
Exploring the Origins of Association of Poly(acrylic acid) Polyelectrolyte with Lysozyme in Aqueous Environment through Molecular Simulations and Experiments

 ,
,  ,
,  and
and 
Abstract

1. Introduction
2. Systems and Methods
2.1. Simulation Details
2.2. Experimental Details
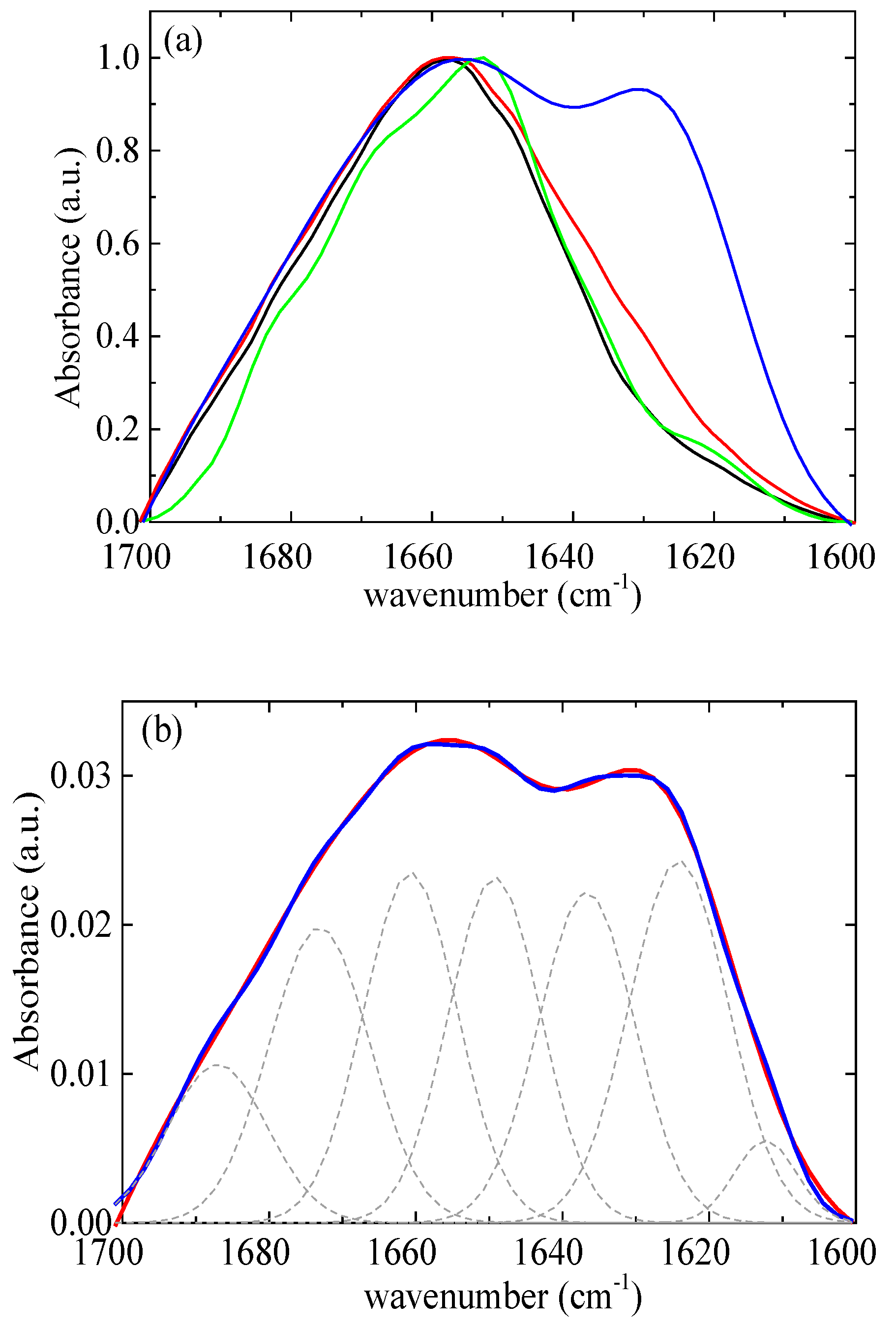
2.2.1. Fourier Transform Infrared Spectroscopy
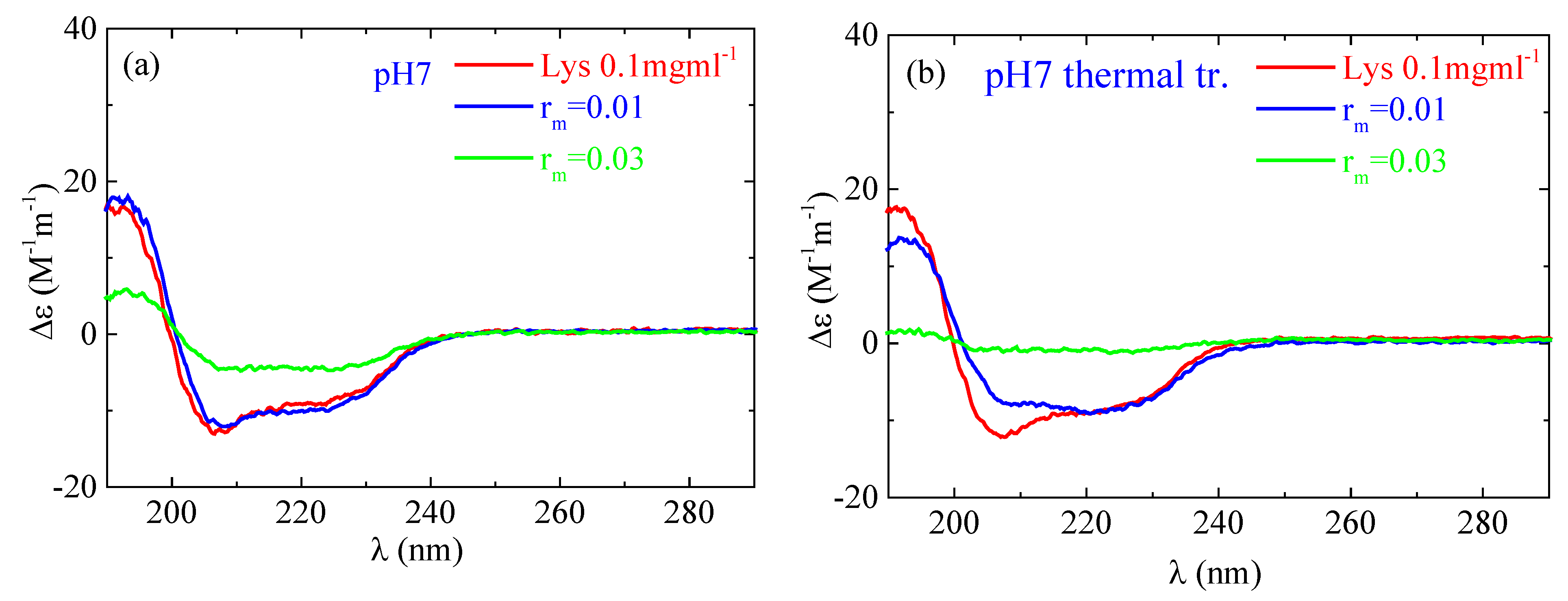
2.2.2. Circular Dichroism
3. Results and Discussion
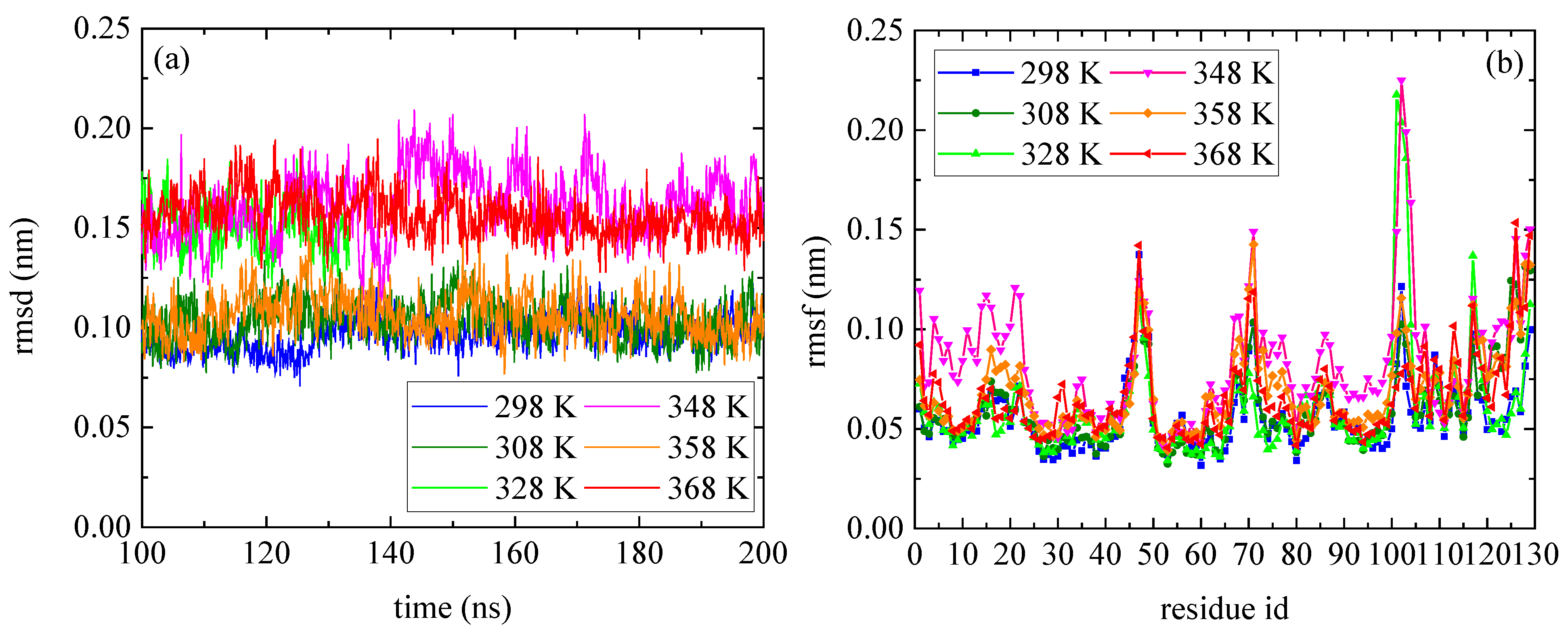
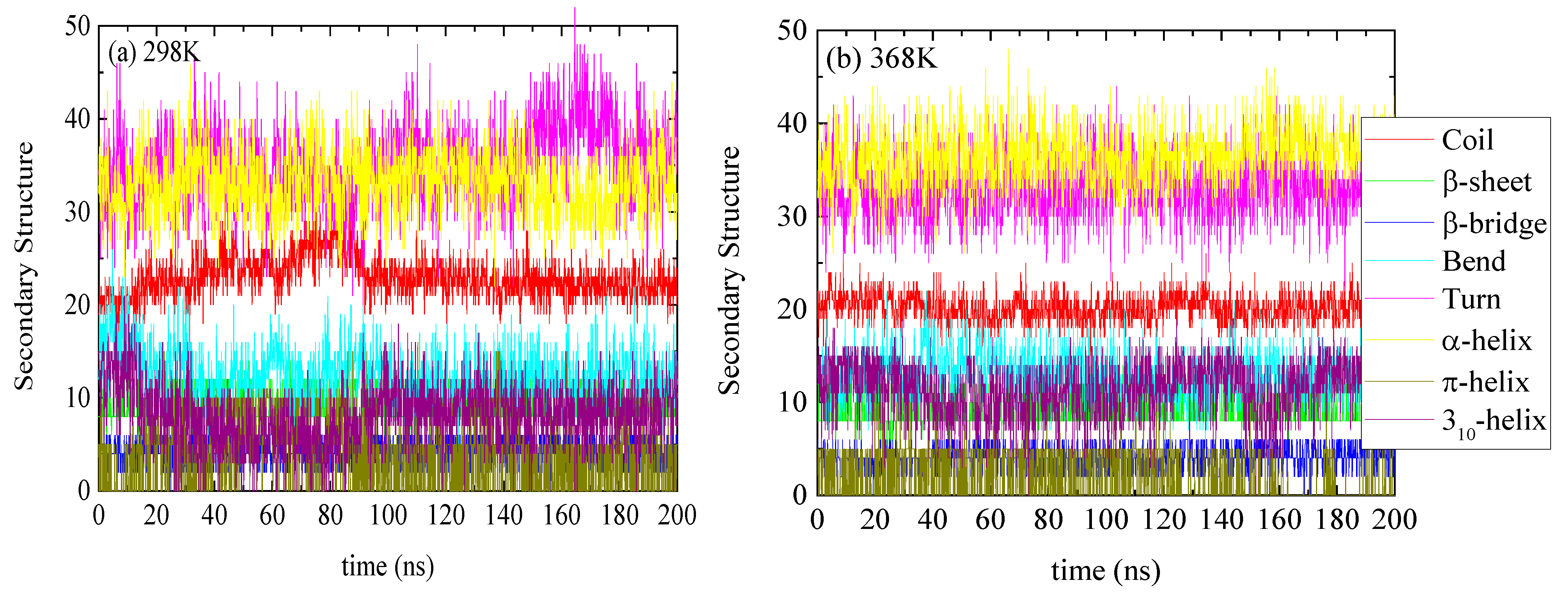
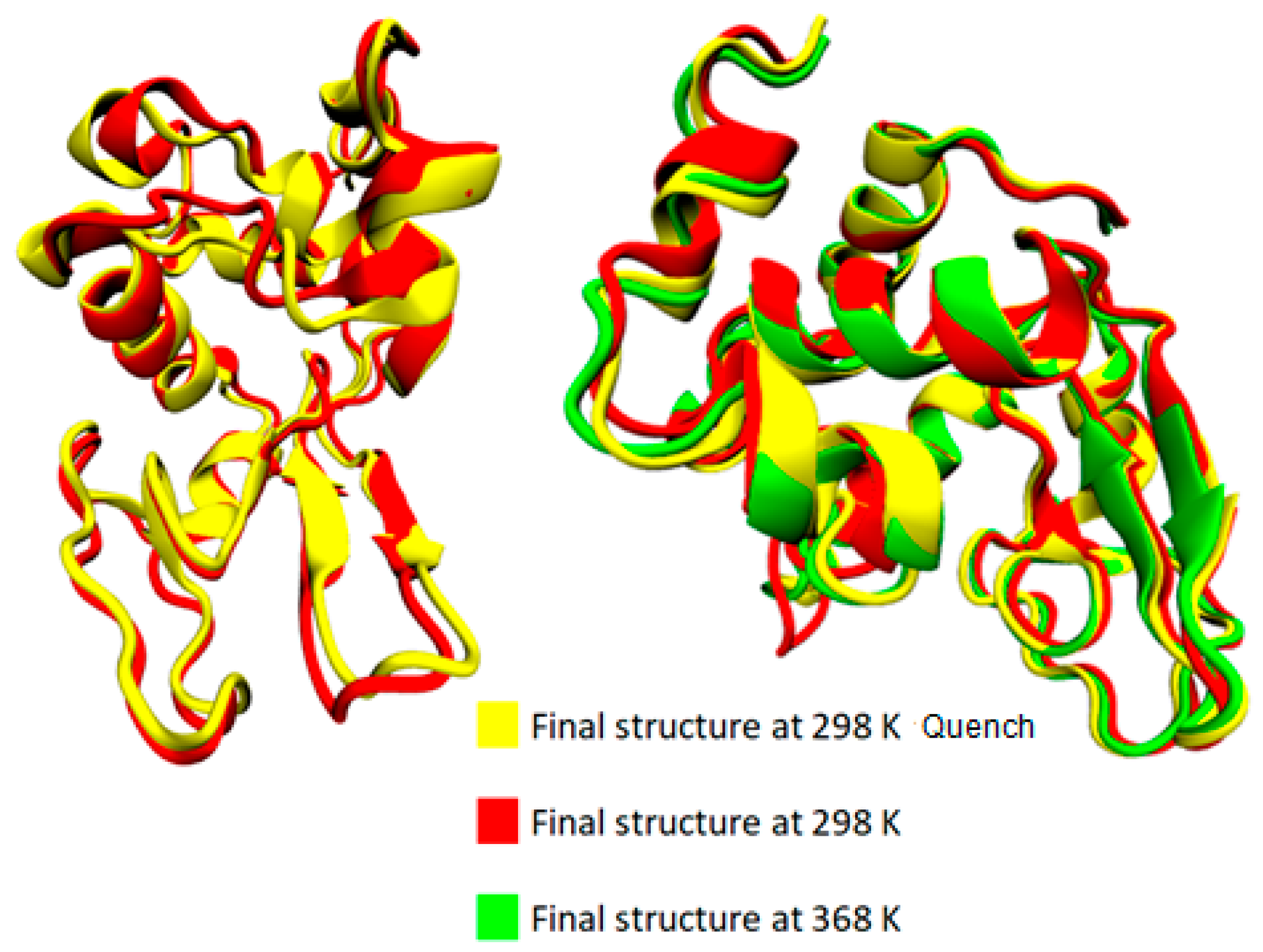
3.1. Effect of Temperature on the Conformational Properties of Lysozyme
3.2. Effect of PAA on the Conformational Properties of Lysozyme
3.3. Conformational Analysis of the Mixed System of Lysozyme and PAA
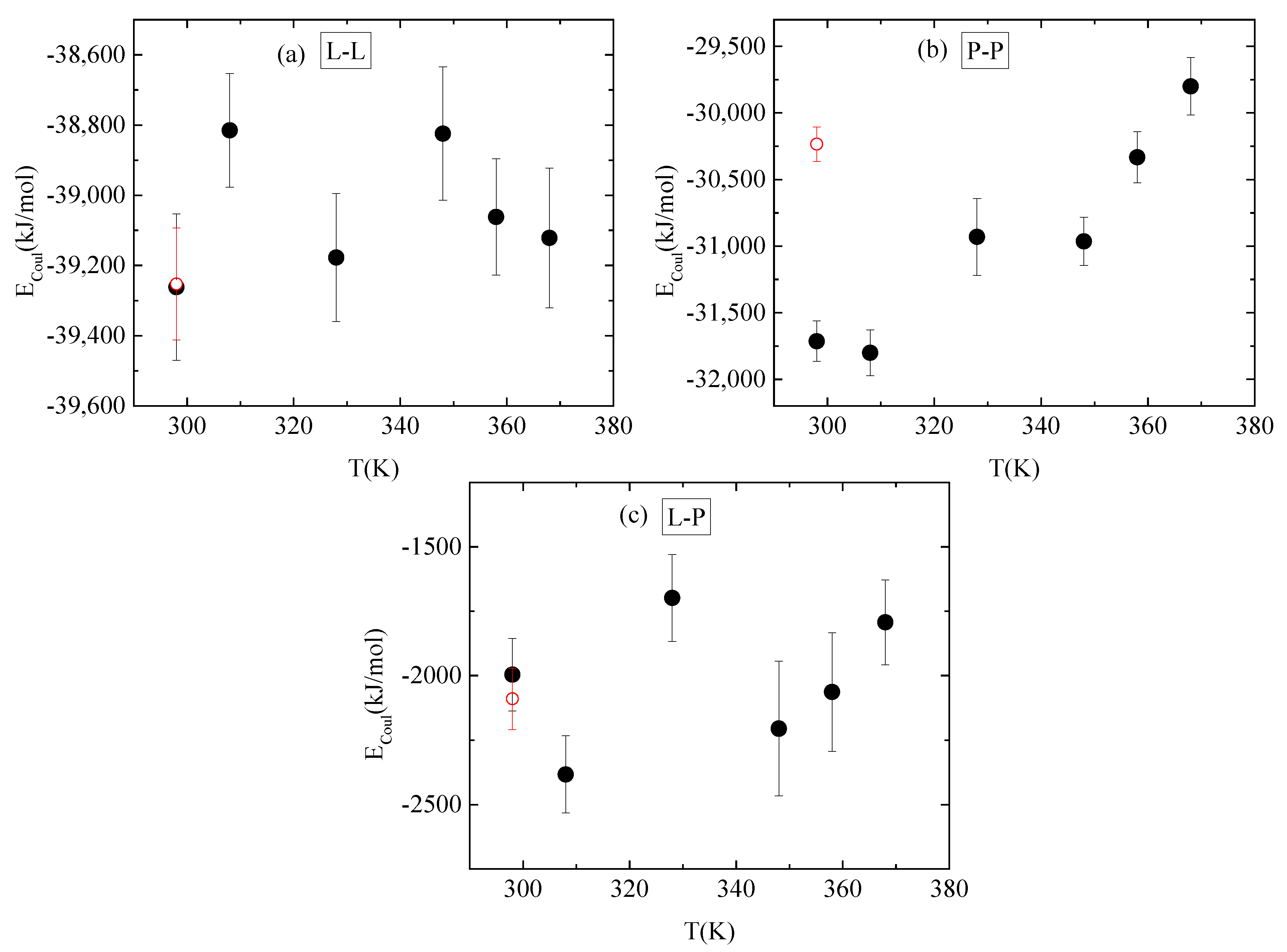
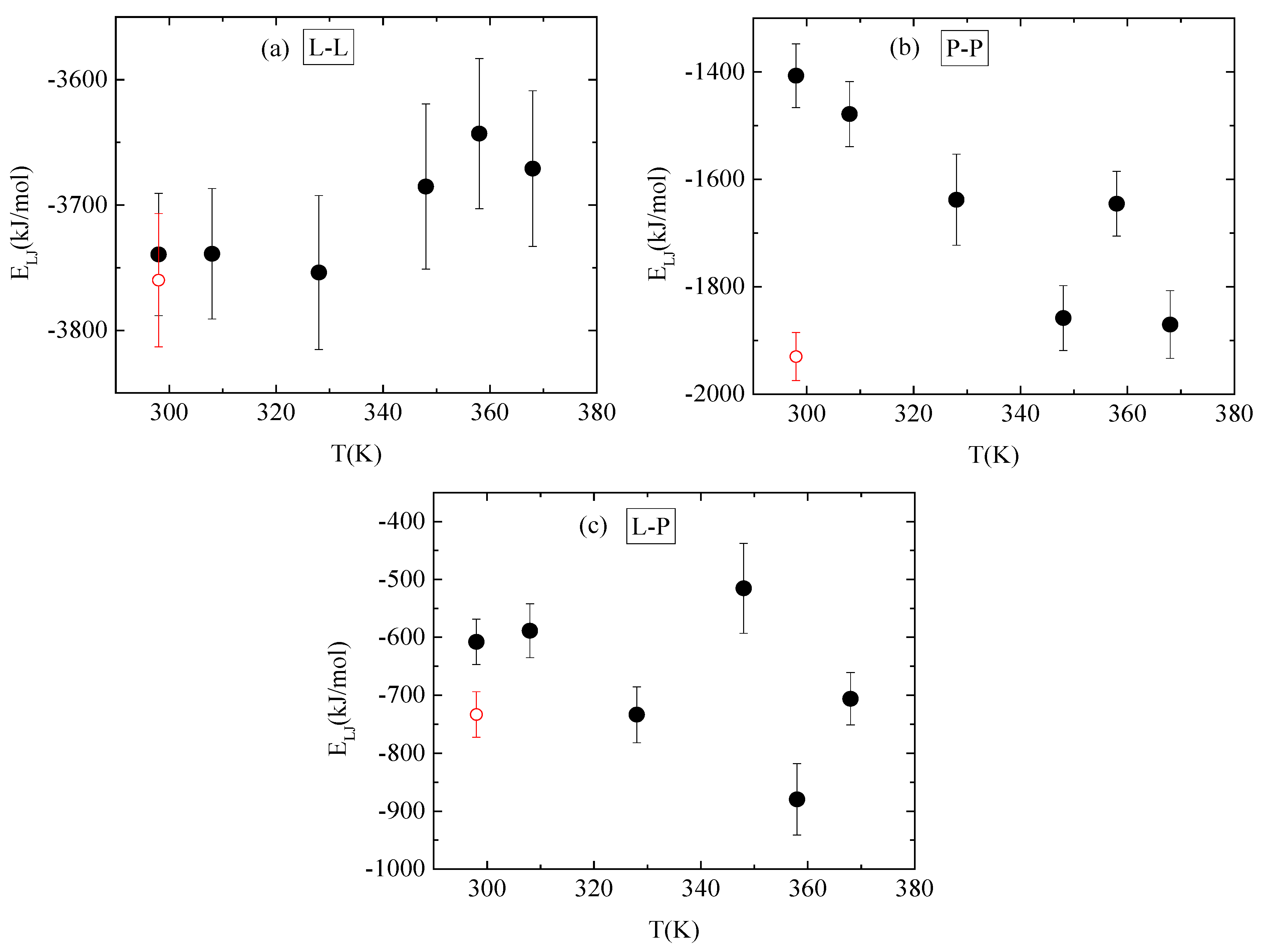
3.4. Energetics–Hydrogen Bonds
4. Experimental Evidence
5. Linking MD Insights to Experimental Data and Broader Scientific Impact
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Biswal, T.; BadJena, S.K.; Pradhan, D. Sustainable biomaterials and their applications: A short review. Mater. Today Proc. 2020, 30, 274–282. [Google Scholar] [CrossRef]
- Qi, W.; Zhang, X.; Wang, H. Self-assembled polymer nanocomposites for biomedical application. Curr. Opin. Colloid Interface Sci. 2018, 35, 36–41. [Google Scholar] [CrossRef]
- Valencia, G.A.; Zare, E.N.; Makvandi, P.; Gutiérrez, T.J. Self-Assembled Carbohydrate Polymers for Food Applications: A Review. Compr. Rev. Food Sci. Food Saf. 2019, 18, 2009–2024. [Google Scholar] [CrossRef] [PubMed]
- Sikder, A.; Pearce, A.K.; Parkinson, S.J.; Napier, R.; O’Reilly, R.K. Recent Trends in Advanced Polymer Materials in Agriculture Related Applications. ACS Appl. Polym. Mater. 2021, 3, 1203–1217. [Google Scholar] [CrossRef]
- Cooper, C.L.; Dubin, P.L.; Kayitmazer, A.B.; Turksen, S. Polyelectrolyte–protein complexes. Curr. Opin. Colloid Interface Sci. 2005, 10, 52–78. [Google Scholar] [CrossRef]
- Comert, F.; Malanowski, A.J.; Azarikia, F.; Dubin, P.L. Coacervation and precipitation in polysaccharide–protein systems. Soft Matter 2016, 12, 4154–4161. [Google Scholar] [CrossRef]
- Murmiliuk, A.; Iwase, H.; Kang, J.-J.; Mohanakumar, S.; Appavou, M.-S.; Wood, K.; Almásy, L.; Len, A.; Schwärzer, K.; Allgaier, J.; et al. Polyelectrolyte-protein synergism: pH-responsive polyelectrolyte/insulin complexes as versatile carriers for targeted protein and drug delivery. J. Colloid Interface Sci. 2024, 665, 801–813. [Google Scholar] [CrossRef]
