
An Additive Manufacturing MicroFactory: Overcoming Brittle Material Failure and Improving Product Performance through Tablet Micro-Structure Control for an Immediate Release Dose Form


Abstract
1. Introduction
1.1. Melt Extrusion Additive Manufacturing
1.2. Fused Filament Fabrication (FFF)
1.3. Feedstock Filament Material Properties for FFF
1.4. Model Drug Mefenamic Acid (MFA)—Formulation Approaches to Improve Drug Product Performance
1.5. Solid Dispersion Formulations for FFF Applications
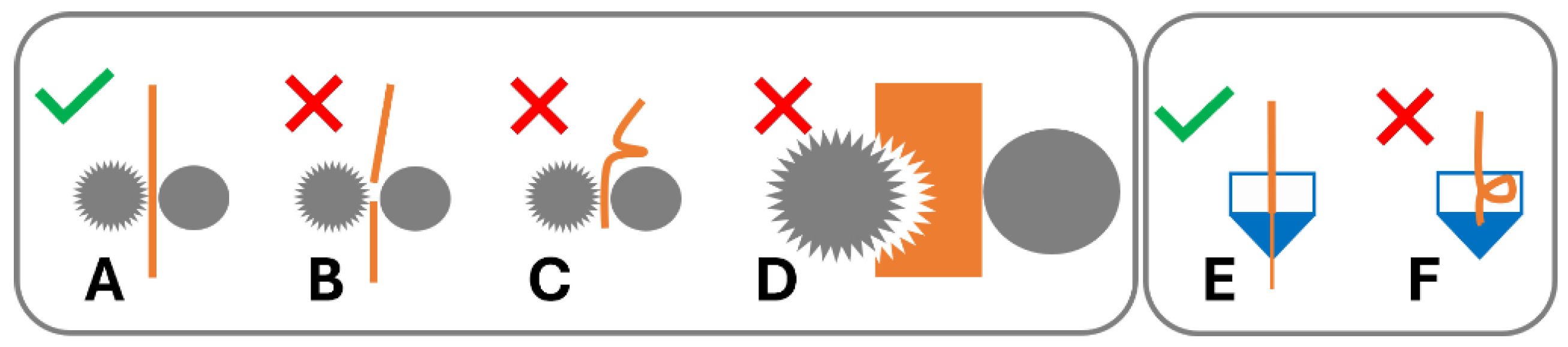
1.6. Overcoming Brittle Filament Feedstock Material Failure in FFF
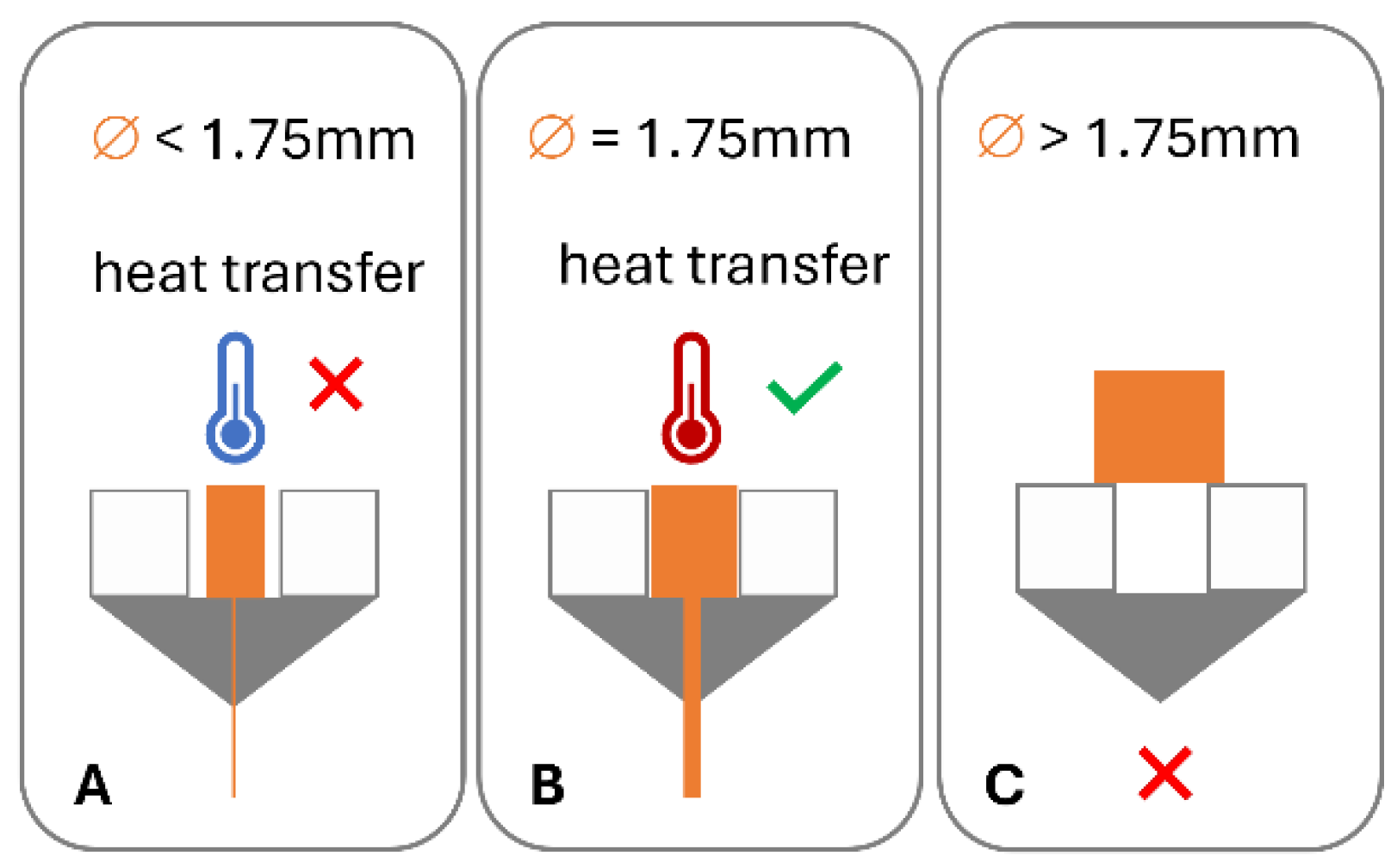
1.7. Filament-Free Material Extrusion
1.8. Aims of This Study
2. Methods and Materials
2.1. Materials
2.2. Formulation Preparation
2.3. HME-3D Printing
2.4. HPLC Content Analysis
2.5. Rheology Analysis
2.6. FTIR Analysis
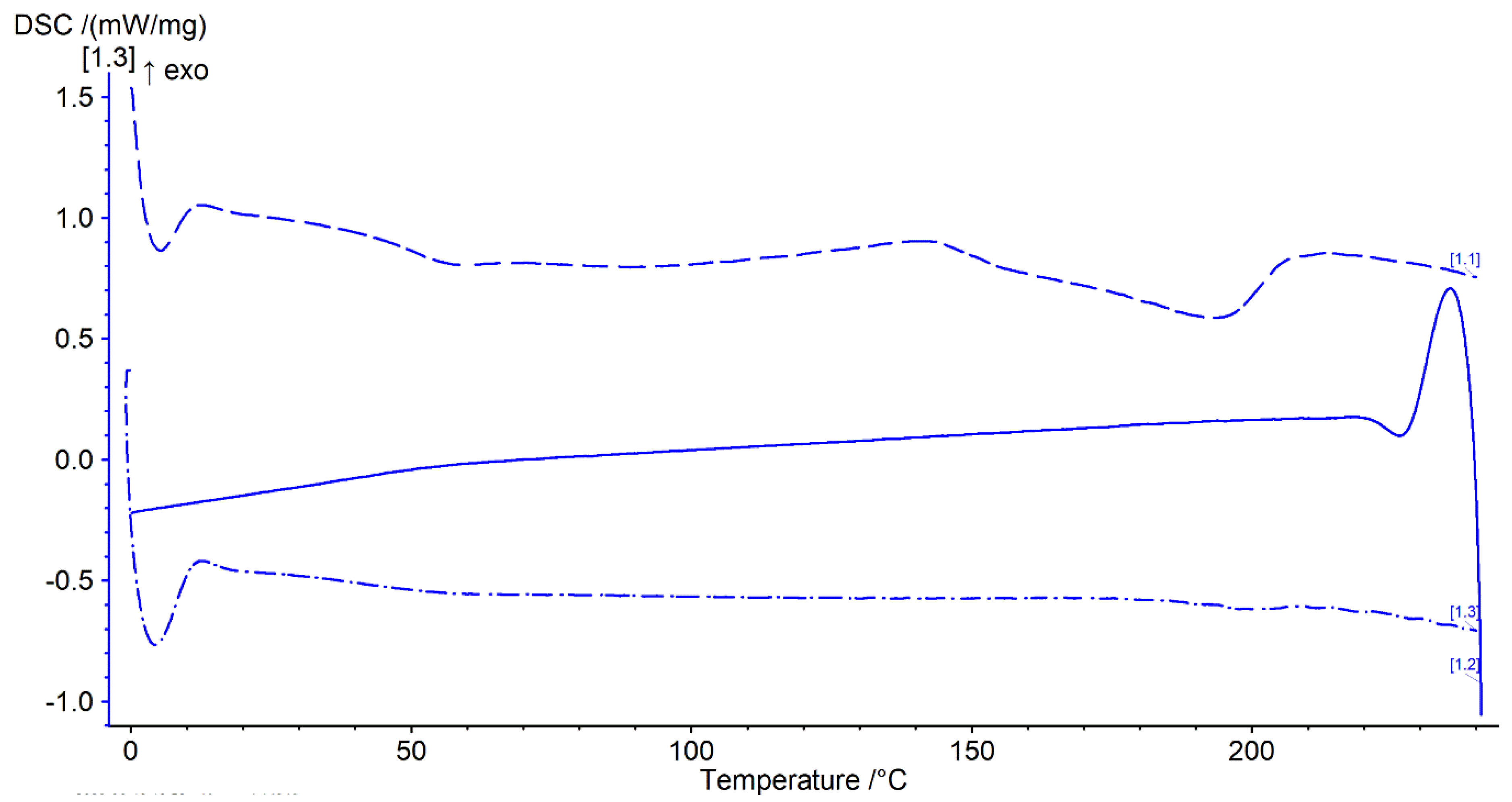
2.7. Thermal Analysis: Differential Scanning Calorimetry (DSC)
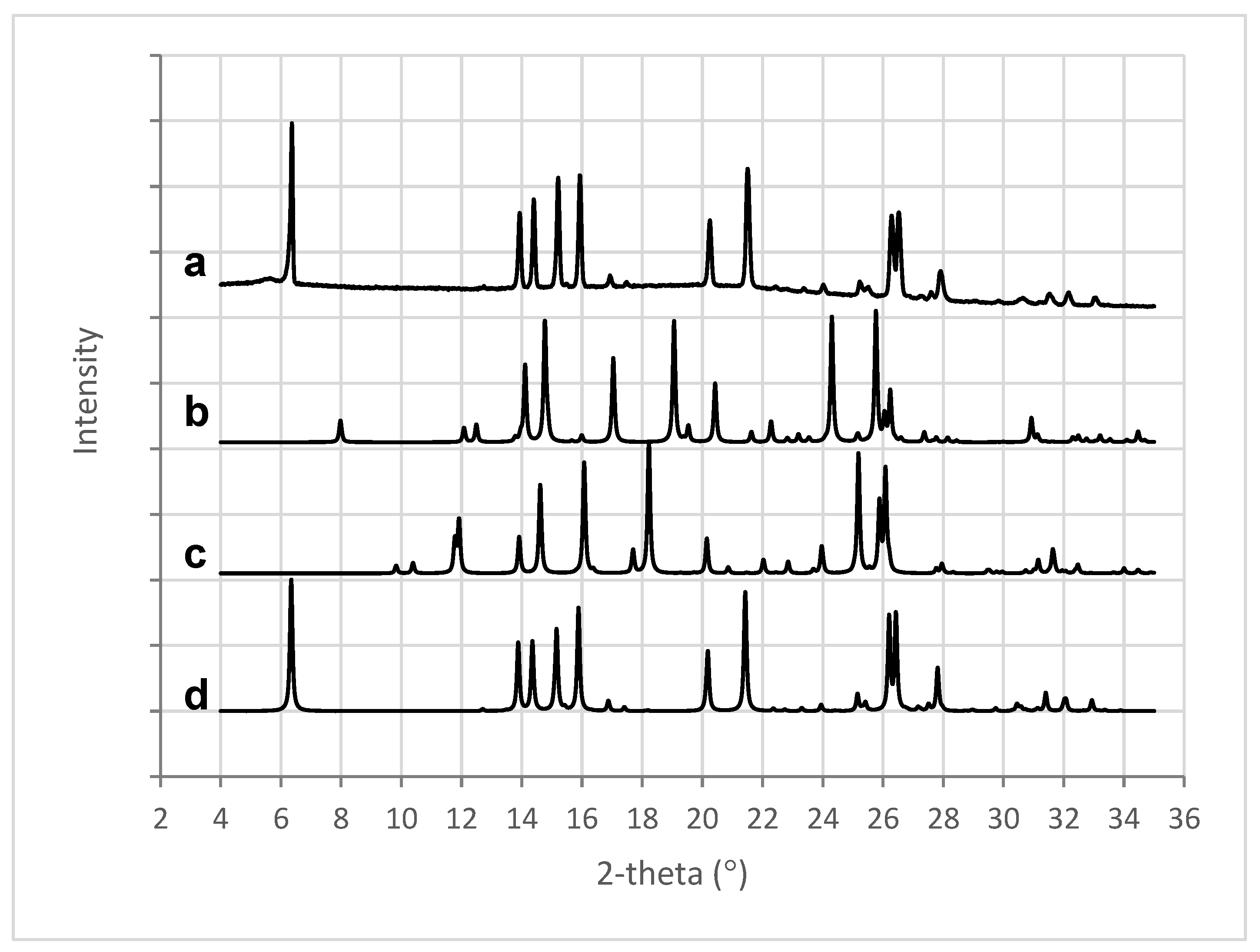
2.8. X-ray Powder Diffraction Analysis (XRPD)
2.9. Dissolution
2.10. Mathematical Description—Weibull Model
3. Results
3.1. 3D Printing of Dose Forms
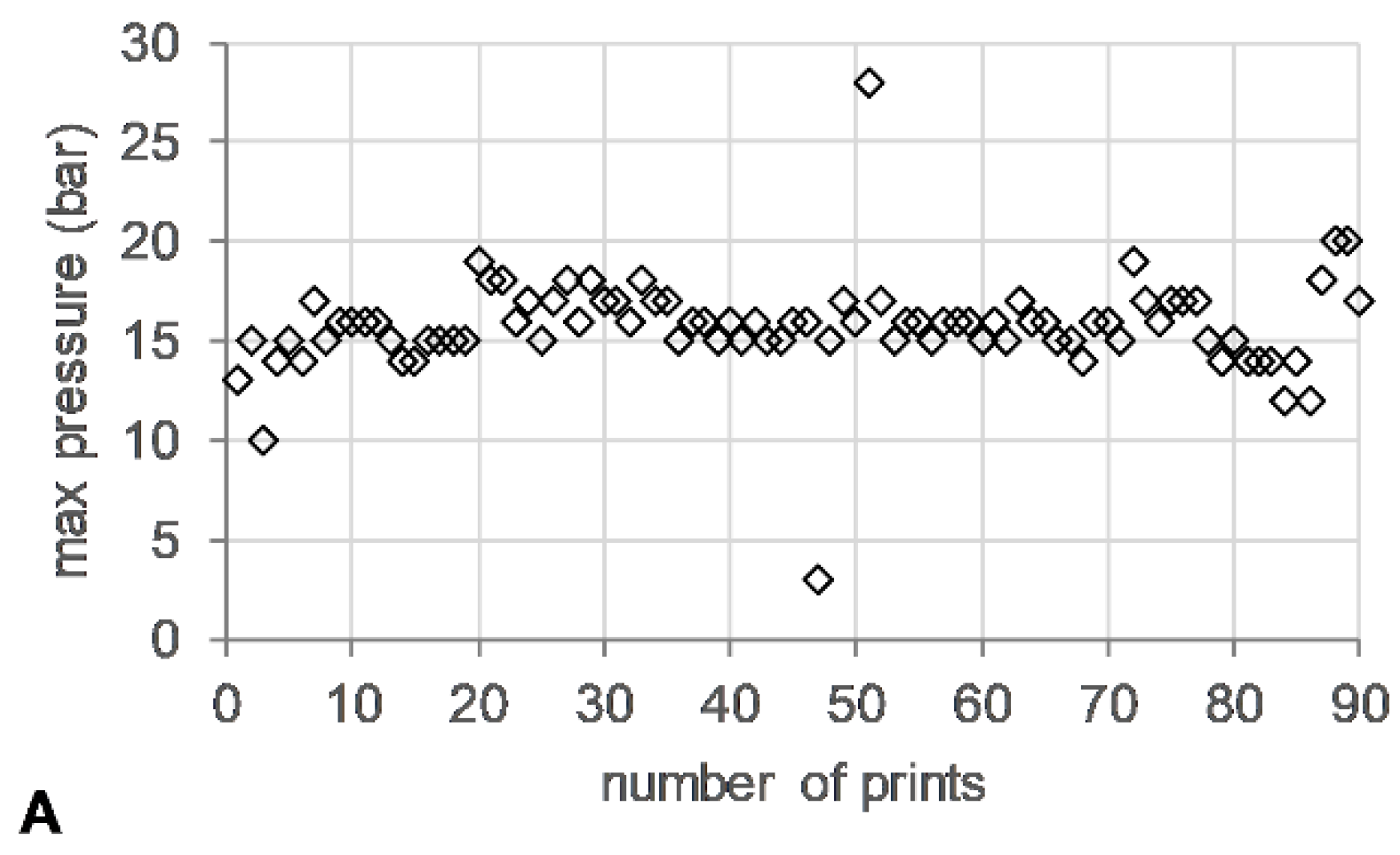
3.2. HME Process Parameters
3.3. HPLC Content
3.4. Rheology
3.5. FTIR
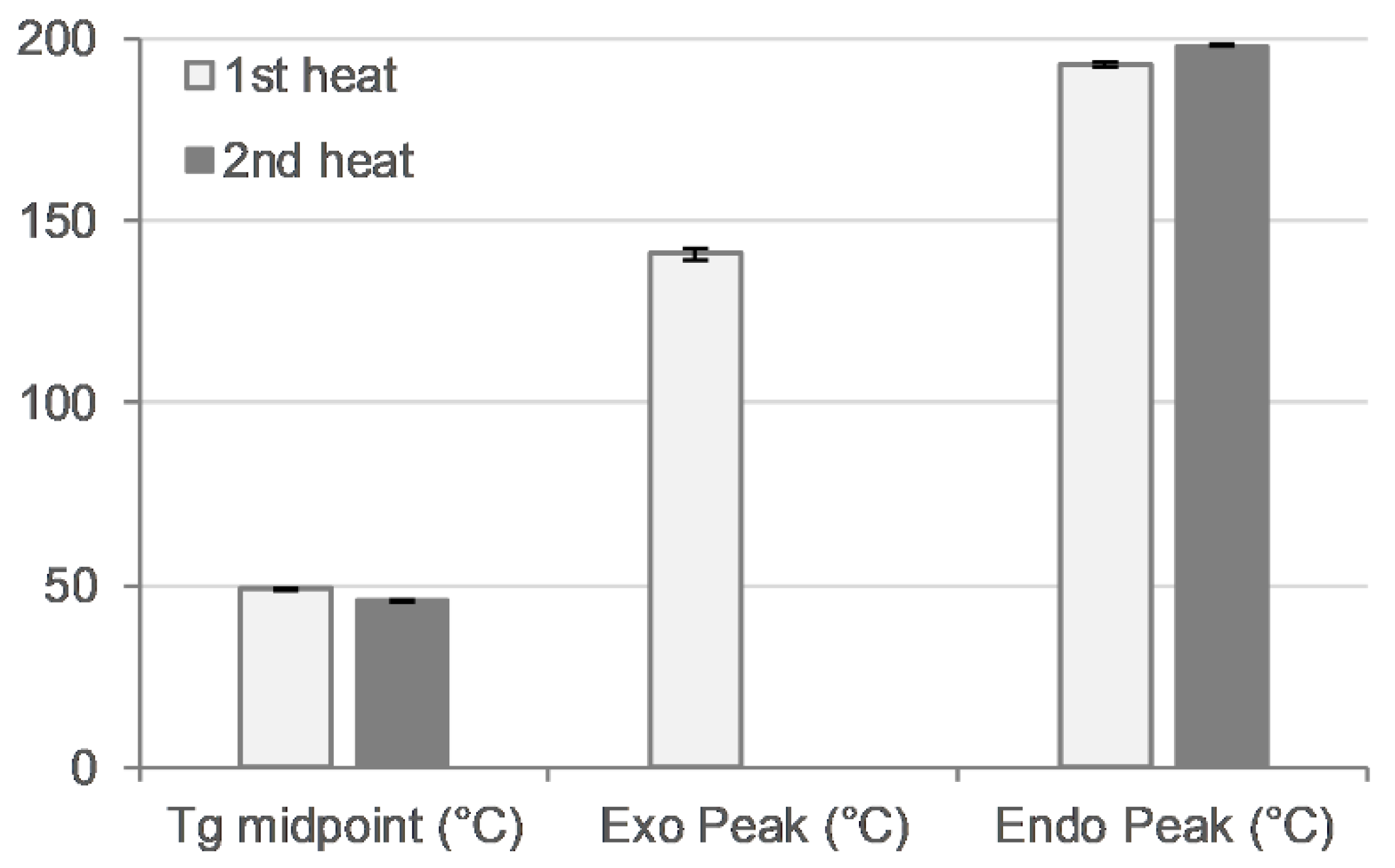
3.6. DSC
3.7. X-ray Powder Diffraction Analysis: XRPD
3.8. Dissolution USP II
3.9. Mathematical Description of Dissolution Data—Weibull Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Glossary
| EPSRC | Engineering and Physical Sciences Research Council (EPSRC) |
| CMAC | Center for Continuous Manufacturing and Advanced Crystallisation at the University of Strathclyde |
| HPLC | High-Performance Liquid Chromatography |
| Ph Eur | European Pharmacopoeia |
| USP | United States Pharmacopeia |
References
- Abaci, A.; Gedeon, C.; Kuna, A.; Guvendiren, M. Additive Manufacturing of Oral Tablets: Technologies, Materials and Printed Tablets. Pharmaceutics 2021, 13, 156. [Google Scholar] [CrossRef] [PubMed]
- Prasad, E.; Robertson, J.; Halbert, G.W. Solid dispersions: Improving drug performance through tablet micro structure design. In Proceedings of the American Association of Pharmaceutical Scientists (AAPS) 2021 PharmSci 360, Virtual and Philadelphia, PA, USA, 17–20 October 2021. [Google Scholar]
- Prasad, E.; Robertson, J.; Halbert, G.W. Opening up the pharmaceutical formulation space for Additive Manufacturing. In Proceedings of the EUPAT 10 (2021) Pan-European Science Conference on QbD & PAT, Virtual, 4–6 October 2021. [Google Scholar]
