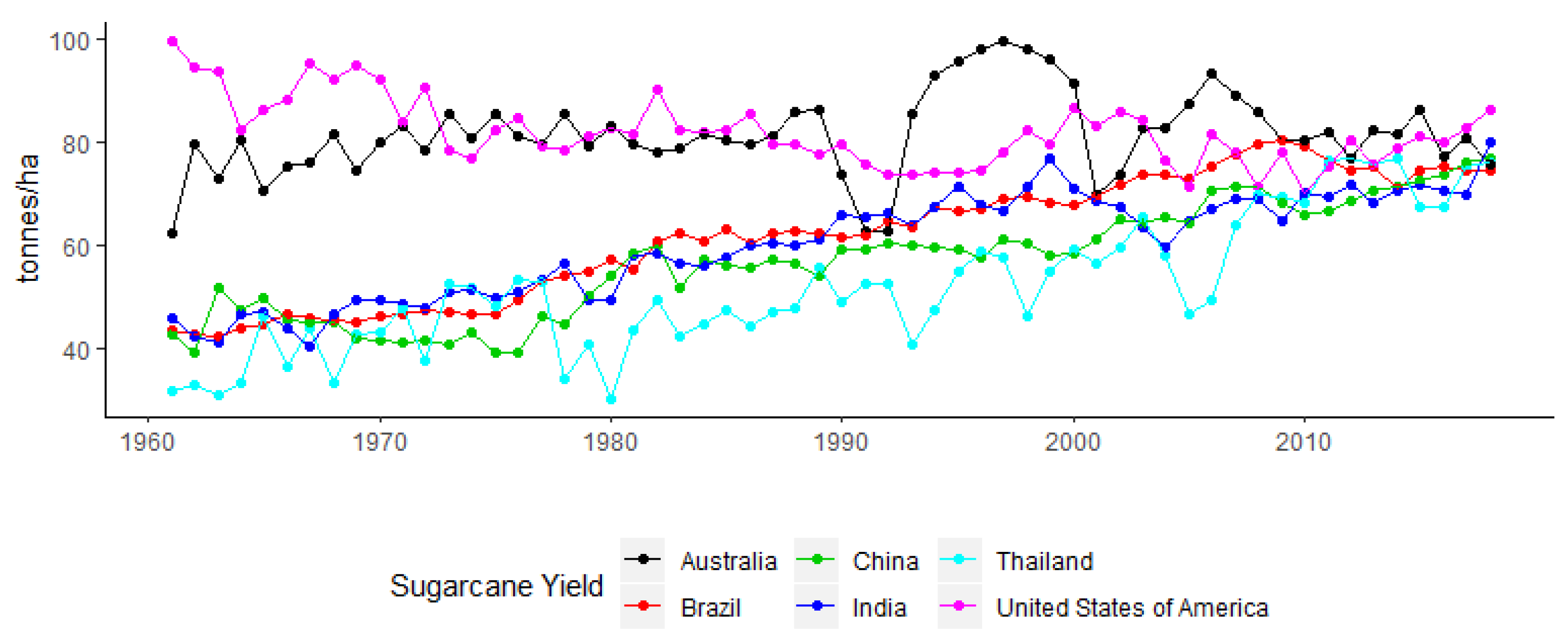
1. The Commercial Importance of Sugarcane and Production Trends
2. Development of Modern Cultivars and Inherent Challenges
3. Identifying and Overcoming Bottlenecks in Breeding Programs Using the Breeder’s Equation
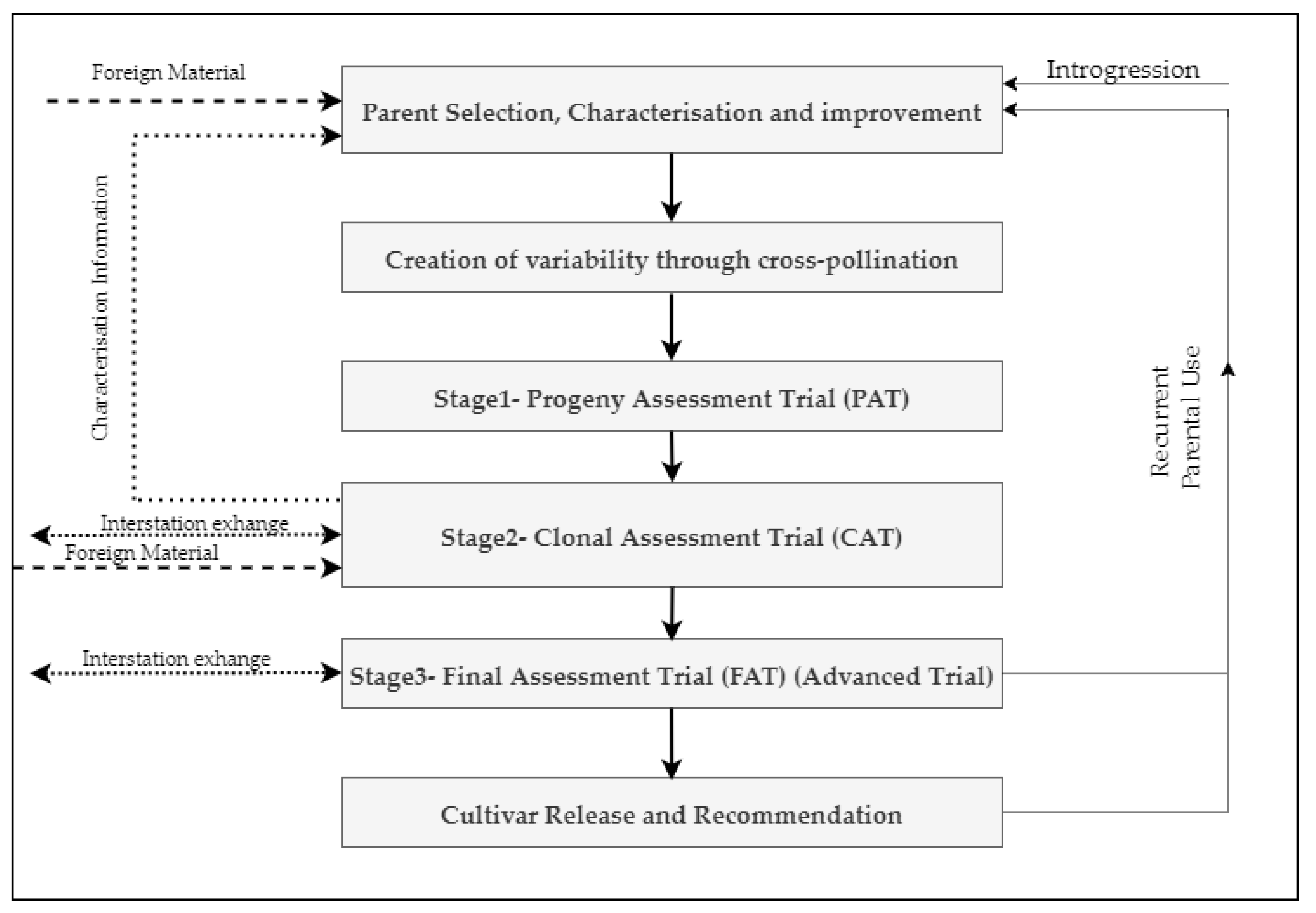
4. Practices and Limitations of Conventional Sugarcane Breeding
5. Genomic Selection: A Powerful New Breeding Tool
6. Implementation of Genomic Selection in Sugarcane Breeding
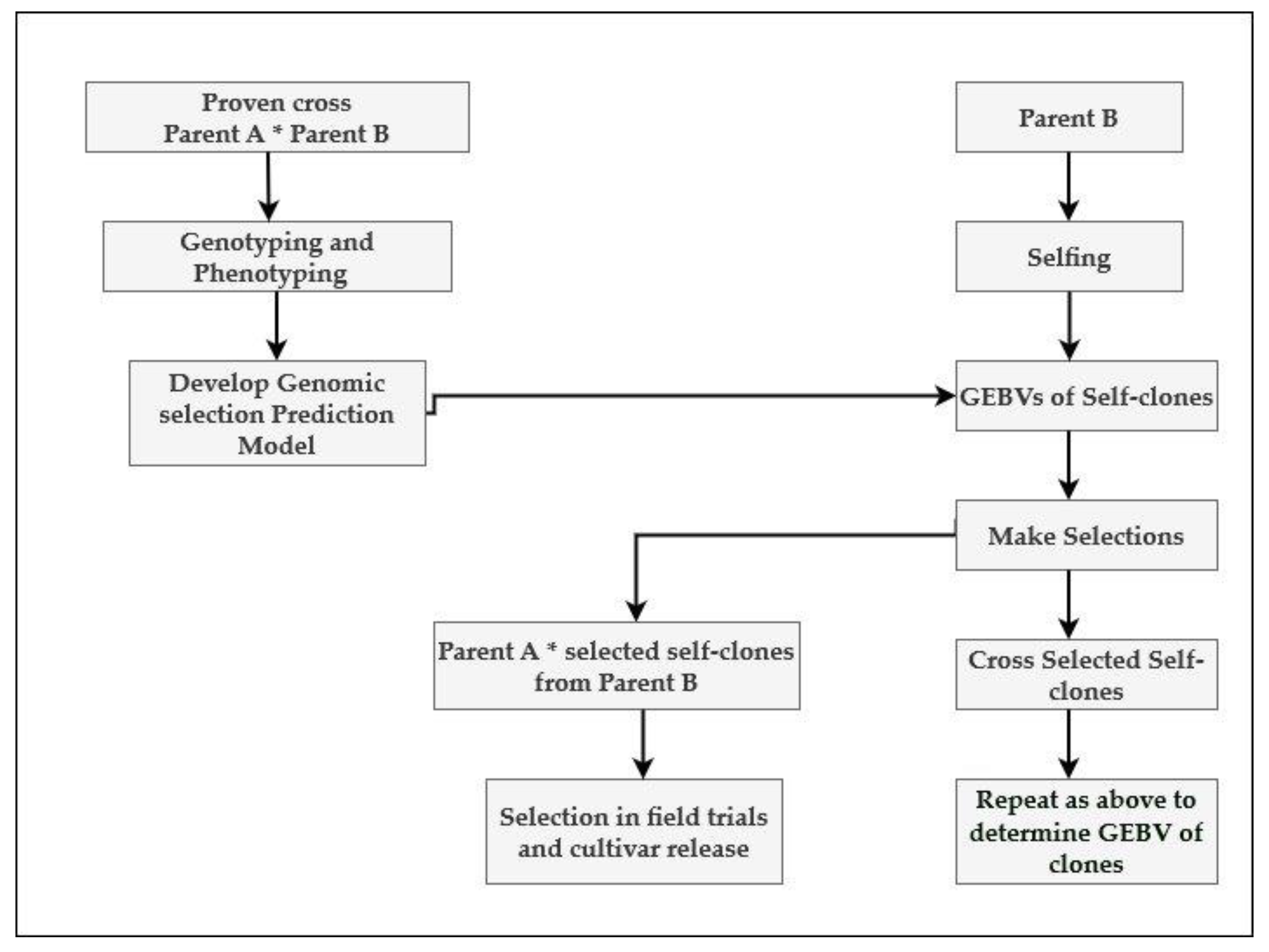
7. Recurrent Genomic Selection and Reciprocal Recurrent Genomic Selection: Two Strategies for the Incorporation of Genomic Selection in Sugarcane Breeding
Author Contributions
Funding
Conflicts of Interest
References
- Hatch, M.D. C4 photosynthesis: Discovery and resolution. In Discoveries in Photosynthesis; Govindjee, B.J.T., Gest, H., Allen, J.F., Eds.; Springer Netherlands: Dordrecht, The Netherlands, 2005; Volume 20, pp. 875–880. ISBN 978-1-4020-3324-7. [Google Scholar]
- Hatch, M.D.; Slack, C.R.; Johnson, H.S. Further studies on a new pathway of photosynthetic carbon dioxide fixation in sugar-cane and its occurrence in other plant species. Biochem. J. 1967, 102, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Ming, R.; Moore, P.H.; Wu, K.K.; D’Hont, A.; Glaszmann, J.C.; Tew, T.L.; Mirkov, T.E.; da Silva, J.; Jifon, J.; Rai, M.; et al. Plant Breeding Reviews; Wiley Blackwell: Hoboken, NJ, USA, 2010; Volume 27, pp. 15–118. ISBN 978-0-4706-5034-9. [Google Scholar]
- Souza, A.; Grandis, A.; Leite, D.; Buckeridge, M. Sugarcane as a bioenergy source: History, performance, and perspectives for second-generation bioethanol. Bioenergy Res. 2014, 7, 24–35. [Google Scholar] [CrossRef]
- Mackintosh, D. Sugar Milling. Available online: http://hdl.handle.net/11079/15541 (accessed on 10 January 2020).
