Analysis of the Phase Stability of LiMO2 Layered Oxides (M = Co, Mn, Ni)


 ,
, 
 and
and 
Abstract
1. Introduction
2. Materials and Methods
3. Results
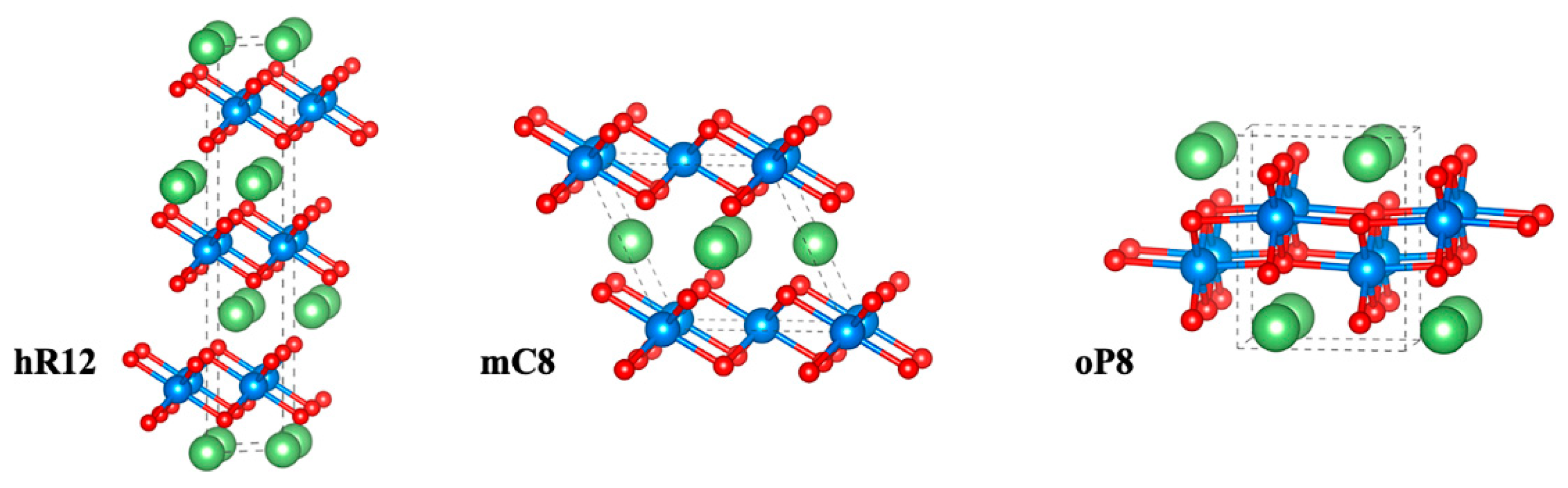
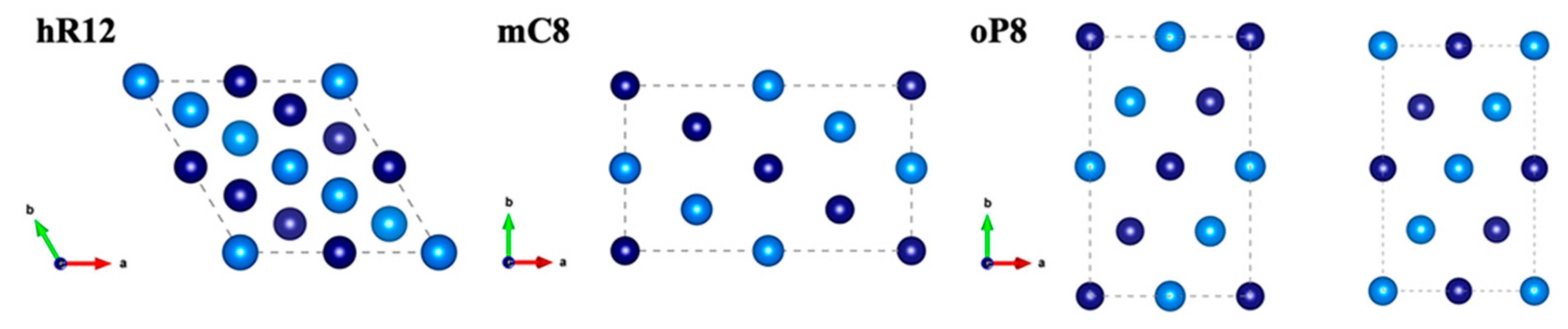
3.1. Structural Analysis
