
DFT Calculation, Hirshfeld Analysis and X-Ray Crystal Structure of Some Synthesized N-alkylated(S-alkylated)-[1,2,4]triazolo[1,5-a]quinazolines

 ,
,  , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. General Information
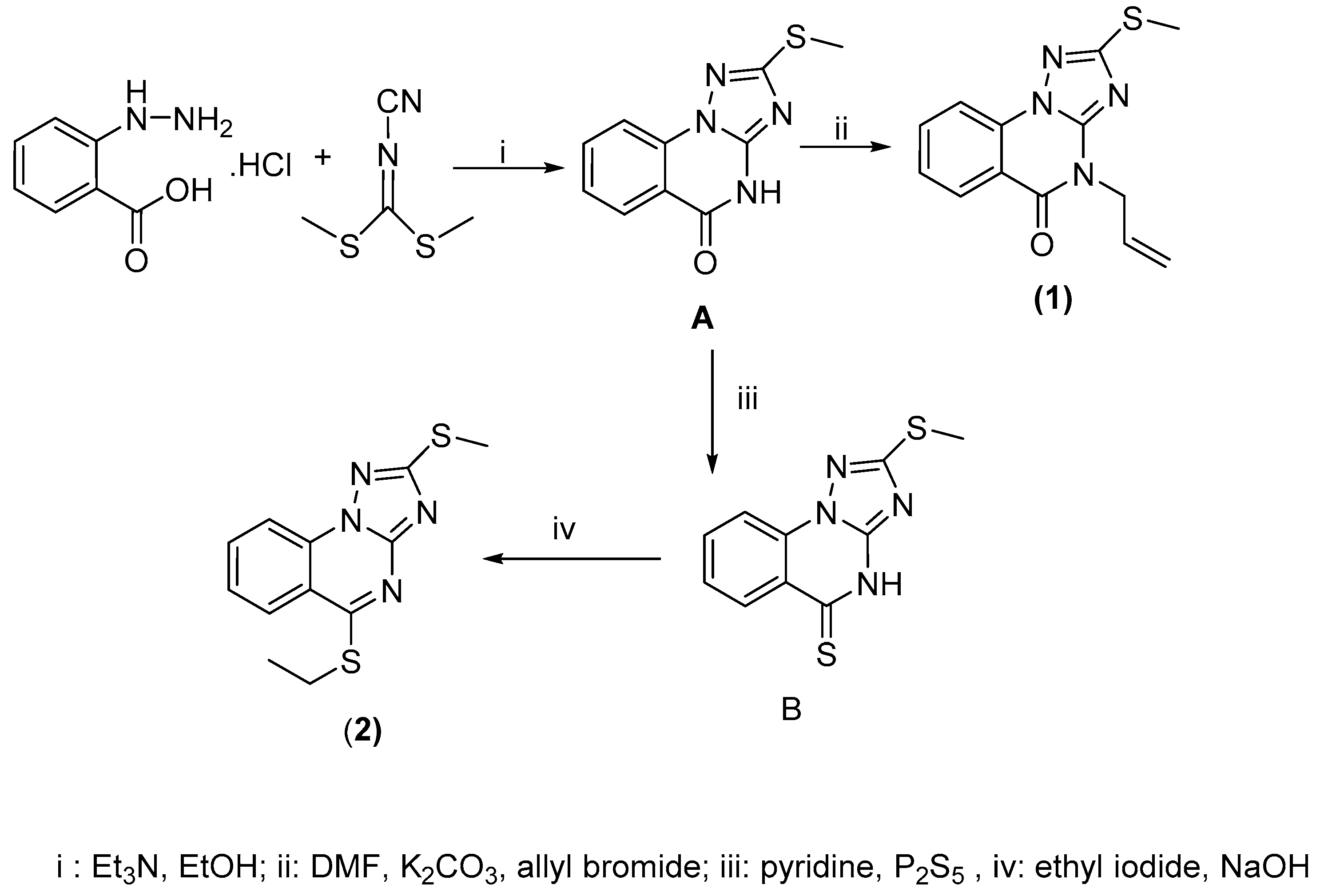
2.2. Synthesis of the Triazoloquinazolines 1 and 2
2.2.1. 4-Allyl-2-methylthio-[1,2,4]triazolo[1,5-a]quinazolin-5-one (1)
2.2.2. 5-Ethylthio-2-methylthio-[1,2,4]triazolo[1,5-a]quinazoline (2)
2.3. X-ray Crystallography Analysis
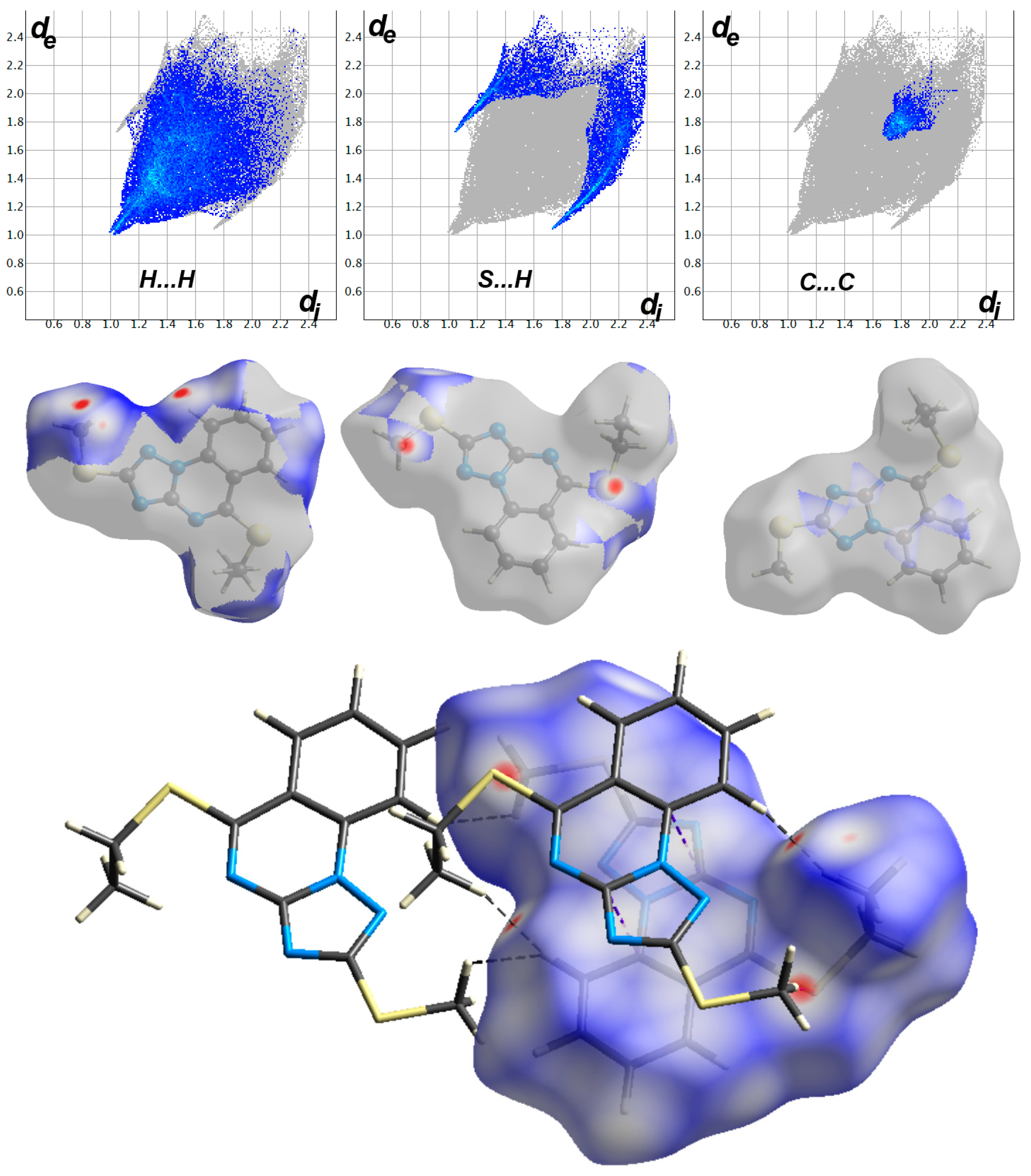
2.4. Hirshfeld Surface Analysis
Computational Methods
3. Results and Discussion
3.1. Chemistry
3.2. Spectroscopic Data
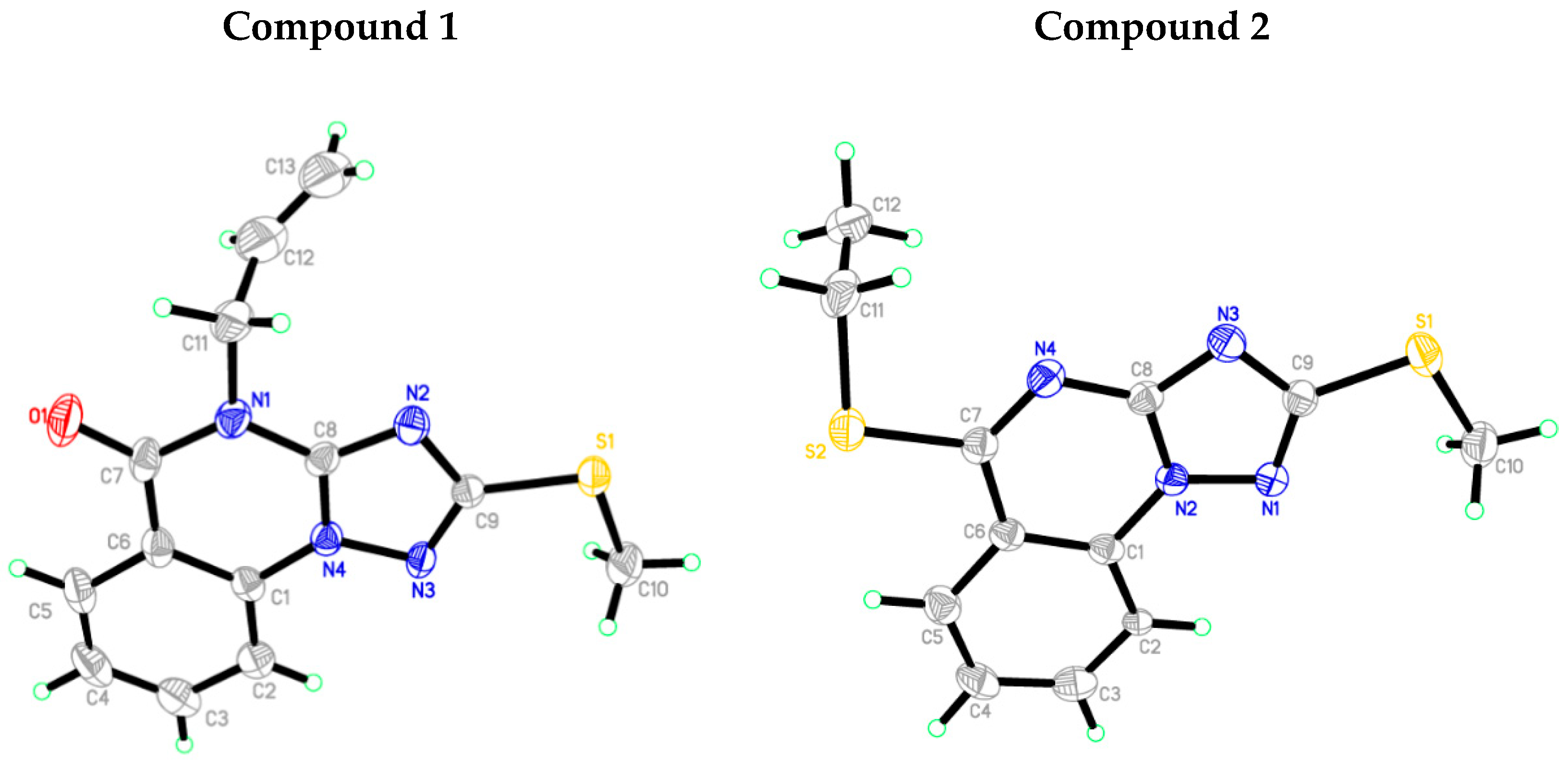
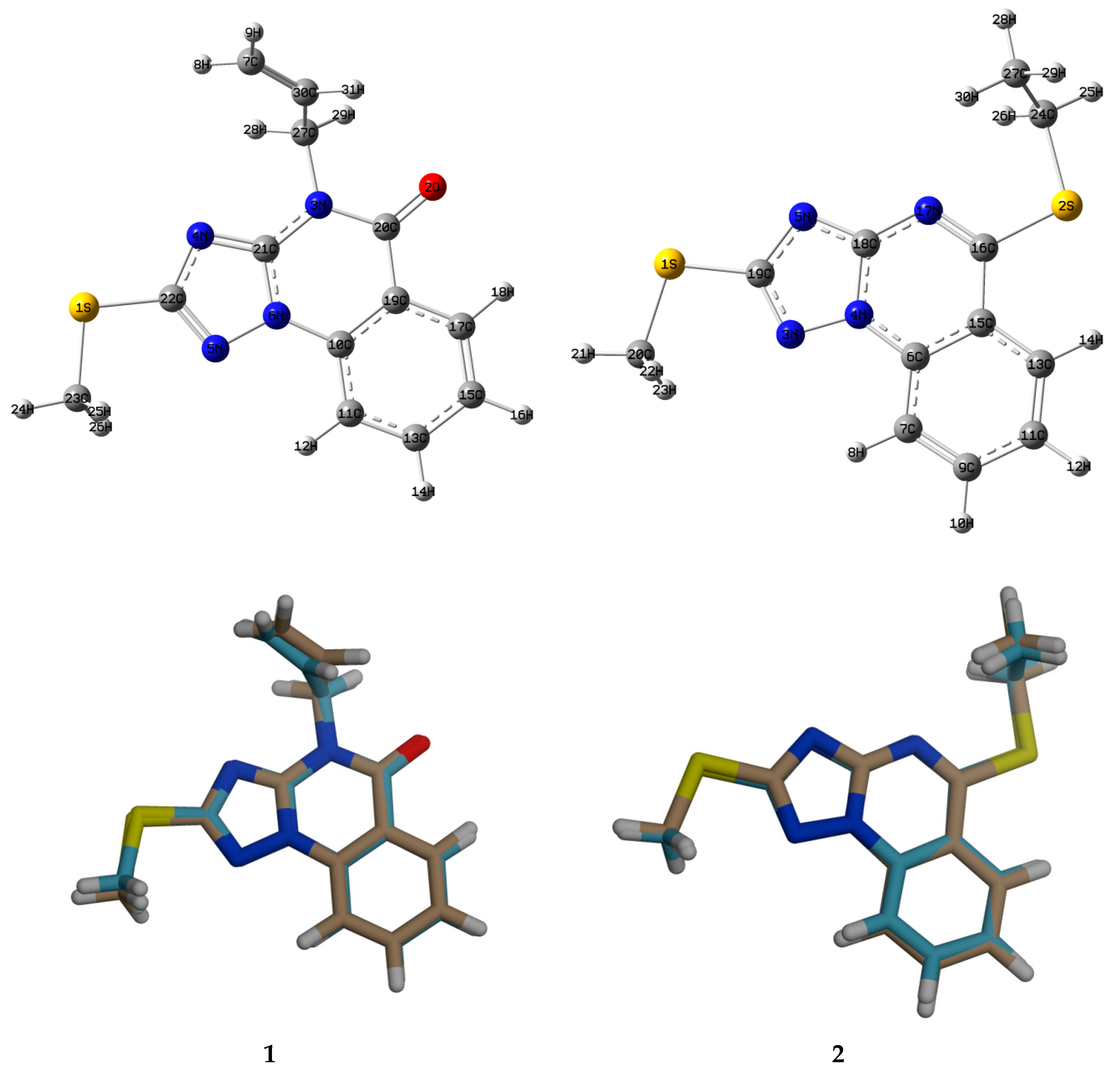
3.3. Crystallographic Data
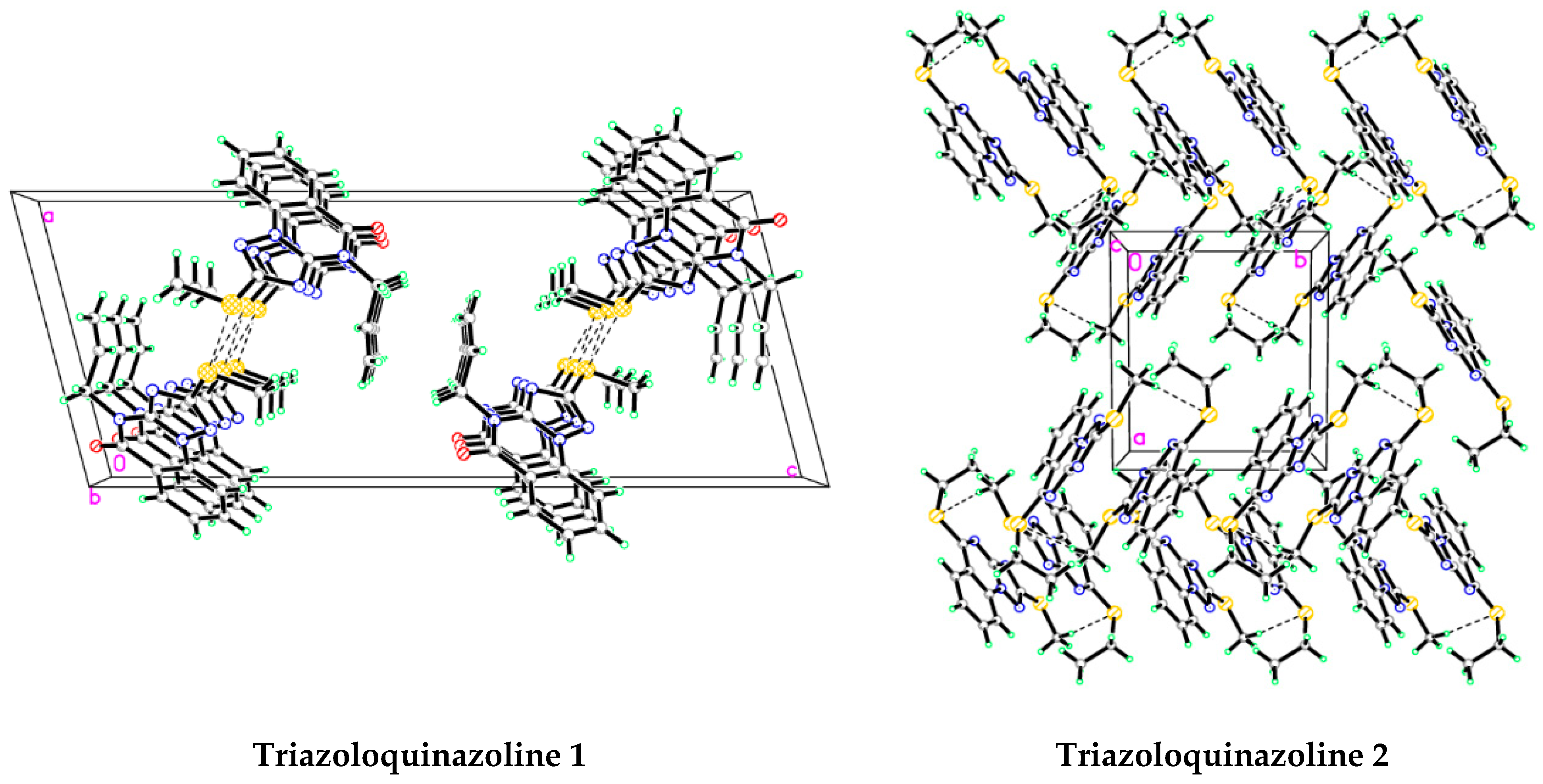
3.4. Analysis of Molecular Packing
4. DFT Analysis
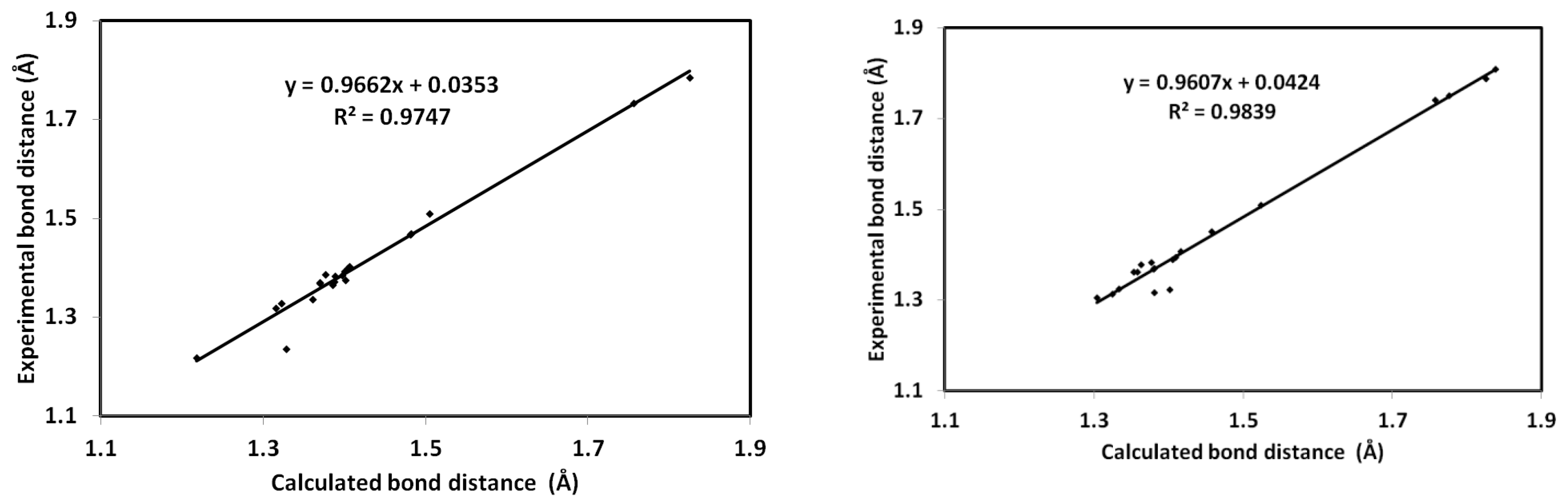
4.1. Geometric Parameters
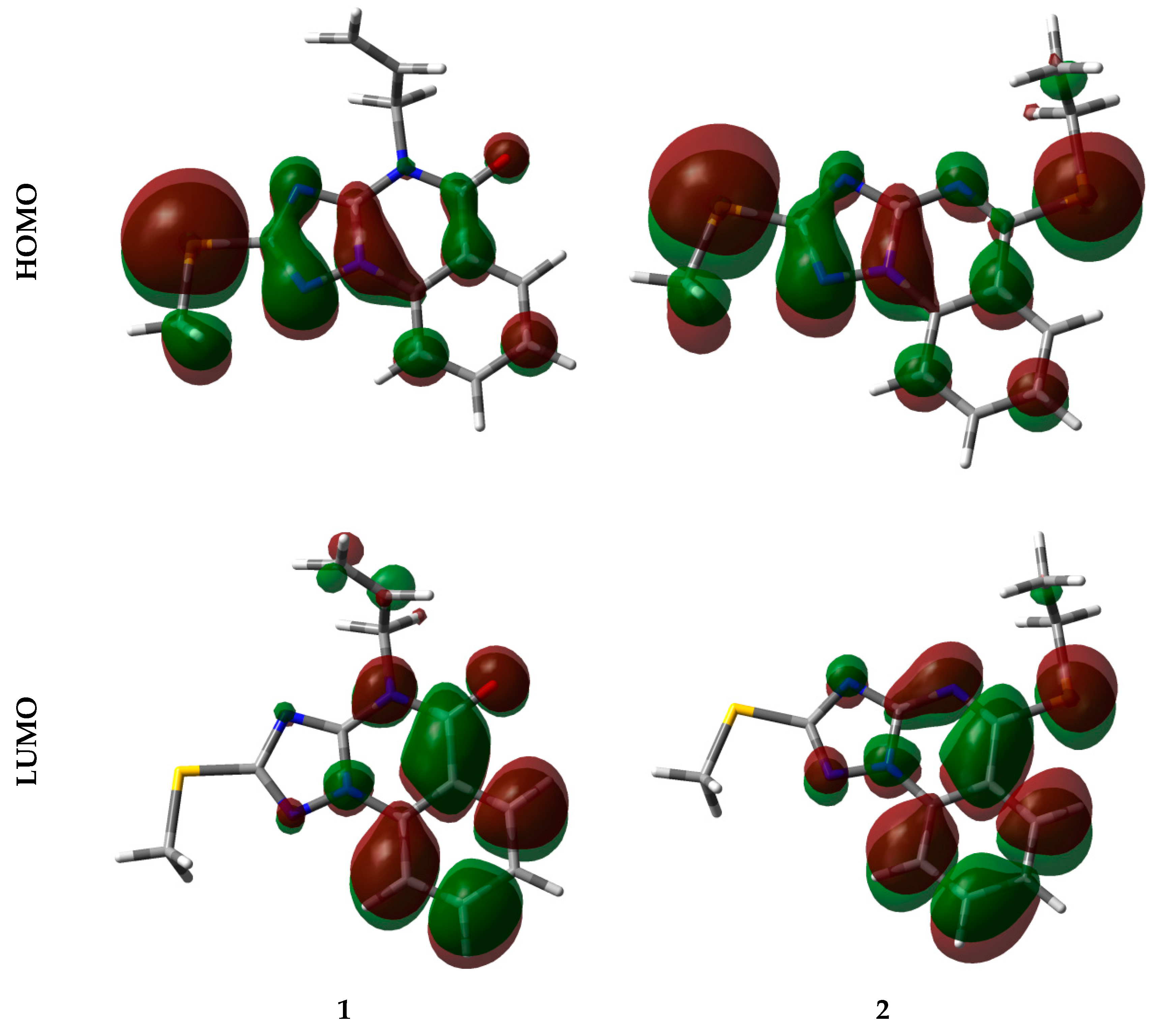
4.2. Analysis of Reactivity
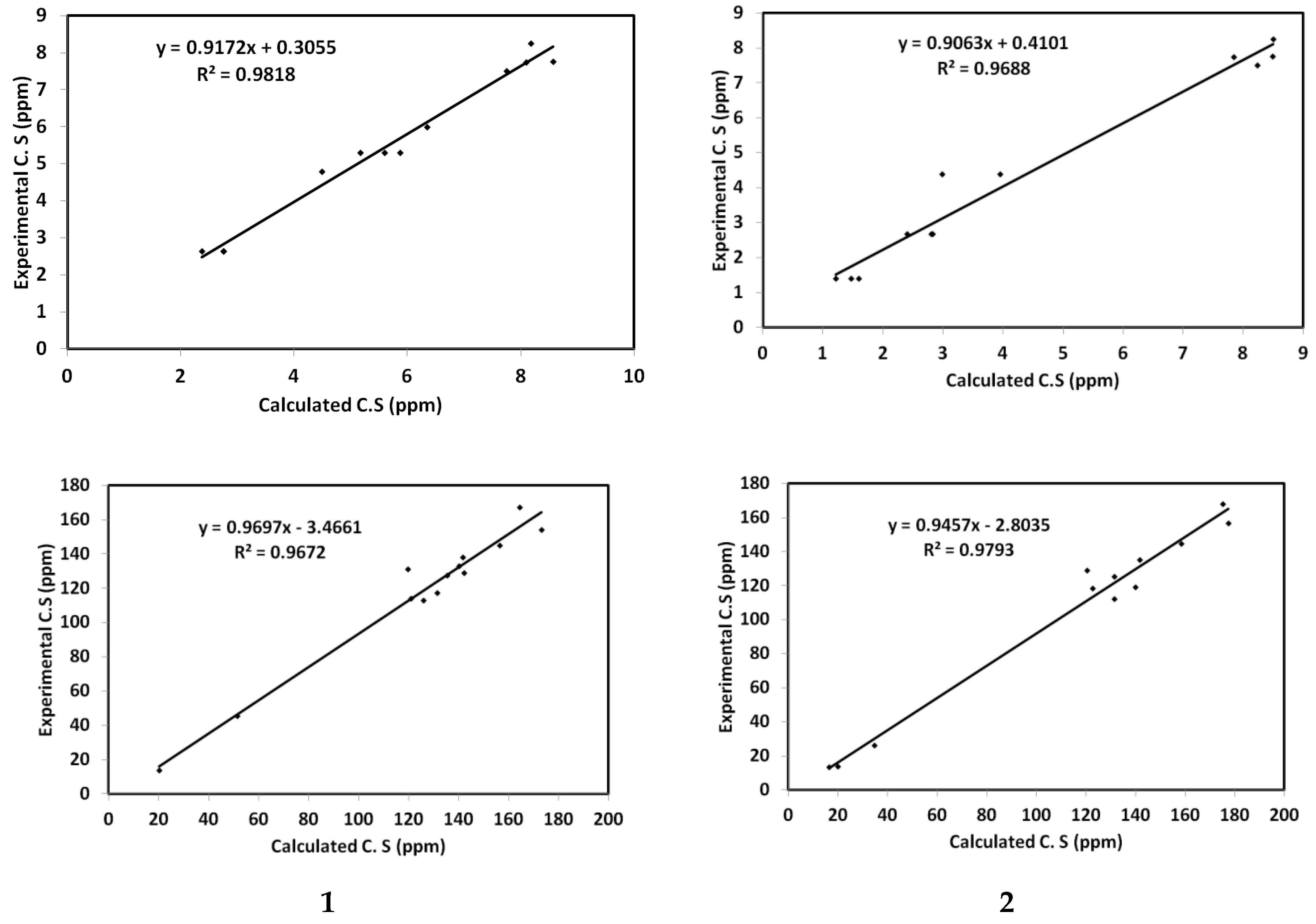
5. NMR Spectra
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Al-Salahi, R.; Gamal-Eldeen, A.M.; Alanazi, A.; Al-Omar, M.; Marzouk, M.; Fouda, M.G. Cytotoxic and anti-inflammatory active methylsulfanyltriazoloq uinazolines. J. Pure Appl. Microbiol. 2013, 7, 189–198. [Google Scholar]
- Abuelizz, H.A.; Al-Salahi, R.; Al-Asri, J.; Mortier, J.; Marzouk, M.; Ezzeldin, E.; Ali, A.A.; Khalil, M.G.; Wolber, G.; Ghabbour, H.A. Synthesis, crystallographic characterization, molecular docking and biological activity of isoquinoline derivatives. Chem. Cent. J. 2017, 11, 103. [Google Scholar] [CrossRef] [Green Version]