- Cheavegatti-Gianotto, A.; Abreu, H.; Arruda, P.; Bespalhok Filho, J.; Burnquist, W.; Creste, S.; Ciero, L.; Ferro, J.; Oliveira Figueira, A.; Sousa Filgueiras, T.; et al. Sugarcane (Saccharum X officinarum): A reference study for the regulation of genetically modified cultivars in Brazil. Trop. Plant Biol. 2011, 4, 62–89. [Google Scholar] [CrossRef] [PubMed]
- Henry, R.J. Evaluation of plant biomass resources available for replacement of fossil oil. Plant Biotechnol. J. 2010, 8, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Byrt, C.S.; Grof, C.; Furbank, R. C4 Plants as biofuel feedstocks: optimising biomass production and feedstock quality from a lignocellulosic perspective. J. Integr. Plant Biol. 2011, 53, 120–135. [Google Scholar] [CrossRef] [PubMed]
- Goldemberg, J.; Coelho, S.T.; Guardabassi, P. The sustainability of ethanol production from sugarcane. Energy Policy 2008, 36, 2086–2097. [Google Scholar] [CrossRef]
- Moore, P.H.; Paterson, A.H.; Tew, T. Sugarcane: The crop, the plant, and domestication. In Sugarcane: Physiology, Biochemistry, and Functional Biology; Moore, P.H., Botha, F.C., Eds.; John Wiley & Sons, Inc: Hoboken, NJ, USA, 2013; pp. 1–17. ISBN 978-1-1187-7128-0. [Google Scholar]
- FAOSTAT. Food and Agricultural Organisation : Sugarcane Production Countries. 2018. Available online: http://www.fao.org/faostat (accessed on 10 January 2020).
- De Morais, L.K.; de Aguiar, M.S.; de Albuquerque e Silva, P.; Câmara, T.M.M.; Cursi, D.E.; Júnior, A.R.F.; Chapola, R.G.; Carneiro, M.S.; Bespalhok Filho, J.C. Breeding of sugarcane. In Industrial Crops: Breeding for BioEnergy and Bioproducts; Cruz, V.M.V., Dierig, D.A., Eds.; Springer: New York, NY, USA, 2015; pp. 29–42. ISBN 978-1-4939-1447-0. [Google Scholar] [CrossRef]
- Zhao, D.; Yang-Rui, L. Climate change and sugarcane production: Potential impact and mitigation strategies. Int. J. Agron. 2015, 2015, 1–10. [Google Scholar] [CrossRef]
- Stirling, G.; Blair, B.; Pattemore, J.; Garside, A.; Bell, M. Changes in nematode populations on sugarcane following fallow, fumigation and crop rotation, and implications for the role of nematodes in yield decline. Australas. Plant Pathol. 2001, 30, 323–335. [Google Scholar] [CrossRef]
- Bailey, R.; Bechet, G. The effect of ratoon stunting disease on the yield of some South African sugarcane varieties under irrigated and rainfed conditions. In Proceedings of the South African Sugar Technologists’ Association: Annual conference, Durban, South Africa, 5–8 June 1995; pp. 74–78. [Google Scholar]
- Johnson, S.S.; Tyagi, A.P. Effect of ratoon stunting disease (RSD) on sugarcane yield in Fiji. South Pac. J. Nat. Appl. Sci. 2010, 28, 69–73. [Google Scholar] [CrossRef]
- Magarey, R.; Croft, B.; Willcox, T. An epidemic of orange rust on sugarcane in Australia. In Proceedings of the International Society of Sugar Cane Technologists: XXIV Congress, Brisbane, Australia, 17–21 September 2001; pp. 410–416. [Google Scholar]
- Magarey, R.; Bull, J.; Sheahan, T.; Denney, D.; Bruce, R. Yield losses caused by sugarcane smut in several crops in Queensland. In Proceedings of the Australian Society of Sugar Cane Technologists: 32nd Annual Conference, Bundaberg, Australia, 11–14 May 2010; pp. 347–354. [Google Scholar]
- Sundar, A.R.; Barnabas, E.L.; Malathi, P.; Viswanathan, R.; Sundar, A.; Barnabas, E.J.B. A mini-review on smut disease of sugarcane caused by Sporisorium Scitamineum. In Botany; Mworia, J.K., Ed.; InTech: Rijeka, Croatia, 2012; Volume 16, pp. 107–128. ISBN 978-953-51-0355-4. [Google Scholar]
- Croft, B.J.; Berding, N.; Cox, M.C.; Bhuiyan, S. Breeding smut-resistant sugarcane varieties in Australia: Progress and future directions. In Proceedings of the Australian Society of Sugar Cane Technologists: XXX Annual Conference, Townsville, Australia, 29 April–2 May 2008; pp. 125–134. [Google Scholar]
- Bhuiyan, S.; Croft, B.; Cox, M. Breeding for sugarcane smut resistance in Australia and industry response: 2006–2011. In Proceedings of the Australian Society of Sugar Cane Technologists : XXXV Annual Confernce, Townsville, QLD, Australia, 16–18 April 2013; pp. 1–9. [Google Scholar]
- Gawander, J. The impact of climate change on sugar cane production in Fiji. Wmo Bull. 2007, 56, 34–39. [Google Scholar]
- Li, Y.-R.; Wei, Y.-A. Sugar industry in China: R & D and policy initiatives to meet sugar and biofuel demand of future. Sugar Tech 2006, 8, 203–216. [Google Scholar] [CrossRef]
- Stevenson, G.C. Genetics and Breeding of Sugar Cane; Longmans: London, UK, 1965. [Google Scholar]
- D’Hont, A.; Paulet, F.; Glaszmann, J. Oligoclonal interspecific origin of ‘North Indian’ and ‘Chinese’ sugarcanes. Chromosome Res. 2002, 10, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Mohan Naidu, K.; Sreenivasan, T. Conservation of sugarcane germoplasm. In Proceedings of the Copersucar International Sugarcane Breeding Workshop, São Paulo, Brazil, May–June 1987; pp. 33–53. [Google Scholar]
- Roach, B. Origin and improvement of the genetic base of sugarcane. In Proceedings of the Australian Society of Sugar Cane Technologists: Annual Conference, Tweed Heads, Australia, 2–5 May 1989; pp. 34–47. [Google Scholar]