- Patel, S.K.; Khoder, M.; Peak, M.; Alhnan, M.A. Controlling drug release with additive manufacturing-based solutions. Adv. Drug Deliv. Rev. 2021, 174, 369–386. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Deng, F.; Wang, B.; Wu, Y.; Luo, Q.; Zuo, X.; Liu, X.; Cao, L.; Li, M.; Lu, H.; et al. Melt extrusion deposition (MED™) 3D printing technology—A paradigm shift in design and development of modified release drug products. Int. J. Pharm. 2021, 602, 120639. [Google Scholar] [CrossRef] [PubMed]
- Prasad, E.; Robertson, J.; Florence, A.J.; Halbert, G.W. Expanding the pharmaceutical formulation space in material extrusion 3D printing applications. Addit. Manuf. 2023, 77, 103803. [Google Scholar] [CrossRef]
- Prasad, E.; Robertson, J.; Halbert, G.W. Mefenamic acid solid dispersions—Impact of formulation composition on processing parameters, product properties and performance. Int. J. Pharm. 2022, 616, 121505. [Google Scholar] [CrossRef]
- Zhang, J.; Feng, X.; Patil, H.; Tiwari, R.V.; Repka, M.A. Coupling 3D printing with hot-melt extrusion to produce controlled-release tablets. Int. J. Pharm. 2017, 519, 186–197. [Google Scholar] [CrossRef]
- Prasad, E.; Robertson, J.; Halbert, G.W. Quality control test for pharmaceutical feedstock material for FDM 3D printers. In Proceedings of the Virtual PharmSci: APS International Conference, Virtual, 7–9 September 2021. [Google Scholar]
- Yang, Y.; Wang, H.; Xu, X.; Yang, G. Strategies and mechanisms to improve the printability of pharmaceutical polymers Eudragit® EPO and Soluplus®. Int. J. Pharm. 2021, 599, 120410. [Google Scholar] [CrossRef]
- Palekar, S.; Nukala, P.K.; Mishra, S.M.; Kipping, T.; Patel, K. Application of 3D printing technology and quality by design approach for development of age-appropriate pediatric formulation of baclofen. Int. J. Pharm. 2019, 556, 106–116. [Google Scholar] [CrossRef]
- Govender, R.; Kissi, E.O.; Larsson, A.; Tho, I. Polymers in pharmaceutical additive manufacturing: A balancing act between printability and product performance. Adv. Drug Deliv. Rev. 2021, 177, 113923. [Google Scholar] [CrossRef]
- Prasad, E.; Islam, M.T.; Goodwin, D.J.; Megarry, A.J.; Halbert, G.W.; Florence, A.J.; Robertson, J. Development of a hot-melt extrusion (HME) process to produce drug loaded Affinisol™ 15LV filaments for fused filament fabrication (FFF) 3D printing. Addit. Manuf. 2019, 29, 100776. [Google Scholar] [CrossRef]
- Prasad, E.; Robertson, J.; Halbert, G.W. Printability of pharmaceutical polymers in FDM 3D printers. In Proceedings of the Virtual PharmSci : APS International Conference, Virtual, 7–9 September 2021. [Google Scholar]