- Abuelizz, H.A.; Al-Salahi, R. An overview of triazoloquinazolines: Pharmacological significance and recent developments. Bioorg. Chem. 2021, 115, 105263. [Google Scholar] [CrossRef]
- Al-Salahi, R.; Marzouk, M. Some 2-aminobenzo[de]isoquinolin1,3-diones as antimicrobial agents. Asian J. Chem. 2014, 26, 8163–8165. [Google Scholar] [CrossRef]
- Alagarsamy, V.; Rupeshkumar, M.; Kavitha, K.; Meena, S.; Shankar, D.; Siddiqui, A.A.; Rajesh, R. Synthesis and pharmacological investigation of novel 4-(2-methylphenyl)-1-substituted-4H-[1,2,4]triazolo[4,3-a]quinazolin-5-ones as new class of H(1)-antihistaminic agents. Eur. J. Med. Chem. 2008, 43, 2331–2337. [Google Scholar] [CrossRef]
- Al-Salahi, R.; Marzouk, M.; El-Eraky, W.I.; Saleh, O.S. Antihistamine Activity of a new set 1,2,4-triazolo[1,5-a]quinazolines. Asian J. Chem. 2014, 26, 8625–8627. [Google Scholar]
- Alagarsamy, V.; Pathak, U.S. Synthesis and antihypertensive activity of novel 3-benzyl-2-substituted-3H-[1,2,4]triazolo[5,1-b]quinazolin-9-ones. Bioorg. Med. Chem. 2007, 15, 3457–3462. [Google Scholar] [CrossRef]
- Al-Salahi, R.; Tahir, K.E.; Alswaidan, I.; Lolak, N.; Hamidaddin, M.; Marzouk, M. Biological effects of a new set 1,2,4-triazolo[1,5-a]quinazolines on heart rate and blood pressure. Chem. Cent. J. 2014, 8, 3. [Google Scholar] [CrossRef] [Green Version]
- Francis, J.E.; Cash, W.D.; Psychoyos, S.; Ghai, G.; Wenk, P.; Friedmann, R.C.; Atkins, C.; Warren, V.; Furness, P.; Hyun, J.L.; et al. Structure–activity profile of a series of novel triazoloquinazoline adenosine antagonists. J. Med. Chem. 1988, 31, 1014–1020. [Google Scholar] [CrossRef]