- Devi, N.; Sarmah, M.; Khatun, B.; Maji, T.K. Encapsulation of active ingredients in polysaccharide–protein complex coacervates. Adv. Colloid Interface Sci. 2017, 239, 136–145. [Google Scholar] [CrossRef]
- Maeng, S.-W.; Ko, J.-Y.; Park, T.Y.; Yun, J.; Park, S.H.; Han, S.J.; Joo, K.I.; Ha, S.; Jee, M.; Im, G.-I.; et al. Adipose stem cell transplantation using adhesive protein-based viscous immiscible liquid for cartilage reconstruction. Chem. Eng. J. 2023, 463, 142379. [Google Scholar] [CrossRef]
- Mao, L.; Pan, Q.; Yuan, F.; Gao, Y. Formation of soy protein isolate-carrageenan complex coacervates for improved viability of Bifidobacterium longum during pasteurization and in vitro digestion. Food Chem. 2019, 276, 307–314. [Google Scholar] [CrossRef]
- Papagiannopoulos, A. Current Research on Polyelectrolyte Nanostructures: From Molecular Interactions to Biomedical Applications. Macromol 2021, 1, 155–172. [Google Scholar] [CrossRef]
- Jones, O.G.; McClements, D.J. Biopolymer Nanoparticles from Heat-Treated Electrostatic Protein–Polysaccharide Complexes: Factors Affecting Particle Characteristics. J. Food Sci. 2010, 75, N36–N43. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Li, W.-W.; Katzir, A.; Raichlin, Y.; Yu, H.-Q.; Mizaikoff, B. Probing the secondary structure of bovine serum albumin during heat-induced denaturation using mid-infrared fiberoptic sensors. Analyst 2015, 140, 765–770. [Google Scholar] [CrossRef]
- Papagiannopoulos, A.; Vlassi, E. Stimuli-responsive nanoparticles by thermal treatment of bovine serum albumin inside its complexes with chondroitin sulfate. Food Hydrocoll. 2019, 87, 602–610. [Google Scholar] [CrossRef]
- Jones, O.G.; Decker, E.A.; McClements, D.J. Formation of biopolymer particles by thermal treatment of β-lactoglobulin–pectin complexes. Food Hydrocoll. 2009, 23, 1312–1321. [Google Scholar] [CrossRef]
- Cousin, F.; Gummel, J.; Combet, S.; Boué, F. The model Lysozyme–PSSNa+ system for electrostatic complexation: Similarities and differences with complex coacervation. Adv. Colloid Interface Sci. 2011, 167, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.G.; Jiang, Y.W.; Chen, Z.; Yu, Z.W. Folding Behaviors of Protein (Lysozyme) Confined in Polyelectrolyte Complex Micelle. Langmuir ACS J. Surf. Colloids 2016, 32, 3655–3664. [Google Scholar] [CrossRef]