- Ilyes, K.; Kovacs, N.K.; Balogh, A.; Borbas, E.; Farkas, B.; Casian, T.; Marosi, G.; Tomuta, I.; Nagy, Z.K. The applicability of pharmaceutical polymeric blends for the fused deposition modelling (FDM) 3D technique: Material considerations-printability-process modulation, with consecutive effects on in vitro release, stability and degradation. Eur. J. Pharm. Sci. 2019, 129, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Genina, N.; Hollander, J.; Jukarainen, H.; Makila, E.; Salonen, J.; Sandler, N. Ethylene vinyl acetate (EVA) as a new drug carrier for 3D printed medical drug delivery devices. Eur. J. Pharm. Sci. 2016, 90, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Venkataraman, N.; Rangarajan, S.; Matthewson, M.J.; Harper, B.; Safari, A.; Danforth, S.C.; Wu, G.; Langrana, N.; Guceri, S.; Yardimci, A. Feedstock material property—process relationships in fused deposition of ceramics (FDC). Rapid Prototyp. J. 2000, 6, 244–253. [Google Scholar] [CrossRef]
- Prasad, E.; Robertson, J.; Halbert, G.W. Improving consistency for a Mefenamic acid immediate release formulation. J. Pharm. Sci. 2020, 109, 3462–3470. [Google Scholar] [CrossRef]
- Patil, P.; Gupta, V.; Udupi, R.; Srikanth, K.; Prasad, B. Development of dissolution medium for poorly water soluble drug mefenamic acid. Res. J. Pharm. Biol. Chem. Sci. 2010, 1, 546–548. [Google Scholar]
- Butler, J.M.; Dressman, J.B. The Developability Classification System: Application of Biopharmaceutics Concepts to Formulation Development. J. Pharm. Sci. 2010, 99, 4940–4954. [Google Scholar] [CrossRef]
- Abdul Mudalip, S.K.; Abu Bakar, M.R.; Jamal, P.; Adam, F. Solubility and Dissolution Thermodynamic Data of Mefenamic Acid Crystals in Different Classes of Organic Solvents. J. Chem. Eng. Data 2013, 58, 3447–3452. [Google Scholar] [CrossRef]
- Ullah, I.; Baloch, M.K.; Ullah, I.; Mustaqeem, M. Enhancement in Aqueous Solubility of Mefenamic Acid using Micellar Solutions of Various Surfactants. J. Solut. Chem. 2014, 43, 1360–1373. [Google Scholar] [CrossRef]
- Derle, D.V.; Bele, M.; Kasliwal, N. In Vitro and In Vivo Evaluation of Mefenamic acid and its complexes with beta-Cyclodextrin and HP-beta-Cyclodextrin. Asian J. Pharm. 2008, 2, 30–34. [Google Scholar] [CrossRef]
- Gursoy, R.N.; Benita, S. Self-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophilic drugs. Biomed. Pharmacother. 2004, 58, 173–182. [Google Scholar] [CrossRef]
- Sriamornsak, P.; Limmatvapirat, S.; Piriyaprasarth, S.; Mansukmanee, P.; Huang, Z. A new self-emulsifying formulation of mefenamic acid with enhanced drug dissolution. Asian J. Pharm. Sci. 2015, 10, 121–127. [Google Scholar] [CrossRef]