- Al-Salahi, R.; Geffken, D.; Koellner, M. A new series of 2-Alkoxy(aralkoxy)-[1,2,4]triazolo[1,5-a]quinazolin-5-ones as Adenosine receptor antagonists. Chem. Pharm. Bull. 2011, 59, 730–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Salahi, R.; Marzouk, M.; Awad, G.; Al-Omar, M.; Ezzeldin, E. Antimicrobial activity of newly synthesized methylsulfanyl-triazoloquinazoline derivatives. J. Pharm. Pharmacol. 2013, 65, 790–797. [Google Scholar] [CrossRef]
- Al-Salahi, R.; Alswiadan, I.; Alomer, M.; Marzouk, M. Synthesis and antimicrobial of new 2-Phenoxy-4H-[1,2,4]triazolo[1,5-a]quinazoline derivatives. Life Sci. J. 2013, 10, 2018–2028. [Google Scholar]
- Al-Salahi, R.; Alswiadan, I.; Alomer, M.; Marzouk, M. Antiviral activity of 2-Phenoxy-4H-[1,2,4]triazolo[1,5-a]quinazoline derivatives. Life Sci. J. 2013, 10, 2164–2169. [Google Scholar]
- Al-Salahi, R.; Alswiadan, I.; Marzouk, M.; Alsenousy, W.; Abd Elgalil, A. Antiviral activities of some methylsulfanyltriazoloquinazolines. Res. Chem. Intermed. 2015, 41, 151–161. [Google Scholar] [CrossRef]
- Abuelizz, H.A.; Anouar, E.H.; Ahmad, R.; Azman, N.I.I.N.; Marzouk, M.; Al-Salahi, R. Triazoloquinazolines as a new class of potent α-glucosidase inhibitors: In vitro evaluation and docking study. PLoS ONE 2019, 14, e0220379. [Google Scholar] [CrossRef] [Green Version]
- Al-Salahi, R.; Anouar, E.H.; Marzouk, M.; Taie, H.A.; Abuelizz, H.A. Screening and evaluation of antioxidant activity of some 1,2,4-triazolo[1,5-a]quinazoline derivatives. Future Med. Chem. 2018, 10, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Sompalle, R.; Roopan, S.M.; Al-Dhabi, N.A.; Suthindhiran, K.; Sarkar, G.; Arasu, M.V. 1,2,4-triazoloquinazoline-thiones: Non-conventional synthetic approach, study of solvatochromism and antioxidant assessment. J. Photochem. Photobiol. B Biol. 2016, 162, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Al-Salahi, R.; Marzouk, M.; Ashour, A.E.; Alswaidan, I. Synthesis and antitumor activity of 1,2,4-triazolo[1,5-a]quinazolines. Asian J. Chem. 2014, 26, 2173–2176. [Google Scholar] [CrossRef]