- Ndour, M.; Janot, J.-M.; Soussan, L.; Bouaziz, Z.; Voiry, D.; Balme, S. Impact of polyelectrolytes on lysozyme properties in colloidal dispersions. Colloids Surf. B Biointerfaces 2019, 183, 110419. [Google Scholar] [CrossRef]
- Johansson, C.; Hansson, P.; Malmsten, M. Interaction between lysozyme and poly(acrylic acid) microgels. J. Colloid Interface Sci. 2007, 316, 350–359. [Google Scholar] [CrossRef]
- Johansson, C.; Gernandt, J.; Bradley, M.; Vincent, B.; Hansson, P. Interaction between lysozyme and colloidal poly(NIPAM-co-acrylic acid) microgels. J. Colloid Interface Sci. 2010, 347, 241–251. [Google Scholar] [CrossRef]
- Xu, W.; Jin, W.; Wang, Y.; Li, J.; Huang, K.; Shah, B.R.; Li, B. Effect of physical interactions on structure of lysozyme in presence of three kinds of polysaccharides. J. Food Sci. Technol. 2018, 55, 3056–3064. [Google Scholar] [CrossRef] [PubMed]
- Makshakova, O.N.; Bogdanova, L.R.; Faizullin, D.A.; Ermakova, E.A.; Zuev, Y.F.; Sedov, I.A. Interaction-induced structural transformation of lysozyme and kappa-carrageenan in binary complexes. Carbohydr. Polym. 2021, 252, 117181. [Google Scholar] [CrossRef] [PubMed]
- Kayitmazer, A.B.; Seeman, D.; Minsky, B.B.; Dubin, P.L.; Xu, Y. Protein–polyelectrolyte interactions. Soft Matter 2013, 9, 2553–2583. [Google Scholar] [CrossRef]
- Yu, B.; Liang, H.; Nealey, P.F.; Tirrell, M.V.; Rumyantsev, A.M.; de Pablo, J.J. Structure and Dynamics of Hybrid Colloid–Polyelectrolyte Coacervates: Insights from Molecular Simulations. Macromolecules 2023, 56, 7256–7270. [Google Scholar] [CrossRef] [PubMed]
- Moses, K.; Van Tassel, P.R. Polyelectrolyte Influence on Beta-Hairpin Peptide Stability: A Simulation Study. J. Phys. Chem. B 2023, 127, 359–370. [Google Scholar] [CrossRef]
- Samanta, R.; Ganesan, V. Direct Simulations of Phase Behavior of Mixtures of Oppositely Charged Proteins/Nanoparticles and Polyelectrolytes. J. Phys. Chem. B 2020, 124, 10943–10951. [Google Scholar] [CrossRef]
- Yigit, C.; Heyda, J.; Ballauff, M.; Dzubiella, J. Like-charged protein-polyelectrolyte complexation driven by charge patches. J. Chem. Phys. 2015, 143, 064905. [Google Scholar] [CrossRef]
- Carlsson, F.; Linse, P.; Malmsten, M. Monte Carlo Simulations of Polyelectrolyte−Protein Complexation. J. Phys. Chem. B 2001, 105, 9040–9049. [Google Scholar] [CrossRef]
- Carlsson, F.; Malmsten, M.; Linse, P. Protein-Polyelectrolyte Cluster Formation and Redissolution: A Monte Carlo Study. J. Am. Chem. Soc. 2003, 125, 3140–3149. [Google Scholar] [CrossRef]