- Kumar, M.; Singh, D.; Bedi, N. Mefenamic acid-loaded solid SMEDDS: An innovative aspect for dose reduction and improved pharmacodynamic profile. Ther. Deliv. 2019, 10, 21–36. [Google Scholar] [CrossRef]
- Alshehri, S.M.; Park, J.B.; Alsulays, B.B.; Tiwari, R.V.; Almutairy, B.; Alshetaili, A.S.; Morott, J.; Shah, S.; Kulkarni, V.; Majumdar, S.; et al. Mefenamic acid taste-masked oral disintegrating tablets with enhanced solubility via molecular interaction produced by hot melt extrusion technology. J. Drug Deliv. Sci. Technol. 2015, 27, 18–27. [Google Scholar] [CrossRef]
- Rao, K.R.; Nagabhushanam, M.V.; Chowdary, K.P. In vitro Dissolution Studies on Solid Dispersions of Mefenamic Acid. Indian. J. Pharm. Sci. 2011, 73, 243–247. [Google Scholar] [CrossRef]
- Darwich, M. Solubility/Bioavailability Enhancement and Modified Release Formulations of Poorly Water-Soluble Drugs. Ph.D. Thesis, Freie Universität Berlin, Berlin, Germany, 2015. [Google Scholar]
- Andrews, G.P.; Zhai, H.; Tipping, S.; Jones, D.S. Characterisation of the thermal, spectroscopic and drug dissolution properties of mefenamic acid and polyoxyethylene-polyoxypropylene solid dispersions. J. Pharm. Sci. 2009, 98, 4545–4556. [Google Scholar] [CrossRef] [PubMed]
- Tambosi, G.; Coelho, P.F.; Luciano, S.; Lenschow, I.C.S.; Zétola, M.; Stulzer, H.K.; Pezzini, B.R. Challenges to improve the biopharmaceutical properties of poorly water-soluble drugs and the application of the solid dispersion technology. Matéria (Rio J.) 2018, 23. [Google Scholar] [CrossRef]
- Vasconcelos, T.; Sarmento, B.; Costa, P. Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Drug Discov. Today 2007, 12, 1068–1075. [Google Scholar] [CrossRef] [PubMed]
- Mahlin, D.; Bergström, C.A.S. Early drug development predictions of glass-forming ability and physical stability of drugs. Eur. J. Pharm. Sci. 2013, 49, 323–332. [Google Scholar] [CrossRef]
- Surov, A.O.; Terekhova, I.V.; Bauer-Brandl, A.; Perlovich, G.L. Thermodynamic and Structural Aspects of Some Fenamate Molecular Crystals. Cryst. Growth Des. 2009, 9, 3265–3272. [Google Scholar] [CrossRef]
- Elbadawi, M.; Gustaffson, T.; Gaisford, S.; Basit, A.W. 3D printing tablets: Predicting printability and drug dissolution from rheological data. Int. J. Pharm. 2020, 590, 119868. [Google Scholar] [CrossRef]