- Al-Salahi, R.; Ashour, A.E.; Marzouk, M.; Ashok, K. In Vitro Cytotoxicity Evaluation of a New Series of Benzo[g][1,2,4]triazolo[1,5-α]quinazolines. Lat. Am. J. Pharm. 2015, 34, 1926–1930. [Google Scholar]
- Al-Salahi, R.; Elsayed, E.A.; El Dib, R.A.; Wadaan, M.; Ezzeldin, E.; Marzouk, M. Synthesis, Characterization and Cytotoxicity Evaluation of 5-Hydrazono-[1,2,4]triazolo[1,5-a]quinazolines (Part I). Lat. Amer. J. Pharm. 2016, 35, 58–65. [Google Scholar]
- Almehizia, A.A.; Abuelizz, H.A.; Taie, H.A.A.; Anouar, E.A.; Marzouk, M.; Al-Salahi, R. Investigation the antioxidant activity of benzo[g]triazoloquinazolines correlated with a DFT study. Saudi Pharm. J. 2019, 27, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Al-Salahi, R.; Marzouk, M.; Abbas, M.; Ng, S.W. 2-Methylsulfonyl-1,2,4-triazolo[1,5-a]quinazolin-5 (4H)-one. Acta Crystallogr. 2012, E68, o1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Salahi, R.; Marzouk, M.; Al-Omar, M.; Amr, A.E.; Ng, S.W.; Tiekink, E.R.T. 2-Methylsulfanyl-1,2,4-triazolo[1,5-a]quinazoline-5(4H)-thione. Acta Crystallogr. 2013, E69, o434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Salahi, R.; Nabih, L.; Al-Omar, M.; Ng, S.W. 2-Phenoxy-1,2,4-triazolo[1,5-a] quinazolin-5(4H)-one. Acta Crystallogr. 2012, E68, o1808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Salahi, R.; Al-Omar, M.; Marzouk, M.; Ng, S.W.; Tiekink, E.R.T. 5-chloro-2-methylsulfonyl-1,2,4-triazolo[1,5-a]quinazoline. Acta Crystallogr. 2012, E68, o1809. [Google Scholar] [CrossRef] [Green Version]