- Sofronova, A.A.; Evstafyeva, D.B.; Izumrudov, V.A.; Muronetz, V.I.; Semenyuk, P.I. Protein-polyelectrolyte complexes: Molecular dynamics simulations and experimental study. Polymer 2017, 113, 39–45. [Google Scholar] [CrossRef]
- Rajpersaud, T.; Tabandeh, S.; Leon, L.; Loverde, S.M. Molecular Dynamics Simulations of Polyelectrolyte Complexes. Biomacromolecules 2024, 25, 1468–1480. [Google Scholar] [CrossRef] [PubMed]
- Arnittali, M.; Rissanou, A.N.; Kefala, A.; Kokkinidis, M.; Harmandaris, V. Structure of amino acid sequence-reversed wtRop protein: Insights from atomistic molecular dynamics simulations. J. Biomol. Struct. Dyn. 2023, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Angioletti-Uberti, S.; Lu, Y.; Dzubiella, J.; Ballauff, M. Interaction of Proteins with Polyelectrolytes: Comparison of Theory to Experiment. Langmuir ACS J. Surf. Colloids 2019, 35, 5373–5391. [Google Scholar] [CrossRef]
- Walkowiak, J.J.; Ballauff, M. Interaction of Polyelectrolytes with Proteins: Quantifying the Role of Water. Adv. Sci. 2021, 8, 2100661. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.-H.; Wang, W.-Q.; Zhou, Y.-W.; Gao, X.-J.; Zhang, Q.; Zhang, M.-H. Research on the Interaction Mechanism and Structural Changes in Human Serum Albumin with Hispidin Using Spectroscopy and Molecular Docking. Molecules 2024, 29, 655. [Google Scholar] [CrossRef]
- Arkaban, H.; Barani, M.; Akbarizadeh, M.R.; Pal Singh Chauhan, N.; Jadoun, S.; Dehghani Soltani, M.; Zarrintaj, P. Polyacrylic Acid Nanoplatforms: Antimicrobial, Tissue Engineering, and Cancer Theranostic Applications. Polymers 2022, 14, 1259. [Google Scholar] [CrossRef]
- Chen, S.; Gil, C.J.; Ning, L.; Jin, L.; Perez, L.; Kabboul, G.; Tomov, M.L.; Serpooshan, V. Adhesive Tissue Engineered Scaffolds: Mechanisms and Applications. Front. Bioeng. Biotechnol. 2021, 9, 683079. [Google Scholar] [CrossRef]
- Swift, T.; Swanson, L.; Geoghegan, M.; Rimmer, S. The pH-responsive behaviour of poly(acrylic acid) in aqueous solution is dependent on molar mass. Soft Matter 2016, 12, 2542–2549. [Google Scholar] [CrossRef]
- Lamch, Ł.; Ronka, S.; Moszyńska, I.; Warszyński, P.; Wilk, K.A. Hydrophobically Functionalized Poly(Acrylic Acid) Comprising the Ester-Type Labile Spacer: Synthesis and Self-Organization in Water. Polymers 2020, 12, 1185. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Grigera, J.R.; Straatsma, T.P. The missing term in effective pair potentials. J. Phys. Chem. 1987, 91, 6269–6271. [Google Scholar] [CrossRef]