- Arafat, B.; Wojsz, M.; Isreb, A.; Forbes, R.T.; Isreb, M.; Ahmed, W.; Arafat, T.; Alhnan, M.A. Tablet fragmentation without a disintegrant: A novel design approach for accelerating disintegration and drug release from 3D printed cellulosic tablets. Eur. J. Pharm. Sci. 2018, 118, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Shi, K.; Salvage, J.P.; Maniruzzaman, M.; Nokhodchi, A. Role of release modifiers to modulate drug release from fused deposition modelling (FDM) 3D printed tablets. Int. J. Pharm. 2021, 597, 120315. [Google Scholar] [CrossRef]
- Nasereddin, J.M.; Wellner, N.; Alhijjaj, M.; Belton, P.; Qi, S. Development of a Simple Mechanical Screening Method for Predicting the Feedability of a Pharmaceutical FDM 3D Printing Filament. Pharm. Res. 2018, 35, 151. [Google Scholar] [CrossRef] [PubMed]
- Korte, C.; Quodbach, J. Formulation development and process analysis of drug-loaded filaments manufactured via hot-melt extrusion for 3D-printing of medicines. Pharm. Dev. Technol. 2018, 23, 1117–1127. [Google Scholar] [CrossRef]
- Skorski, M.R.; Esenther, J.M.; Ahmed, Z.; Miller, A.E.; Hartings, M.R. The chemical, mechanical, and physical properties of 3D printed materials composed of TiO2-ABS nanocomposites. Sci. Technol. Adv. Mater. 2016, 17, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Gottschalk, N.; Bogdahn, M.; Harms, M.; Quodbach, J. Brittle polymers in Fused Deposition Modeling: An improved feeding approach to enable the printing of highly drug loaded filament. Int. J. Pharm. 2021, 597, 120216. [Google Scholar] [CrossRef] [PubMed]
- Tabriz, A.G.; Scoutaris, N.; Gong, Y.; Hui, H.-W.; Kumar, S.; Douroumis, D. Investigation on hot melt extrusion and prediction on 3D printability of pharmaceutical grade polymers. Int. J. Pharm. 2021, 604, 120755. [Google Scholar] [CrossRef]
- Okwuosa, T.C.; Stefaniak, D.; Arafat, B.; Isreb, A.; Wan, K.W.; Alhnan, M.A. A Lower Temperature FDM 3D Printing for the Manufacture of Patient-Specific Immediate Release Tablets. Pharm. Res. 2016, 33, 2704–2712. [Google Scholar] [CrossRef]
- Alhijjaj, M.; Belton, P.; Qi, S. An investigation into the use of polymer blends to improve the printability of and regulate drug release from pharmaceutical solid dispersions prepared via fused deposition modeling (FDM) 3D printing. Eur. J. Pharm. Biopharm. 2016, 108, 111–125. [Google Scholar] [CrossRef]
- Sadia, M.; Sosnicka, A.; Arafat, B.; Isreb, A.; Ahmed, W.; Kelarakis, A.; Alhnan, M.A. Adaptation of pharmaceutical excipients to FDM 3D printing for the fabrication of patient-tailored immediate release tablets. Int. J. Pharm. 2016, 513, 659–668. [Google Scholar] [CrossRef]
- Fanous, M.; Gold, S.; Muller, S.; Hirsch, S.; Ogorka, J.; Imanidis, G. Simplification of fused deposition modeling 3D-printing paradigm: Feasibility of 1-step direct powder printing for immediate release dosage form production. Int. J. Pharm. 2020, 578, 119124. [Google Scholar] [CrossRef] [PubMed]