- Jeswiet, J. The development of selection and breeding of the sugar cane in Java. In Proceedings of the International Society of Sugarcane Technologists, Soerabaia, Indonesia, 7–19 June 1929; pp. 44–57. [Google Scholar]
- Mangelsdorf, A.J. Sugar-Cane breeding methods. In Proceedings of the International Society of Sugar Cane Technologists: X Congress, Honolulu, HI, USA, 3–22 May 1959. [Google Scholar]
- Hogarth, D.M. New varieties lift sugar production. Prod. Rev. 1976, 66, 21–22. [Google Scholar]
- Cock, J.H. Sugarcane growth and development. Int. SugarJ. 2001, 105, 5–15. [Google Scholar]
- Edme, S.J.; Miller, J.D.; Glaz, B.; Tai, P.Y.P.; Comstock, J.C. Genetic contribution to yield gains in the Florida sugarcane industry across 33 years. Crop Sci. 2005, 45, 92–97. [Google Scholar] [CrossRef]
- Jackson, P.A. Breeding for improved sugar content in sugarcane. Field Crop. Res. 2005, 92, 277–290. [Google Scholar] [CrossRef]
- Wei, X.; Jackson, P. Addressing slow rates of long-term genetic gain in sugarcane. In Proceedings of the International Society of Sugar Cane Technologists: XXIX Congress, Chiang Mai, Thailand, 5–9 December 2016; pp. 480–484. [Google Scholar]
- Aitken, K.; Jackson, P.; McIntyre, C. Quantitative trait loci identified for sugar related traits in a sugarcane (Saccharum spp.) cultivar× Saccharum officinarum population. Appl. Genet. 2006, 112, 1306–1317. [Google Scholar] [CrossRef]
- Raboin, L.-M.; Pauquet, J.; Butterfield, M.; D’Hont, A.; Glaszmann, J.-C. Analysis of genome-wide linkage disequilibrium in the highly polyploid sugarcane. Appl. Genet. 2008, 116, 701–714. [Google Scholar] [CrossRef]
- Price, S. Cytogenetics of modern sugar canes. Econ. Bot. 1963, 17, 97–106. [Google Scholar] [CrossRef]
- Bremer, G. Problems in breeding and cytology of sugar cane. Euphytica 1961, 10, 59–78. [Google Scholar] [CrossRef]
- Sreenivasan, T.; Ahloowalia, B. Cytogenetics. In Sugarcane Improvement Through Breeding; Heinz, D.J., Ed.; Elsevier: Amsterdam, The Netherlands, 1987; pp. 211–256. ISBN 978-1-4832-8998-4. [Google Scholar]
- D’Hont, A.; Grivet, L.; Feldmann, P.; Glaszmann, J.C.; Rao, S.; Berding, N. Characterisation of the double genome structure of modern sugarcane cultivars (Saccharum spp.) by molecular cytogenetics. Mol. Genet. Genom. 1996, 250, 405–413. [Google Scholar] [CrossRef]
- D’Hont, A.; Ison, D.; Alix, K.; Roux, C.; Glaszmann, J. Determination of basic chromosome numbers in the genus Saccharum by physical mapping of ribosomal RNA genes. Genome 1998, 41, 221–225. [Google Scholar] [CrossRef]
- Aitken, K.S.; McNeil, M.D.; Hermann, S.; Bundock, P.C.; Kilian, A.; Heller-Uszynska, K.; Henry, R.J.; Li, J. A comprehensive genetic map of sugarcane that provides enhanced map coverage and integrates high-throughput Diversity Array Technology (DArT) markers. BMC Genom. 2014, 15, 1–152. [Google Scholar] [CrossRef] [PubMed]
- Garsmeur, O.; Droc, G.; Antonise, R.; Grimwood, J.; Potier, B.; Aitken, K.; Jenkins, J.; Martin, G.; Charron, C.; Hervouet, C.; et al. A mosaic monoploid reference sequence for the highly complex genome of sugarcane. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Grivet, L.; Arruda, P. Sugarcane genomics: Depicting the complex genome of an important tropical crop. Curr. Opin. Plant Biol. 2002, 5, 122–127. [Google Scholar] [CrossRef]
- Souza, G.; Berges, H.; Bocs, S.; Casu, R.; D’Hont, A.; Ferreira, J.; Henry, R.; Ming, R.; Potier, B.; Sluys, M.-A.; et al. The sugarcane genome challenge: Strategies for sequencing a highly complex genome. Trop. Plant Biol. 2011, 4, 145–156. [Google Scholar] [CrossRef]
- D’Hont, A.; Glaszmann, J.C. Sugarcane genome analysis with molecular markers—A first decade of research. In Proceedings of the International Society of Sugar Cane Technologists: XXIV Congress, Brisbane, Australia, 17–21 September 2001; pp. 556–559. [Google Scholar]
- Le Cunff, L.; Garsmeur, O.; Raboin, L.M.; Pauquet, J.; Telismart, H.; Selvi, A.; Grivet, L.; Philippe, R.; Begum, D.; Deu, M.J.G. Diploid/polyploid syntenic shuttle mapping and haplotype-specific chromosome walking toward a rust resistance gene (Bru1) in highly polyploid sugarcane (2n ~12x ~115). Genetics 2008, 180, 649–660. [Google Scholar] [CrossRef]
- Aitken, K.; Berkman, P.; Rae, A. The first sugarcane genome assembly: How can we use it. In Proceedings of the Australian Society of Sugar Cane Technologists: 38th Annual Conference, Mackay, Australia, 27–29 April 2016. [Google Scholar]
- Ming, R.; Liu, S.C.; Lin, Y.; Da Silva, J.; Wilson, W.; Braga, D.; van Deynze, A.; Wenslaff, T.; Wu, K.; Moore, P.; et al. Detailed alignment of Saccharum and sorghum chromosomes: Comparative organization of closely related diploid and polyploid genomes. Genetics 1998, 150, 1663–1682. [Google Scholar]
- Lush, J.L. Animal Breeding Plans. Ames; Collegiate Press: Ames, IA, USA, 1937; ISBN 978-0-8138-2345-4. [Google Scholar]
- Park, S.; Jackson, P.; Berding, N.; Inman-Bamber, G. Conventional breeding practices within the Australian sugarcane breeding program. In Proceedings of the Australian Society of Sugar Cane Technologists: 29th Annual Conference, Cairns, Australia, 8–11 May 2007; pp. 113–121. [Google Scholar]