- Chatterjee, S.; Debenedetti, P.G.; Stillinger, F.H.; Lynden-Bell, R.M. A computational investigation of thermodynamics, structure, dynamics and solvation behavior in modified water models. J. Chem. Phys. 2008, 128, 124511. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Papagiannopoulos, A.; Sklapani, A.; Spiliopoulos, N. Thermally stabilized chondroitin sulfate-hemoglobin nanoparticles and their interaction with bioactive compounds. Biophys. Chem. 2024, 304, 107127. [Google Scholar] [CrossRef] [PubMed]
- Matsunami, H.; Kikuchi, R.; Ogawa, K.; Kokufuta, E. Light scattering study of complex formation between protein and polyelectrolyte at various ionic strengths. Colloids Surf. B Biointerfaces 2007, 56, 142–148. [Google Scholar] [CrossRef]
- Micsonai, A.; Wien, F.; Kernya, L.; Lee, Y.H.; Goto, Y.; Réfrégiers, M.; Kardos, J. Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc. Natl. Acad. Sci. USA 2015, 112, E3095–E3103. [Google Scholar] [CrossRef]
- Bolhuis, P.G. Sampling Kinetic Protein Folding Pathways using All-Atom Models. In Computer Simulations in Condensed Matter Systems: From Materials to Chemical Biology Volume 1; Ferrario, M., Ciccotti, G., Binder, K., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; pp. 393–433. [Google Scholar]
- Carugo, O. How root-mean-square distance (r.m.s.d.) values depend on the resolution of protein structures that are compared. J. Appl. Crystallogr. 2003, 36, 125–128. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Heinig, M.; Frishman, D. STRIDE: A web server for secondary structure assignment from known atomic coordinates of proteins. Nucleic Acids Res. 2004, 32 (Suppl. S2), W500–W502. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Wu, T.; Jiang, Q.; Wu, D.; Hu, Y.; Chen, S.; Ding, T.; Ye, X.; Liu, D.; Chen, J. What is new in lysozyme research and its application in food industry? A review. Food Chem. 2019, 274, 698–709. [Google Scholar] [CrossRef]
- Ascoli, G.A.; Pergami, P.; Luu, K.X.; Alkon, D.L.; Bramanti, E.; Bertucci, C.; Di Bari, L.; Salvadori, P. Use of CD and FT-IR to determine the secondary structure of purified proteins in the low-microgram range. Enantiomer 1998, 3, 371–381. [Google Scholar]
- Pribić, R.; van Stokkum, I.H.; Chapman, D.; Haris, P.I.; Bloemendal, M. Protein secondary structure from Fourier transform infrared and/or circular dichroism spectra. Anal. Biochem. 1993, 214, 366–378. [Google Scholar] [CrossRef]