- FabRx. Pharmaceutical 3D Printing for Personalised Medicine. Available online: https://www.fabrx.co.uk/home (accessed on 20 August 2024).
- Goyanes, A.; Allahham, N.; Trenfield, S.J.; Stoyanov, E.; Gaisford, S.; Basit, A.W. Direct powder extrusion 3D printing: Fabrication of drug products using a novel single-step process. Int. J. Pharm. 2019, 567, 118471. [Google Scholar] [CrossRef] [PubMed]
- Pistone, M.; Racaniello, G.F.; Arduino, I.; Laquintana, V.; Lopalco, A.; Cutrignelli, A.; Rizzi, R.; Franco, M.; Lopedota, A.; Denora, N. Direct cyclodextrin-based powder extrusion 3D printing for one-step production of the BCS class II model drug niclosamide. Drug Deliv. Transl. Res. 2022, 12, 1895–1910. [Google Scholar] [CrossRef]
- Costa, P.; Sousa Lobo, J.M. Modeling and comparison of dissolution profiles. Eur. J. Pharm. Sci. 2001, 13, 123–133. [Google Scholar] [CrossRef]
- Windolf, H.; Chamberlain, R.; Quodbach, J. Predicting Drug Release from 3D Printed Oral Medicines Based on the Surface Area to Volume Ratio of Tablet Geometry. Pharmaceutics 2021, 13, 379. [Google Scholar] [CrossRef] [PubMed]
- Bruschi, M.L. (Ed.) 5—Mathematical models of drug release. In Strategies to Modify the Drug Release from Pharmaceutical Systems; Woodhead Publishing: Sawston, UK, 2015; pp. 63–86. [Google Scholar]
- Dokoumetzidis, A.; Papadopoulou, V.; Macheras, P. Analysis of Dissolution Data Using Modified Versions of Noyes–Whitney Equation and the Weibull Function. Pharm. Res. 2006, 23, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Kolter, K.; Karl, M.; Gryczke, A. Hot-Melt Extrusion with BASF Pharma Polymers, 2nd Revised and Enlarged Edition ed.; BASF The Chemical Company: Ludwigshafen, Germany, 2012. [Google Scholar]
- Abbas, N.; Oswald, I.D.H.; Pulham, C.R. Accessing Mefenamic Acid Form II through High-Pressure Recrystallisation. Pharmaceutics 2017, 9, 16. [Google Scholar] [CrossRef]
- SeethaLekshmi, S.; Guru Row, T.N. Conformational Polymorphism in a Non-steroidal Anti-inflammatory Drug, Mefenamic Acid. Cryst. Growth Des. 2012, 12, 4283–4289. [Google Scholar] [CrossRef]
- Lee, E.H.; Byrn, S.R.; Carvajal, M.T. Additive-Induced Metastable Single Crystal of Mefenamic Acid. Pharm. Res. 2006, 23, 2375–2380. [Google Scholar] [CrossRef]
- McConnell, J.F.; Company, F.Z. N-(2,3-xylyl) anthranilic acid, C15H15NO2. Mefenamic acid. J. Cryst. Struct. Commun. 1976, 5, 861–864. [Google Scholar]
- Dissolution Testing of Immediate Release Solid Oral Dosage Forms; Food and Drug Administration: Rockville, MD, USA, 1997.