- Al-Salahi, R.; Geffken, D.; Bari, A. 2-benzyloxy-1,2,4-triazolo[1,5-a] quinazolin-5(4H)-one. Acta Crystallogr. 2011, E67, o1861. [Google Scholar] [CrossRef] [Green Version]
- Al-Salahi, R.; Geffken, D. Synthesis of Novel 2-methylsulfanyl-4H-[1,2,4]triazolo[1,5-a]quinazolin-5-one and derivatives. Synth. Commun. 2011, 41, 3512–3523. [Google Scholar] [CrossRef]
- Al-Salahi, R.; Geffken, D. Synthesis and reactivity of [1,2,4]triazolo-annelated quinazolines. Molecules 2010, 15, 7016–7034. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. SHELXT-integrated space-group and crystal-structure determination. Acta Crystallogr. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Allen, F.H.; Kennard, O.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. Tables of bond Lengths determined by X-ray and neutron diffraction. Part 1. Bond lengths in organic compounds. J. Chem. Soc. Perkin Trans. 1987, II, S1–S19. [Google Scholar] [CrossRef]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. Crystal Explorer. University of Western Australia. 2017. Available online: http://hirshfeldsurface.net (accessed on 2 September 2021).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Fox, GAUSSIAN 09. Revision A02; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Dennington, R., II; Keith, T.; Millam, J. GaussView; Version 4.1; Semichem Inc.: Shawnee Mission, KS, USA, 2007. [Google Scholar]
- Reed, A.E.; Curtiss, L.A.F.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Marten, B.; Kim, K.; Cortis, C.; Friesner, R.A.; Murphy, R.B.; Ringnalda, M.N.; Sitkoff, D.; Honig, B. New model for calculation of solvation free energies: correction of self-consistent reaction field continuum dielectric theory for short-range hydrogen-bonding effects. J. Phys. Chem. 1996, 100, 11775–11788. [Google Scholar] [CrossRef]