- Chang, J.-Y.; Li, L. The unfolding mechanism and the disulfide structures of denatured lysozyme. FEBS Lett. 2002, 511, 73–78. [Google Scholar] [CrossRef]
- Givens, B.E.; Xu, Z.; Fiegel, J.; Grassian, V.H. Bovine serum albumin adsorption on SiO2 and TiO2 nanoparticle surfaces at circumneutral and acidic pH: A tale of two nano-bio surface interactions. J. Colloid Interface Sci. 2017, 493, 334–341. [Google Scholar] [CrossRef]
- Guerrero, P.; Kerry, J.P.; de la Caba, K. FTIR characterization of protein-polysaccharide interactions in extruded blends. Carbohydr. Polym. 2014, 111, 598–605. [Google Scholar] [CrossRef]
- Fu, K.; Griebenow, K.; Hsieh, L.; Klibanov, A.M.; Langer, R. FTIR characterization of the secondary structure of proteins encapsulated within PLGA microspheres. J. Control. Release 1999, 58, 357–366. [Google Scholar] [CrossRef]
- Sadat, A.; Joye, I.J. Peak Fitting Applied to Fourier Transform Infrared and Raman Spectroscopic Analysis of Proteins. Appl. Sci. 2020, 10, 5918. [Google Scholar] [CrossRef]
- Krimm, S.; Bandekar, J. Vibrational Spectroscopy and Conformation of Peptides, Polypeptides, and Proteins. In Advances in Protein Chemistry; Anfinsen, C.B., Edsall, J.T., Richards, F.M., Eds.; Academic Press: Cambridge, MA, USA, 1986; Volume 38, pp. 181–364. [Google Scholar]
- Vasita, R.; Katti, D.S. Structural and functional characterization of proteins adsorbed on hydrophilized polylactide-co-glycolide microfibers. Int. J. Nanomed. 2012, 7, 61–71. [Google Scholar]
- Kong, J.; Yu, S. Fourier transform infrared spectroscopic analysis of protein secondary structures. Acta Biochim. Biophys. Sin. 2007, 39, 549–559. [Google Scholar] [CrossRef]
- Papagiannopoulos, A.; Vlassi, E.; Radulescu, A. Reorganizations inside thermally stabilized protein/polysaccharide nanocarriers investigated by small angle neutron scattering. Carbohydr. Polym. 2019, 218, 218–225. [Google Scholar] [CrossRef]
- Vlassi, E.; Papagiannopoulos, A. Nanoformulation of fibrinogen by thermal stabilization of its electrostatic complexes with hyaluronic acid. Int. J. Biol. Macromol. 2020, 158, 251–257. [Google Scholar] [CrossRef]
- Tatulian, S.A. Structural Characterization of Membrane Proteins and Peptides by FTIR and ATR-FTIR Spectroscopy. In Lipid-Protein Interactions: Methods and Protocols; Kleinschmidt, J.H., Ed.; Humana Press: Totowa, NJ, USA, 2013; pp. 177–218. [Google Scholar]