- Isreb, A.; Baj, K.; Wojsz, M.; Isreb, M.; Peak, M.; Alhnan, M.A. 3D printed oral theophylline doses with innovative ‘radiator-like’ design: Impact of polyethylene oxide (PEO) molecular weight. Int. J. Pharm. 2019, 564, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Qahtani, M.; Wu, F.; Misra, M.; Gregori, S.; Mielewski, D.F.; Mohanty, A.K. Experimental Design of Sustainable 3D printed Poly(Lactic Acid)/Biobased Poly(Butylene Succinate) Blends via Fused Deposition Modeling. ACS Sustain. Chem. Eng. 2019, 7, 14460–14470. [Google Scholar] [CrossRef]
- Ali, S.; Kolter, K.; Karl, M. Evaluation of Different Polymers in 3D Printing Technologies. Am. Pharm. Rev. 2019. [Google Scholar]
- Samaro, A.; Janssens, P.; Vanhoorne, V.; Van Renterghem, J.; Eeckhout, M.; Cardon, L.; De Beer, T.; Vervaet, C. Screening of pharmaceutical polymers for extrusion-Based Additive Manufacturing of patient-tailored tablets. Int. J. Pharm. 2020, 586, 119591. [Google Scholar] [CrossRef]
- Azad, M.A.; Olawuni, D.; Kimbell, G.; Badruddoza, A.Z.M.; Hossain, M.S.; Sultana, T. Polymers for Extrusion-Based 3D Printing of Pharmaceuticals: A Holistic Materials–Process Perspective. Pharmaceutics 2020, 12, 124. [Google Scholar] [CrossRef]
- Smith, A.; Bordos, E.; Robertson, J.; Florence, A.J. The application of SIFT-MS for examining the degradation of polymers. In Proceedings of the CMAC Summer School, Dunblane, UK, 14–16 June 2023. [Google Scholar]
- Prasad, E.; Bordos, E.; Langford, V.; Perkins, M.; Robertson, J.; Halbert, G. Linking 3DP manufacturing with SIFT-MS Volatiles Organic Compound Analysis. In Proceedings of the AAPS 2023 PHARMSCI 360, Orlando, FL, USA, 22–25 October 2023. [Google Scholar]
- Vivattanaseth, P.; Chong, M.; Prasad, E.; Halbert, G.; Robertson, J.; McFarlan, C.; Nordon, A. Application of multivariate curve resolution to in situ THz—Raman spectroscopy of amorphous solid dispersions in pharmaceutical products. In Proceedings of the CMAC Annual Open Day 2022, Glasgow, UK, 16 May 2022; p. 42. [Google Scholar]
- Solanki, N.; Gupta, S.S.; Serajuddin, A.T.M. Rheological analysis of itraconazole-polymer mixtures to determine optimal melt extrusion temperature for development of amorphous solid dispersion. Eur. J. Pharm. Sci. 2018, 111, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Hornsby, P.R. The application of magnesium hydroxide as a fire retardant and smoke-suppressing additive for polymers. Fire Mater. 1994, 18, 269–276. [Google Scholar] [CrossRef]
- Ekblad, N. Melt Processability of Amorphous Solid Dispersions during Hot-Melt Extrusion. Screening Using Vacuum Compression Moulding and Evaluation by Rheology and Solid-State Analysis. Master’s Thesis, UiT The Arctic University of Norway, Tromso, Norway, 2018. [Google Scholar]
- PubChem. Compound Summary Mefenamic Acid. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Mefenamic-Acid (accessed on 27 November 2023).
- Giri, B.R.; Kwon, J.; Vo, A.Q.; Bhagurkar, A.M.; Bandari, S.; Kim, D.W. Hot-Melt Extruded Amorphous Solid Dispersion for Solubility, Stability, and Bioavailability Enhancement of Telmisartan. Pharmaceuticals 2021, 14, 73. [Google Scholar] [CrossRef]
- Stiani, S.N.; Rusdiana, T.; Subarnas, A. Improving Solubility and Dissolution of a Natural Product Apigenin via Preparation of Solid Dispersion by Hot Melt Extrusion. Int. J. Appl. Pharm. 2021, 13, 47–52. [Google Scholar] [CrossRef]
- Song, B.; Wang, J.; Lu, S.-J.; Shan, L.-N. Andrographolide solid dispersions formulated by Soluplus to enhance interface wetting, dissolution, and absorption. J. Appl. Polym. Sci. 2020, 137, 48354. [Google Scholar] [CrossRef]
- Ma, X.; Müller, F.; Huang, S.; Lowinger, M.; Liu, X.; Schooler, R.; Williams Iii, R.O. Influence of Carbamazepine Dihydrate on the Preparation of Amorphous Solid Dispersions by Hot Melt Extrusion. Pharmaceutics 2020, 12, 379. [Google Scholar] [CrossRef] [PubMed]
- Restrepo-Uribe, L.; Ioannidis, N.; Noriega Escobar, M.d.P. Influence of screw configuration and processing parameters on the dissolution of ketoprofen in polymer blends. J. Appl. Polym. Sci. 2020, 137, 49407. [Google Scholar] [CrossRef]
- Guntaka, P.R.; Lankalapalli, S. Solubility and Dissolution Enhancement of Ivacaftor Tablets by Using Solid Dispersion Technique of Hot-Melt Extrusion—A Design of Experimental Approach. Asian J. Pharm. Clin. Res. 2019, 12, 356–363. [Google Scholar] [CrossRef]
- Mateos, H.; Gentile, L.; Murgia, S.; Colafemmina, G.; Collu, M.; Smets, J.; Palazzo, G. Understanding the self-assembly of the polymeric drug solubilizer Soluplus®. J. Colloid Interface Sci. 2022, 611, 224–234. [Google Scholar] [CrossRef]
- Shamma, R.N.; Basha, M. Soluplus®: A novel polymeric solubilizer for optimization of Carvedilol solid dispersions: Formulation design and effect of method of preparation. Powder Technol. 2013, 237, 406–414. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, L.; Ma, D.; Tang, X.; Zhang, Y.; Yin, T.; Gou, J.; Wang, Y.; He, H. Characterizing and Exploring the Differences in Dissolution and Stability between Crystalline Solid Dispersion and Amorphous Solid Dispersion. AAPS PharmSciTech 2020, 21, 262. [Google Scholar] [CrossRef]
- Cellet, T.S.P.; Pereira, G.M.; Muniz, E.C.; Silva, R.; Rubira, A.F. Hydroxyapatite nanowhiskers embedded in chondroitin sulfate microspheres as colon targeted drug delivery systems. J. Mater. Chem. B 2015, 3, 6837–6846. [Google Scholar] [CrossRef]
- Goyanes, A.; Robles Martinez, P.; Buanz, A.; Basit, A.W.; Gaisford, S. Effect of geometry on drug release from 3D printed tablets. Int. J. Pharm. 2015, 494, 657–663. [Google Scholar] [CrossRef]
- Welsh, N.R.; Malcolm, R.K.; Devlin, B.; Boyd, P. Dapivirine-releasing vaginal rings produced by plastic freeforming additive manufacturing. Int. J. Pharm. 2019, 572, 118725. [Google Scholar] [CrossRef]