- Jackson, P.A.; Hogarth, D.M. Genotype x environment interactions in sugarcane, 1. Patterns of response across sites and crop-years in north Queensland. Aust. J. Agric. Res. 1992, 43, 1447–1459. [Google Scholar] [CrossRef]
- Heffner, E.; Sorrells, M.E.; Jannink, J. Genomic selection for crop improvement. Crop Sci. 2009, 49, 1–12. [Google Scholar] [CrossRef]
- Quinton, M.; Smith, C.; Goddard, M. Comparison of selection methods at the same level of inbreeding. J. Anim. Sci. 1992, 70, 1060–1067. [Google Scholar] [CrossRef] [PubMed]
- Hogarth, D. Quantitative inheritance studies in sugar-cane. I. Estimation of variance components. Aust. J. Agric. Res. 1971, 22, 93–102. [Google Scholar] [CrossRef]
- Barbosa, M.H.P.; Resende, M.D.V.; Bressiani, J.A.; Silveira, L.C.I.; Peternelli, L.A. Selection of sugarcane families and parents by Reml/Blup. Crop Breed. Appl. Biotechnol. 2005, 5, 443–450. [Google Scholar] [CrossRef]
- Pisaroglo De Carvalho, M.; Gezan, S.A.; Peternelli, L.A.; Pereira Barbosa, M.H. Estimation of additive and nonadditive genetic components of sugarcane families using multitrait analysis. Agron. J. 2014, 106, 800–808. [Google Scholar] [CrossRef]
- Hogarth, D.M.; Wu, K.K.; Heinz, D.J. Estimating genetic variance in sugarcane using a factorial cross design. Crop Sci. 1981, 21, 21–25. [Google Scholar] [CrossRef]
- O’Reilly, K. An Investigation Into the Heritability of Commercially Important Traits in a Sugarcane Population Under Dryland Conditions. Master’s Thesis of Science in Agriculture, University of Natal, Pietermaritzburg, South Africa, 1995. [Google Scholar]
- Moore, P.; Nuss, K. Flowering and flower synchronization. In Sugarcane Improvement Through Breeding; Heinz, D.J., Ed.; Elsevier Science: Amsterdam, The Netherlands, 1987; pp. 273–311. ISBN 978-1-4832-8998-4. [Google Scholar]
- Gosnell, J. Some factors affecting flowering in sugarcane. In Proceedings of the South African Sugar Technologists’ Association, Durban and Mount Edgecombe, South Africa, 6–9 June 1983; pp. 144–147. [Google Scholar]
- Berding, N.; Hogarth, D. Poor and variable flowering in tropical sugarcane improvement programs: Diagnosis and resolution of a major breeding impediment. In Proceedings of the International Society Sugar Cane Technologists: XXV congress, Guatemala City, Guatemala, 30 January–4 February 2005. [Google Scholar]
- Jackson, P.; McRae, T. Selection of sugarcane clones in small plots effects of plot size and selection criteria. Crop Sci. 2001, 41, 315–322. [Google Scholar] [CrossRef]
- Milligan, S.B.; Balzarini, M.; Gravois, K.A.; Bischoff, K.P. Early stage sugarcane selection using different plot sizes. Crop Sci. 2007, 47, 1859–1864. [Google Scholar] [CrossRef]
- Tai, P.Y.P.; Rice, E.R.; Chew, V.; Miller, J.D. Phenotypic stability analyses of sugarcane cultivar performance tests1. Crop Sci. 1982, 22, 1179–1184. [Google Scholar] [CrossRef]
- Casu, R.E.; Manners, J.M.; Bonnett, G.D.; Jackson, P.A.; McIntyre, C.L.; Dunne, R.; Chapman, S.C.; Rae, A.L.; Grof, C.P.L. Genomics approaches for the identification of genes determining important traits in sugarcane. Field Crop. Res. 2005, 92, 137–147. [Google Scholar] [CrossRef]
- Kang, M.; Miller, J. Genotype✕ environment interactions for cane and sugar yield and their implications in sugarcane breeding 1. Crop Sci. 1984, 24, 435–440. [Google Scholar] [CrossRef]
- Ramburan, S.; Zhou, M.; Labuschagne, M. Interpretation of genotype × environment interactions of sugarcane: Identifying significant environmental factors. Field Crop. Res. 2011, 124, 392–399. [Google Scholar] [CrossRef]
- Hill, J. Genotype-environment interaction—A challenge for plant breeding. J. Agric. Sci. 1975, 85, 477–493. [Google Scholar] [CrossRef]
- Freeman, G. Statistical methods for the analysis of genotype-environment interactions 2. Heredity 1973, 31, 339–354. [Google Scholar] [CrossRef] [PubMed]
- Hammer, G.; Messina, C.; Wu, A.; Cooper, M. Biological reality and parsimony in crop models—Why we need both in crop improvement! Silico Plants 2019, 1, 1–21. [Google Scholar] [CrossRef]
- Meuwissen, T.; Hayes, B.; Goddard, M. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [PubMed]
- Voss-Fels, K.; Cooper, M.; Hayes, B. Accelerating crop genetic gains with genomic selection. Appl. Genet. 2019, 132, 669–686. [Google Scholar] [CrossRef]
- Xu, Y.; Liu, X.; Fu, J.; Wang, H.; Wang, J.; Huang, C.; Prasanna, B.M.; Olsen, M.S.; Wang, G.; Zhang, A. Enhancing genetic gain through genomic selection: From livestock to plants. Plant Commun. 2020, 1, 2641–2666. [Google Scholar] [CrossRef]
- Jannink, J.-L.; Lorenz, A.J.; Iwata, H. Genomic selection in plant breeding: From theory to practice. Brief. Funct. Genom. 2010, 9, 166–177. [Google Scholar] [CrossRef]
- Heslot, N.; Yang, H.-P.; Sorrells, M.E.; Jannink, J.-L. Genomic selection in plant breeding: A comparison of models. Crop Sci. 2012, 52, 146–160. [Google Scholar] [CrossRef]