- Micsonai, A.; Wien, F.; Bulyáki, É.; Kun, J.; Moussong, É.; Lee, Y.-H.; Goto, Y.; Réfrégiers, M.; Kardos, J. BeStSel: A web server for accurate protein secondary structure prediction and fold recognition from the circular dichroism spectra. Nucleic Acids Res. 2018, 46, W315–W322. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Systems | PAA | Lysozyme | Total Atoms | Water Molecules | Ions (Na+) |
|---|---|---|---|---|---|
| Reference | - | 1 | 33,798 | 10,610 | (Cl−) 8 |
| Mixture | 5 | 1 | 50,490 | 15,576 | (Na+) 92 |
| Systems | Coil | β-Sheet | β-Bridge | Bend | Turn | α-Helix | π-Helix | 310-Helix |
|---|---|---|---|---|---|---|---|---|
| L298 | 20.18 ± 1.46 | 9.25 ± 1.87 | 4.15 ± 1.41 | 12.46 ± 2.39 | 32.92 ± 3.22 | 36.75 ± 2.85 | 1.32 ± 2.35 | 11.96 ± 2.90 |
| L308 | 20.01 ± 1.50 | 9.06 ± 1.80 | 3.85 ± 1.18 | 13.98 ± 2.69 | 30.28 ± 2.89 | 36.44 ± 2.68 | 1.45 ± 2.42 | 13.93 ± 2.33 |
| L328 | 22.91 ± 1.72 | 8.11 ± 0.84 | 4.56 ± 1.19 | 13.35 ± 2.27 | 31.97 ± 2.96 | 37.79 ± 3.43 | 4.09 ± 3.28 | 6.22 ± 2.45 |
| L348 | 20.68 ± 1.80 | 9.02 ± 1.78 | 4.02 ± 1.36 | 14.43 ± 2.87 | 33.25 ± 3.92 | 34.53 ± 3.49 | 2.02 ± 2.54 | 11.04 ± 3.28 |
| L358 | 19.73 ± 1.55 | 8.29 ± 1.08 | 4.39 ± 1.11 | 14.79 ± 2.76 | 32.35 ± 3.73 | 35.31 ± 2.60 | 1.26 ± 2.31 | 12.88 ± 2.92 |
| L368 | 22.38 ± 1.64 | 9.04 ± 1.82 | 4.30 ± 1.41 | 11.85 ± 2.61 | 36.35 ± 4.31 | 33.11 ± 3.39 | 3.33 ± 3.42 | 8.64 ± 2.86 |
| L298Quench | 22.03 ± 1.46 | 9.35 ± 1.91 | 4.17 ± 1.39 | 12.07 ± 2.29 | 32.77 ± 2.99 | 39.07 ± 2.68 | 0.53 ± 1.59 | 9.00 ± 2.99 |
| Variation in conformations (V2 − V1)/V1 | ||||||||
| V1: 298 vs. V2: 368 | 0.11 | −0.02 | 0.04 | −0.05 | 0.10 | −0.10 | 1.52 | −0.28 |
| V1: 368 vs. V2: 298Quench | −0.02 | 0.03 | −0.03 | 0.02 | −0.09 | 0.18 | −0.84 | 0.04 |
| V1: 298 vs. V2: 298Quench | 0.09 | 0.01 | 0.005 | −0.03 | −0.004 | 0.06 | −0.59 | −0.25 |
| Variation in Conformations (Mixed-Pure)/Pure | ||||||||
|---|---|---|---|---|---|---|---|---|
| Systems | Coil | β-Sheet | β-Bridge | Bend | Turn | α-Helix | π-Helix | 310-Helix |
| Pure vs. Mixed @298 K | 0.016 | 0.025 | −0.177 | −0.151 | 0.066 | 0.035 | −0.444 | 0.028 |
| Pure vs. Mixed @368 K | 0.012 | −0.024 | 0.109 | −0.186 | 0.061 | −0.014 | 0.695 | −0.078 |
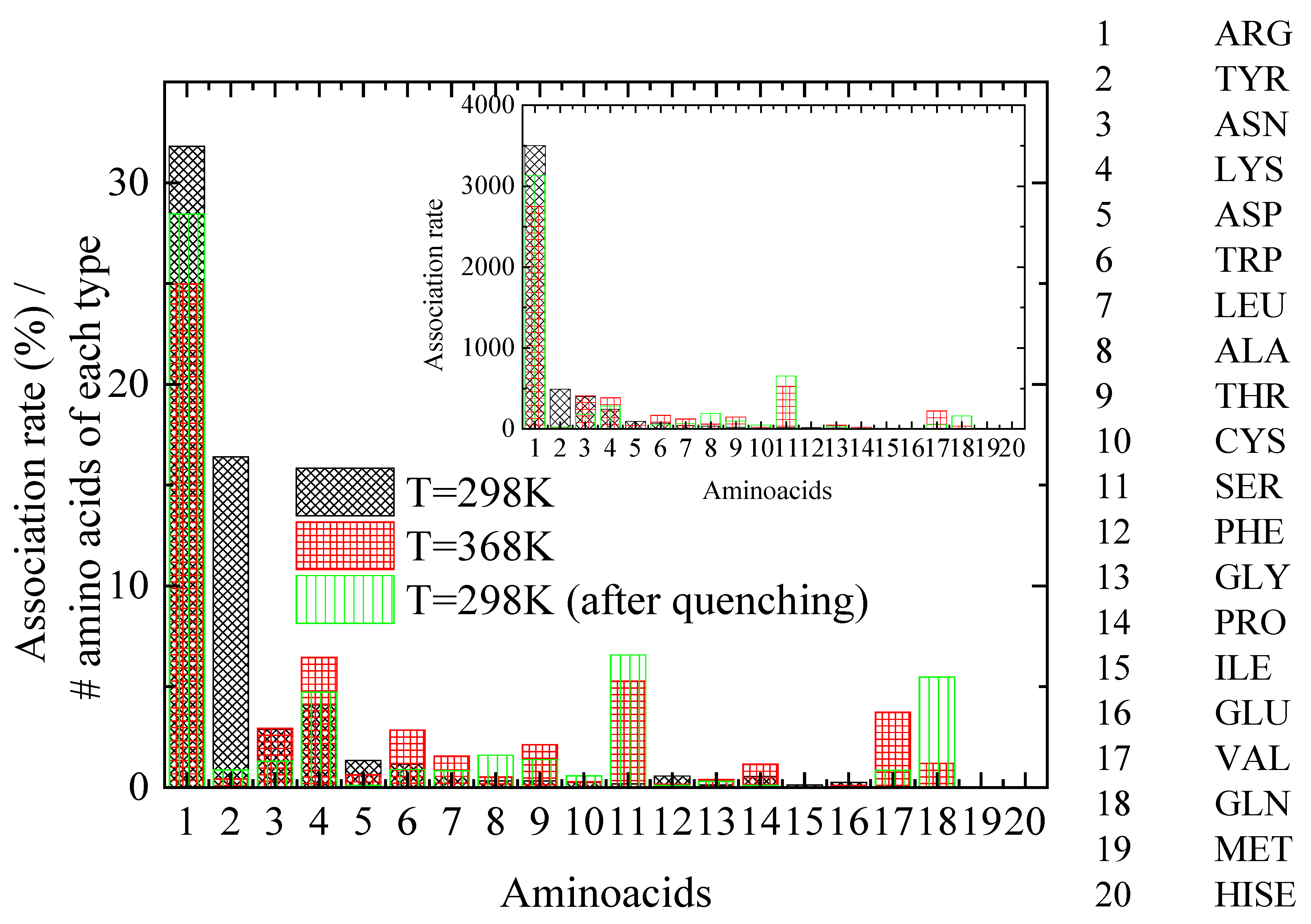
| Association Rate (%) | ||||||
|---|---|---|---|---|---|---|
| #Residues within Lysozyme | T = 298 K | T = 368 K | T = 298 K (after Quenching) | |||