- Tannor, D.J.; Marten, B.; Murphy, R.; Friesner, R.A.; Sitkoff, D.; Nicholls, A.; Ringnalda, M.; Goddard, W.A.; Honig, B. Accurate first principles calculation of molecular charge distributions and solvation energies from ab initio quantum mechanics and continuum dielectric theory. J. Am. Chem. Soc. 1994, 116, 11875–11882. [Google Scholar] [CrossRef]
- Cheeseman, J.R.; Trucks, G.W.; Keith, T.A.; Frisch, M.J. A Comparison of Models for Calculating Nuclear Magnetic Resonance Shielding Tensors. J. Chem. Phys. 1996, 104, 5497–5509. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foresman, J.B.; Frisch, A.E. Exploring Chemistry with Electronic Structure Methods, 2nd ed.; Gaussian: Pittsburgh, PA, USA, 1996. [Google Scholar]
- Chang, R. Chemistry, 7th ed.; McGraw-Hill: New York, NY, USA, 2001. [Google Scholar]
- Kosar, B.; Albayrak, C. Spectroscopic investigations and quantum chemical computational study of (E)-4-methoxy-2-[(p-tolylimino)methyl]phenol. Spectrochim. Acta 2011, 78, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Koopmans, T. About the assignment of wave functions and eigenvalues to the individual electrons of an atom. Physica 1934, 1, 104–113. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Parr, G.; Von Szentpály, L.; Liu, S.B. Electrophilicity Index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Singh, R.N.; Kumar, A.; Tiwari, R.K.; Rawat, P.; Gupta, V.P. A combined experimental and quantum chemical (DFT and AIM) study on molecular structure, spectroscopic properties, NBO and multiple interaction analysis in a novel ethyl 4-[2-(carbamoyl)hydrazinylidene]-3,5-dimethyl-1H-pyrrole-2-carboxylate and its dimer. J. Mol. Struct. 2013, 1035, 427–440. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crystal Data | Triazoloquinazoline 1 | Triazoloquinazoline 2 |