- Campos, G.D.L.; Hickey, J.M.; Pong-Wong, R.; Daetwyler, H.D.; Calus, M.P.L. Whole-genome regression and prediction methods applied to plant and animal breeding. Genetics 2013, 193, 327–345. [Google Scholar] [CrossRef]
- Erbe, M.; Hayes, B.J.; Matukumalli, L.K.; Goswami, S.; Bowman, P.J.; Reich, C.M.; Mason, B.A.; Goddard, M.E. Improving accuracy of genomic predictions within and between dairy cattle breeds with imputed high-density single nucleotide polymorphism panels. J. Dairy Sci. 2012, 95, 4114–4129. [Google Scholar] [CrossRef] [PubMed]
- Gianola, D.; van Kaam, J.B.C.H.M. Reproducing kernel Hilbert spaces regression methods for genomic assisted prediction of quantitative traits. Genetics 2008, 178, 2289–2303. [Google Scholar] [CrossRef] [PubMed]
- Sousa, M.; Cuevas, J.; Couto, E.; Perez-Rodriguez, P.; Jarquin, D.; Fritsche-Neto, R.; Burgueno, J.; Crossa, J. Genomic-enabled prediction in maize using kernel models with genotype x environment interaction. G3 Bethesda 2017, 7, 1995–2014. [Google Scholar] [CrossRef] [PubMed]
- Calus, M.P.L. Genomic breeding value prediction: Methods and procedures. Animal 2010, 4, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Jenko, J.; Wiggans, G.R.; Cooper, T.A.; Eaglen, S.A.E.; Luff, W.G.d.L.; Bichard, M.; Pong-Wong, R.; Woolliams, J.A. Cow genotyping strategies for genomic selection in a small dairy cattle population. J. Dairy Sci. 2017, 100, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Wang, H.; Beyene, Y.; Semagn, K.; Liu, Y.; Cao, S.; Cui, Z.; Ruan, Y.; Burgueno, J.; Vicente, F.S.; et al. Effect of trait heritability, training population size and marker density on genomic prediction accuracy estimation in 22 bi-parental tropical maize populations. Front. Plant Sci. 2017, 8, 1–12. [Google Scholar] [CrossRef]
- Daetwyler, H.D.; Pong-Wong, R.; Villanueva, B.; Woolliams, J.A. The impact of genetic architecture on genome-wide evaluation methods. Genetics 2010, 185, 1021–1031. [Google Scholar] [CrossRef]
- Cericola, F.; Jahoor, A.; Orabi, J.; Andersen, J.R.; Janss, L.L.; Jensen, J. Optimizing training population size and genotyping strategy for genomic prediction using association study results and pedigree information. A case of study in advanced wheat breeding lines. PLoS ONE 2017, 12, e0169606. [Google Scholar] [CrossRef]
- Daetwyler, H.D.; Villanueva, B.; Woolliams, J.A. Accuracy of predicting the genetic risk of disease using a genome-wide approach. PLoS ONE 2008, 3, e3395. [Google Scholar] [CrossRef]
- Hayes Ben, J.; Bowman Phillip, J.; Chamberlain Amanda, C.; Verbyla, K.; Goddard Mike, E. Accuracy of genomic breeding values in multi-breed dairy cattle populations. Genet. Sel. Evol. 2009, 41, 1–9. [Google Scholar] [CrossRef]
- Pszczola, M.; Strabel, T.; Mulder, H.A.; Calus, M.P.L. Reliability of direct genomic values for animals with different relationships within and to the reference population. J. Dairy Sci. 2012, 95, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Goddard, M.E.; Hayes, B.J.; Meuwissen, T.H.E. Using the genomic relationship matrix to predict the accuracy of genomic selection. J. Anim. Breed. Genet. 2011, 128, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Hayes, B.J.; Visscher, P.M.; Goddard, M.E. Increased accuracy of artificial selection by using the realized relationship matrix. Genet. Res. 2009, 91, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Podlich, D.W.; Winkler, C.R.; Cooper, M. Mapping as you go: An effective approach for marker-assisted selection of complex traits. Crop Sci. 2004, 44, 1560–1571. [Google Scholar] [CrossRef]
- Elliot, L.H.; Jean-Luc, J.; Mark, E.S. Genomic selection accuracy using multifamily prediction models in a wheat breeding program. Plant Genome 2011, 4, 65–75. [Google Scholar] [CrossRef]
- Auinger, H.-J.; Schönleben, M.; Lehermeier, C.; Schmidt, M.; Korzun, V.; Geiger, H.; Piepho, H.-P.; Gordillo, A.; Wilde, P.; Bauer, E.; et al. Model training across multiple breeding cycles significantly improves genomic prediction accuracy in rye (Secale cereale L.). Appl. Genet. 2016, 129, 2043–2053. [Google Scholar] [CrossRef]
- García-Ruiz, A.; Cole, J.B.; Vanraden, P.M.; Wiggans, G.R.; Ruiz-López, F.J.; Van Tassell, C.P. Changes in genetic selection differentials and generation intervals in US Holstein dairy cattle as a result of genomic selection. Proc. Natl. Acad. Sci. USA 2016, 113, E3995–E4004. [Google Scholar] [CrossRef]
- Hickey, J.M.; Chiurugwi, T.; Mackay, I.; Powell, W. Genomic prediction unifies animal and plant breeding programs to form platforms for biological discovery. Nat. Genet. 2017, 49, 1297–1303. [Google Scholar] [CrossRef]
- Meuwissen, T.; Hayes, B.; Goddard, M. Genomic selection: A paradigm shift in animal breeding. Anim. Front. 2016, 6, 6–14. [Google Scholar] [CrossRef]
- Mrode, R.; Ojango, J.; Okeyo, A.; Mwacharo, J. Genomic selection and use of molecular tools in breeding programs for indigenous and crossbred cattle in developing countries: Current status and future prospects. Front. Genet. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Bernardo, R.; Yu, J. Prospects for genomewide selection for quantitative traits in maize. Crop Sci. 2007, 47, 1082–1090. [Google Scholar] [CrossRef]