| 11 | ARG | 31.81 | ARG | 24.98 | ARG | 28.45 |
| 3 | TYR | 16.4 | TYR | 0.43 | TYR | 0.87 |
| 14 | ASN | 2.87 | ASN | 2.94 | ASN | 1.31 |
| 6 | LYS | 4.1 | LYS | 6.45 | LYS | 4.73 |
| 10 | SER | 0.18 | SER | 5.28 | SER | 6.57 |
| Hydrogen Bonds Residue—PAA/Residue | |||
|---|---|---|---|
| Residue | T = 298 K | T = 368 K | T = 298 K (after Quenching) |
| ARG | 2.51 ± 0.27 | 1.96 ± 0.28 | 2.48 ± 0.21 |
| TYR | 0.48 ± 0.18 | 0.10 ± 0.18 | 0.004 ± 0.04 |
| ASN | 0.32 ± 0.12 | 0.24 ± 0.11 | 0.14 ± 0.06 |
| LYS | 0.39 ± 0.14 | 0.64 ± 0.19 | 0.47 ± 0.21 |
| SER | 0.06 ± 0.05 | 0.27 ± 0.12 | 0.32 ± 0.14 |
| Systems | SASA (nm2)—PAA | SASA (nm2)—Lysozyme | T (K) |
|---|---|---|---|
| L298 | 162.48 ± 3.28 | 70.76 ± 1.13 | 298 |
| L308 | 159.48 ± 3.90 | 72.74 ± 1.12 | 308 |
| L328 | 146.70 ± 4.34 | 70.06 ± 1.16 | 328 |
| L348 | 131.12 ± 2.44 | 70.54 ± 1.28 | 348 |
| L358 | 137.10 ± 3.54 | 72.23 ± 1.24 | 358 |
| L368 | 130.31 ± 3.69 | 70.92 ± 1.33 | 368 |
| Lquenching | 132.52 ± 1.41 | 70.34 ± 1.08 | 298 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arnittali, M.; Tegopoulos, S.N.; Kyritsis, A.; Harmandaris, V.; Papagiannopoulos, A.; Rissanou, A.N. Exploring the Origins of Association of Poly(acrylic acid) Polyelectrolyte with Lysozyme in Aqueous Environment through Molecular Simulations and Experiments. Polymers 2024, 16, 2565. https://doi.org/10.3390/polym16182565
Arnittali M, Tegopoulos SN, Kyritsis A, Harmandaris V, Papagiannopoulos A, Rissanou AN. Exploring the Origins of Association of Poly(acrylic acid) Polyelectrolyte with Lysozyme in Aqueous Environment through Molecular Simulations and Experiments. Polymers. 2024; 16(18):2565. https://doi.org/10.3390/polym16182565
Chicago/Turabian StyleArnittali, Maria, Sokratis N. Tegopoulos, Apostolos Kyritsis, Vagelis Harmandaris, Aristeidis Papagiannopoulos, and Anastassia N. Rissanou. 2024. "Exploring the Origins of Association of Poly(acrylic acid) Polyelectrolyte with Lysozyme in Aqueous Environment through Molecular Simulations and Experiments" Polymers 16, no. 18: 2565. https://doi.org/10.3390/polym16182565
APA StyleArnittali, M., Tegopoulos, S. N., Kyritsis, A., Harmandaris, V., Papagiannopoulos, A., & Rissanou, A. N. (2024). Exploring the Origins of Association of Poly(acrylic acid) Polyelectrolyte with Lysozyme in Aqueous Environment through Molecular Simulations and Experiments. Polymers, 16(18), 2565. https://doi.org/10.3390/polym16182565