- Kyobula, M.; Adedeji, A.; Alexander, M.R.; Saleh, E.; Wildman, R.; Ashcroft, I.; Gellert, P.R.; Roberts, C.J. 3D inkjet printing of tablets exploiting bespoke complex geometries for controlled and tuneable drug release. J. Control. Release 2017, 261, 207–215. [Google Scholar] [CrossRef]
- Ebrahimi, F.; Xu, H.; Fuenmayor, E.; Major, I. A comparison of droplet deposition modelling, fused filament fabrication, and injection moulding for the production of oral dosage forms containing hydrochlorothiazide. Int. J. Pharm. 2023, 645, 123400. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Nasereddin, J.; McDonagh, T.; von Zeppelin, D.; Gleadall, A.; Alqahtani, F.; Bibb, R.; Belton, P.; Qi, S. Effects of porosity on drug release kinetics of swellable and erodible porous pharmaceutical solid dosage forms fabricated by hot melt droplet deposition 3D printing. Int. J. Pharm. 2021, 604, 120626. [Google Scholar] [CrossRef] [PubMed]
- Lapidus, H.; Lordi, N.G. Some Factors Affecting the Release of a Water-Soluble Drug from a Compressed Hydrophilic Matrix. J. Pharm. Sci. 1966, 55, 840–843. [Google Scholar] [CrossRef] [PubMed]
- Grijseels, H.; de Blaey, C.J. Dissolution at porous interfaces. Int. J. Pharm. 1981, 9, 337–347. [Google Scholar] [CrossRef]
- el-Arini, S.K.; Leuenberger, H. Dissolution properties of praziquantel—PVP systems. Pharm. Acta Helv. 1998, 73, 89–94. [Google Scholar] [CrossRef]
- Uboldi, M.; Chiappa, A.; Pertile, M.; Piazza, A.; Tagliabue, S.; Foppoli, A.; Palugan, L.; Gazzaniga, A.; Zema, L.; Melocchi, A. Investigation on the use of fused deposition modeling for the production of IR dosage forms containing Timapiprant. Int. J. Pharm. X 2023, 5, 100152. [Google Scholar] [CrossRef]
- Henry, S. Evaluation of Fused Filament Fabrication for Oral Solid Dosage Form Production; Ghent University, Faculty of Pharmaceutical Sciences: Ghent, Belgium, 2023. [Google Scholar]
- Nandi, U.; Ajiboye, A.L.; Patel, P.; Douroumis, D.; Trivedi, V. Preparation of Solid Dispersions of Simvastatin and Soluplus Using a Single-Step Organic Solvent-Free Supercritical Fluid Process for the Drug Solubility and Dissolution Rate Enhancement. Pharmaceuticals 2021, 14, 846. [Google Scholar] [CrossRef]
- Milovanovic, S.; Djuris, J.; Dapčević, A.; Medarevic, D.; Ibric, S.; Zizovic, I. Soluplus®, Eudragit®, HPMC-AS foams and solid dispersions for enhancement of Carvedilol dissolution rate prepared by a supercritical CO2 process. Polym. Test. 2019, 76, 54–64. [Google Scholar] [CrossRef]
- Altamimi, M.A.; Neau, S.H. Investigation of the in vitro performance difference of drug-Soluplus® and drug-PEG 6000 dispersions when prepared using spray drying or lyophilization. Saudi Pharm. J. 2017, 25, 419–439. [Google Scholar] [CrossRef]
- Feuerbach, T.; Kock, S.; Thommes, M. Characterisation of fused deposition modeling 3D printers for pharmaceutical and medical applications. Pharm. Dev. Technol. 2018, 23, 1136–1145. [Google Scholar] [CrossRef]
- Shrivastava, A. 5—Plastics Processing. In Introduction to Plastics Engineering; Shrivastava, A., Ed.; William Andrew Publishing: Norwich, NY, USA, 2018; pp. 143–177. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tablet Structure | Infill % | SA/Pore (mm2) | Total SA Pores (mm2) | SA Shell (mm2) | Total SA (mm2) | SA/V (mm−1) | Number of Large Pores |
|---|---|---|---|---|---|---|---|
| A | 47.3 | 17.0 | 2652.0 | 370.1 | 3022.1 | 3.3 | 312 |
| B | 40.6 | 19.8 | 2732.4 | 464.6 | 3197.0 | 3.5 | 138 |
| C | 35.0 | 22.8 | 0 | 559.0 | 559.0 | 0.6 | 0 |
| Microstructure Infill % | A 47.3% | B 40.6% | C 35.0% |
|---|---|---|---|
| Weight variation (% RSD) | 0.9 | 0.6 | 1.1 |
| Max % difference from average weight | 1.7 | −1.0 | −2.2 |
| Uniformity of mass | PASS | PASS | PASS |
| Length variation (% RSD) | 0.73 | 0.13 | 0.12 |
| Width variation (% RSD) | 0.13 | 0.29 | 0.45 |
| Height variation (% RSD) | 0.49 | 1.39 | 0.99 |
| Tablet Shape | f1 Difference Factor | f2 Similarity Factor |
|---|---|---|
| A | 28.6 | 41.9 |
| B | 43.4 | 39.1 |
| C | 23.6 | 52.0 |
| limits | 0–15 | 50–100 |
| Tablet A vs. B | Tablet A vs. C | Tablet B vs. C | |
|---|---|---|---|
| f1 (0–15) | 12.7 | 24.7 | 15.4 |
| f2 (50–100) | 55.0 | 41.1 | 49.6 |
| Formulation | Process Temperature | Complex Viscosity (Pa∙s) | Max. Print Speed (mm/s) | Min. Layer Height (mm) |
|---|---|---|---|---|
| 50MFA | 140 °C | 2 × 103 | 40 | 0.2 |
| 30% w/w Paracetamol, HPMC (Affinisol 15LV) | 145 °C | 6.1 × 104 | 20 | 0.4 |
| 165 °C | 1.9 × 104 | 20 | 0.3 |
| f1 Similarity Factor (0–15) | f2 Difference Factor (50–100) | |
|---|---|---|
| Tablet A vs. B | 12.7 | 55.0 |
| Tablet A vs. C | 24.7 | 41.1 |
| Tablet B vs. C | 15.4 | 49.6 |
| Extrudate vs. A | 21.6 | 48.4 |
| Extrudate vs. B | 7.6 | 66.0 |
| Extrudate vs. C | 9.4 | 60.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prasad, E.; Robertson, J.; Halbert, G.W. An Additive Manufacturing MicroFactory: Overcoming Brittle Material Failure and Improving Product Performance through Tablet Micro-Structure Control for an Immediate Release Dose Form. Polymers 2024, 16, 2566. https://doi.org/10.3390/polym16182566
Prasad E, Robertson J, Halbert GW. An Additive Manufacturing MicroFactory: Overcoming Brittle Material Failure and Improving Product Performance through Tablet Micro-Structure Control for an Immediate Release Dose Form. Polymers. 2024; 16(18):2566. https://doi.org/10.3390/polym16182566
Chicago/Turabian StylePrasad, Elke, John Robertson, and Gavin W. Halbert. 2024. "An Additive Manufacturing MicroFactory: Overcoming Brittle Material Failure and Improving Product Performance through Tablet Micro-Structure Control for an Immediate Release Dose Form" Polymers 16, no. 18: 2566. https://doi.org/10.3390/polym16182566
APA StylePrasad, E., Robertson, J., & Halbert, G. W. (2024). An Additive Manufacturing MicroFactory: Overcoming Brittle Material Failure and Improving Product Performance through Tablet Micro-Structure Control for an Immediate Release Dose Form. Polymers, 16(18), 2566. https://doi.org/10.3390/polym16182566