|---|---|---|
| Chemical formula | C13H12N4OS | C12H12N4S2 |
| Mr | 272.33 | 276.38 |
| Crystal system, space group | Monoclinic, P21/c | Monoclinic, P21/n |
| Temperature (K) | 293 | 293 |
| a, b, c (Å) | 10.3567 (12), 5.0392 (5), 25.013 (3) | 9.5153 (7), 8.1162 (6), 16.1568 (12) |
| β (°) | 104.850 (5) | 102.852 (3) |
| V (Å3) | 1261.8 (2) | 1216.50 (16) |
| Z | 4 | 4 |
| Type of radiation | Mo Kα | Mo Kα |
| µ (mm−1) | 0.25 | 0.42 |
| Size of crystal (mm) | 0.42 × 0.21 × 0.04 | 0.60 × 0.13 × 0.09 |
| Data Collection | ||
| Diffractometer | Bruker APEX-II D8 venture diffractometer | Bruker APEX-II D8 venture diffractometer |
| Absorption correction | Multi-scan SADABS Bruker 2014 | Multi-scan SADABS Bruker 2014 |
| Tmin, Tmax | 0.810, 0.874 | 0.806, 0.812 |
| The measured, independent and observed [I > 2σ(I)] reflections No. | 16671, 2614, 1481 | 27487, 2796, 1979 |
| Rint | 0.176 | 0.094 |
| Refinement | ||
| R[F2 > 2σ( F2)], wR( F2), S | 0.064, 0.155, 1.02 | 0.054, 0.150, 1.03 |
| Reflections No. | 2614 | 2796 |
| Parameters No. | 167 | 165 |
| Restraints No. | 0 | 0 |
| Δρmax, Δρmin (e Å−3) | 0.55, −0.50 | 0.57, −0.86 |
| CCDC No. | 1826859 | 1827320 |
| Triazoloquinazoline 1 | ||||
|---|---|---|---|---|
| D—H···A | D—H | H···A | D···A | D—H···A |
| C5—H5A···O1i | 0.9300 | 2.5900 | 3.500 (5) | 164.00 |
| Symmetry codes: (i) −x + 2, −y − 2, −z + 1. | ||||
| Triazoloquinazoline 2 | ||||
| D—H···A | D—H | H···A | D···A | D—H···A |
| C5—H5A···S2 | 0.9500 | 2.6900 | 3.072 (3) | 105.00 |
| C11—H11B···N4 | 0.9900 | 2.4500 | 2.854 (4) | 104.00 |
| 1 | 2 | ||
|---|---|---|---|