- Lorenz, A.J.; Smith, K.P.; Jannink, J.L. Potential and optimization of genomic selection for fusarium head blight resistance in six-row barley. Crop Sci. 2012, 52, 1609–1621. [Google Scholar] [CrossRef]
- Spindel, J.; Begum, H.; Akdemir, D.; Virk, P.; Collard, B.; Redoña, E.; Atlin, G.; Jannink, J.-L.; McCouch, S.R. Genomic selection and association mapping in rice (Oryza Sativa): Effect of trait genetic architecture, training population composition, marker number and statistical model on accuracy of rice genomic selection in elite, tropical rice breeding lines. PLoS Genet. 2015, 11, 1–25. [Google Scholar] [CrossRef]
- Muleta, K.T.; Pressoir, G.; Morris, G.P. Optimizing genomic selection for a sorghum breeding program in Haiti: A simulation study. G3 Bethesda 2019, 9, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.; Gho, C.; Leafgren, R.; Tang, T.; Messina, C. Breeding drought-tolerant maize hybrids for the US corn-belt: Discovery to product. J. Exp. Bot. 2014, 65, 6191–6204. [Google Scholar] [CrossRef]
- Gaffney, J.; Schussler, J.; Löffler, C.; Cai, W.; Paszkiewicz, S.; Messina, C.; Groeteke, J.; Keaschall, J.; Cooper, M. Industry-scale evaluation of maize hybrids selected for increased yield in drought-stress conditions of the US corn belt. Crop Sci. 2015, 55, 1608–1618. [Google Scholar] [CrossRef]
- Lee, M. DNA markers and plant breeding programs. Adv. Agron. 1995, 55, 265–344. [Google Scholar] [CrossRef]
- Pastina, M.M.; Pinto, L.R.; Oliveira, K.M.; De Souza, A.P.; Garcia, A.A.F. Molecular mapping of complex traits. In Genetics, Genomics and Breeding of Sugarcane; Henry, R., Kole, C., Eds.; CRC Press: Boca Raton, FL, USA, 2010; pp. 117–148. ISBN 978-1-4398-4860-9. [Google Scholar] [CrossRef]
- Beavis, W.D. QTL analyses: Power, precision, and accuracy. In Molecular dissection of complex traits; Patterson, A.H., Ed.; CRC Press: Boca Raton, FL, USA, 1998; Volume 1998, pp. 145–162. ISBN 978-0-8493-7686-3. [Google Scholar]
- Xu, Y.; Crouch, J.H. Marker-assisted selection in plant breeding: From publications to practice. Crop Sci. 2008, 48, 391–407. [Google Scholar] [CrossRef]
- Dekkers, J.C.M.; Hospital, F. The use of molecular genetics in the improvement of agricultural populations. Nat. Rev. Genet. 2002, 3, 22–32. [Google Scholar] [CrossRef]
- Zingaretti, M.L.; Monfort, A.; Pérez-Enciso, M. pSBVB: A versatile simulation tool to evaluate genomic selection in polyploid species. G3 Bethesda 2019, 9, 327–334. [Google Scholar] [CrossRef]
- Salvador, A.G.; Luis, F.O.; Verma, S.; Whitaker, V.M. An experimental validation of genomic selection in octoploid strawberry. Hortic. Res. 2017, 4, 1–9. [Google Scholar] [CrossRef]
- Gouy, M.; Rousselle, Y.; Bastianelli, D.; Lecomte, P.; Bonnal, L.; Roques, D.; Efile, J.C.; Rocher, S.; Daugrois, J.; Toubi, L.; et al. Experimental assessment of the accuracy of genomic selection in sugarcane. Appl. Genet. 2013, 126, 2575–2586. [Google Scholar] [CrossRef] [PubMed]
- Deomano, E.; Jakson, P.; Wei, X.; Aitken, K.; Kota, R.; Perez-Rodriguez, P. Genomic Prediction of sugar content an cane yield in sugar cane clones in different stages of selection in a breeding program, with and without pedigree information. Mol. Breed. 2020, 40. [Google Scholar] [CrossRef]
- Van Eeuwijk, F.A.; Bustos-Korts, D.; Millet, E.J.; Boer, M.P.; Kruijer, W.; Thompson, A.; Malosetti, M.; Iwata, H.; Quiroz, R.; Kuppe, C.; et al. Modelling strategies for assessing and increasing the effectiveness of new phenotyping techniques in plant breeding. Plant Sci. 2019, 282, 23–39. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.; Technow, F.; Messina, C.; Gho, C.; Totir, L.R. Use of crop growth models with whole-genome prediction: Application to a maize multienvironment trial. Crop Sci. 2016, 56, 2141–2156. [Google Scholar] [CrossRef]
- Hill, W.G.; Goddard, M.E.; Visscher, P.M. Data and theory point to mainly additive genetic variance for complex traits. PLoS Genet. 2005, 4, 1–10. [Google Scholar] [CrossRef]
- Cheverud, J.M.; Routman, E.J. Epistasis and its contribution to genetic variance components. Genetics 1995, 139, 1455–1461. [Google Scholar]
- Cheverud, J.M.; Routman, E.J. Epistasis as a source of increased additive genetic variance at population bottlenecks. Evolution 1996, 50, 1042–1051. [Google Scholar] [CrossRef]
- Varona, L.; Legarra, A.; Toro, M.A.; Vitezica, Z.G. Non-additive effects in genomic selection. Front. Genet. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Osborn, T.C.; Chris Pires, J.; Birchler, J.A.; Auger, D.L.; Jeffery Chen, Z.; Lee, H.-S.; Comai, L.; Madlung, A.; Doerge, R.W.; Colot, V.; et al. Understanding mechanisms of novel gene expression in polyploids. Trends Genet. 2003, 19, 141–147. [Google Scholar] [CrossRef]
- Endelman, J.B.; Carley, C.A.S.; Bethke, P.C.; Coombs, J.J.; Clough, M.E.; Da Silva, W.L.; De Jong, W.S.; Douches, D.S.; Frederick, C.M.; Haynes, K.G.; et al. Genetic variance partitioning and genome-wide prediction with allele dosage information in autotetraploid potato. Genetics 2018, 209, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Gaynor, R.C.; Gorjanc, G.; Bentley, A.R.; Ober, E.S.; Howell, P.; Jackson, R.; Mackay, I.J.; Hickey, J.M. A two-part strategy for using genomic selection to develop inbred lines. Crop Sci. 2017, 57, 2372–2386. [Google Scholar] [CrossRef]