| Contact | Distance | Contact | Distance |
| O1 H5A | 2.448 | H2A H10C | 2.024 |
| S1 S1 | 3.448 | S2 H10B | 2.777 |
| C6 C9 | 3.382 | C1 C8 | 3.388 |
| Parameter | 1 | 2 |
|---|---|---|
| HOMO | −6.0921 | −6.0026 |
| LUMO | −1.7671 | −2.1973 |
| I = −EHOMO | 6.0921 | 6.0026 |
| A = −ELUMO | 1.7671 | 2.1973 |
| η = (I − A)/2 | 4.3250 | 3.8053 |
| μ = −(I + A)/2 | −3.9296 | −4.1000 |
| ω = μ2/2η | 1.7852 | 2.2087 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abuelizz, H.A.; Soliman, S.M.; Ghabbour, H.A.; Marzouk, M.; Abdellatif, M.M.; Al-Salahi, R. DFT Calculation, Hirshfeld Analysis and X-Ray Crystal Structure of Some Synthesized N-alkylated(S-alkylated)-[1,2,4]triazolo[1,5-a]quinazolines. Crystals 2021, 11, 1195. https://doi.org/10.3390/cryst11101195
Abuelizz HA, Soliman SM, Ghabbour HA, Marzouk M, Abdellatif MM, Al-Salahi R. DFT Calculation, Hirshfeld Analysis and X-Ray Crystal Structure of Some Synthesized N-alkylated(S-alkylated)-[1,2,4]triazolo[1,5-a]quinazolines. Crystals. 2021; 11(10):1195. https://doi.org/10.3390/cryst11101195
Chicago/Turabian StyleAbuelizz, Hatem A., Saied M. Soliman, Hazem A. Ghabbour, Mohamed Marzouk, Mohamed M. Abdellatif, and Rashad Al-Salahi. 2021. "DFT Calculation, Hirshfeld Analysis and X-Ray Crystal Structure of Some Synthesized N-alkylated(S-alkylated)-[1,2,4]triazolo[1,5-a]quinazolines" Crystals 11, no. 10: 1195. https://doi.org/10.3390/cryst11101195
APA StyleAbuelizz, H. A., Soliman, S. M., Ghabbour, H. A., Marzouk, M., Abdellatif, M. M., & Al-Salahi, R. (2021). DFT Calculation, Hirshfeld Analysis and X-Ray Crystal Structure of Some Synthesized N-alkylated(S-alkylated)-[1,2,4]triazolo[1,5-a]quinazolines. Crystals, 11(10), 1195. https://doi.org/10.3390/cryst11101195