- Comstock, R.E.; Robinson, H.F.; Harvey, P.H. A breeding procedure designed to make maximum use of both general and specific combining ability1. Agron. J. 1949, 41, 360–367. [Google Scholar] [CrossRef]
- Hecker, R.J. Recurrent and reciprocal recurrent selection in sugarbeet1. Crop Sci. 1978, 18, 805–809. [Google Scholar] [CrossRef]
- Santos, M.F.; Moro, G.V.; Aguiar, A.M.; Souza, C.L.D., Jr. Responses to reciprocal recurrent selection and changes in genetic variability in IG-1 and IG-2 maize populations. Genet. Mol. Biol. 2005, 28, 781–788. [Google Scholar] [CrossRef][Green Version]
- Cooper, M.; Messina, C.D.; Podlich, D.; Totir, L.R.; Baumgarten, A.; Hausmann, N.J.; Wright, D.; Graham, G. Predicting the future of plant breeding: complementing empirical evaluation with genetic prediction. Crop Pasture Sci. 2014, 65, 311–336. [Google Scholar] [CrossRef]
- Cros, D.; Denis, M.; Bouvet, J.-M.; Sánchez, L. Long-term genomic selection for heterosis without dominance in multiplicative traits: Case study of bunch production in oil palm. BMC Genom. 2015, 16, 1–17. [Google Scholar] [CrossRef]
- Rembe, M.; Zhao, Y.; Jiang, Y.; Reif, J. Reciprocal recurrent genomic selection: An attractive tool to leverage hybrid wheat breeding. Appl. Genet. 2019, 132, 687–698. [Google Scholar] [CrossRef]
- Crossa, J.; Pérez-Rodríguez, P.; Cuevas, J.; Montesinos-López, O.; Jarquín, D.; de Los Campos, G.; Burgueño, J.; González-Camacho, J.M.; Pérez-Elizalde, S.; Beyene, Y.; et al. Genomic selection in plant breeding: Methods, models, and perspectives. Trends Plant Sci. 2017, 22, 961–975. [Google Scholar] [CrossRef]
- Ødegård, J.; Sonesson Anna, K.; Yazdi, M.H.; Meuwissen Theo, H. Introgression of a major QTL from an inferior into a superior population using genomic selection. Genet. Sel. Evol. 2009, 41, 1–10. [Google Scholar] [CrossRef]
- Olatoye, M.; Clark, L.V.; Wang, J.P.; Yang, X.; Yamada, T.; Sacks, E.; Lipka, A.E. Evaluation of genomic selection and marker-assisted selection in Miscanthus and energycane. Mol. Breed. 2019, 39, 1–16. [Google Scholar] [CrossRef]
- Daetwyler, H.D.; Villanueva, B.; Bijma, P.; Woolliams, J.A. Inbreeding in genome-wide selection. J. Anim. Breed. Genet. 2007, 124, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Bouquet, A.; Juga, J. Integrating genomic selection into dairy cattle breeding programmes: A review. Animal 2013, 7, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Meuwissen, T.H.E. Maximizing the response of selection with a predefined rate of inbreeding. J. Anim. Sci. 1997, 75, 934–940. [Google Scholar] [CrossRef] [PubMed]
- Goddard, M. Genomic selection: prediction of accuracy and maximisation of long term response. Genetica 2009, 136, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Sonesson, A.K.; Woolliams, J.A.; Meuwissen, T.H.E. Genomic selection requires genomic control of inbreeding. Genet. Sel. Evol. 2012, 44, 1–10. [Google Scholar] [CrossRef]
- Daetwyler, H.D.; Hayden, M.J.; Spangenberg, G.C.; Hayes, B.J. Selection on optimal haploid value increases genetic gain and preserves more genetic diversity relative to genomic selection. Genetics 2015, 200, 1341–1348. [Google Scholar] [CrossRef]
- Müller, D.; Schopp, P.; Melchinger, A.E. Selection on expected maximum haploid breeding values can increase genetic gain in recurrent genomic selection. G3 Bethesda 2018, 8, 1173–1181. [Google Scholar] [CrossRef]
- Toro, M.A.; Varona, L. A note on mate allocation for dominance handling in genomic selection. Genet. Sel. Evol. 2010, 42, 1–9. [Google Scholar] [CrossRef]
- Li, X.; Zhu, C.; Wang, J.; Yu, J. Computer Simulation in plant breeding. In Advances in Agronomy; Sparks, D.L., Ed.; Elsevier Science & Technology: San Diego, CA, USA, 2012; Volume 116, pp. 219–264. ISBN 978-0-12-394277-7. [Google Scholar]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yadav, S.; Jackson, P.; Wei, X.; Ross, E.M.; Aitken, K.; Deomano, E.; Atkin, F.; Hayes, B.J.; Voss-Fels, K.P. Accelerating Genetic Gain in Sugarcane Breeding Using Genomic Selection. Agronomy 2020, 10, 585. https://doi.org/10.3390/agronomy10040585
Yadav S, Jackson P, Wei X, Ross EM, Aitken K, Deomano E, Atkin F, Hayes BJ, Voss-Fels KP. Accelerating Genetic Gain in Sugarcane Breeding Using Genomic Selection. Agronomy. 2020; 10(4):585. https://doi.org/10.3390/agronomy10040585
Chicago/Turabian StyleYadav, Seema, Phillip Jackson, Xianming Wei, Elizabeth M. Ross, Karen Aitken, Emily Deomano, Felicity Atkin, Ben J. Hayes, and Kai P. Voss-Fels. 2020. "Accelerating Genetic Gain in Sugarcane Breeding Using Genomic Selection" Agronomy 10, no. 4: 585. https://doi.org/10.3390/agronomy10040585
APA StyleYadav, S., Jackson, P., Wei, X., Ross, E. M., Aitken, K., Deomano, E., Atkin, F., Hayes, B. J., & Voss-Fels, K. P. (2020). Accelerating Genetic Gain in Sugarcane Breeding Using Genomic Selection. Agronomy, 10(4), 585. https://doi.org/10.3390/agronomy10040585