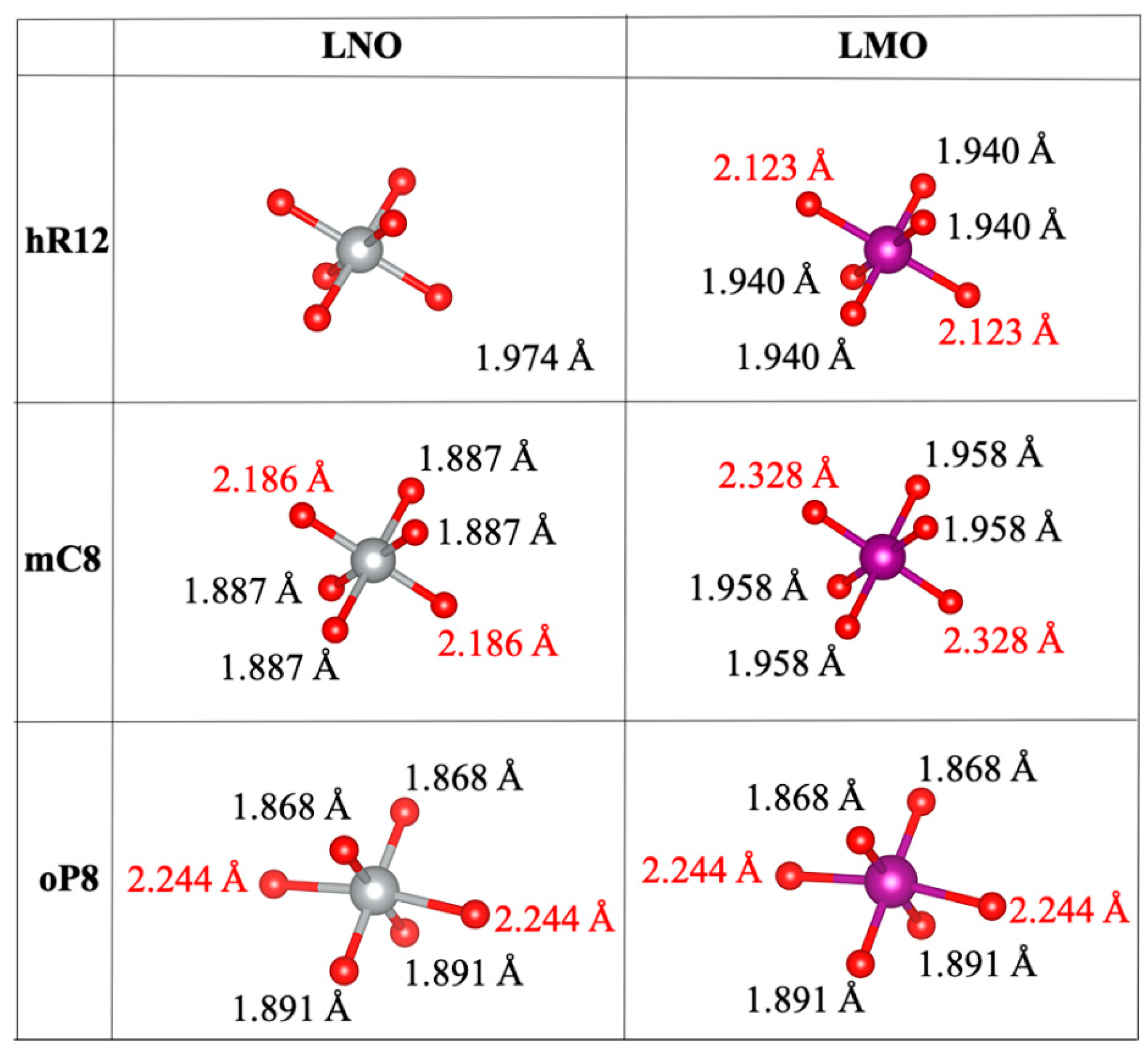
3.2. Bond Distances and Jahn-Teller Distortions
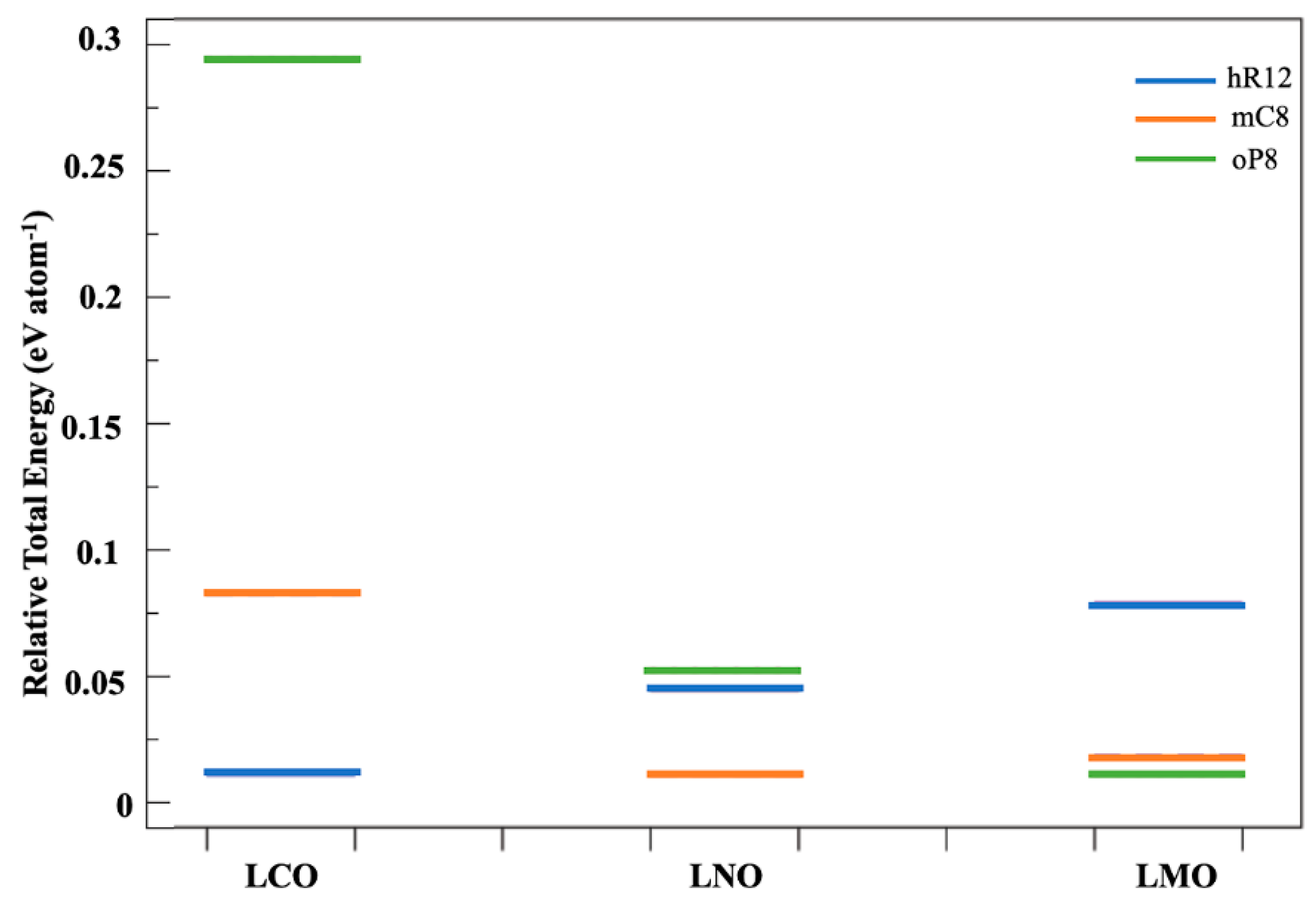
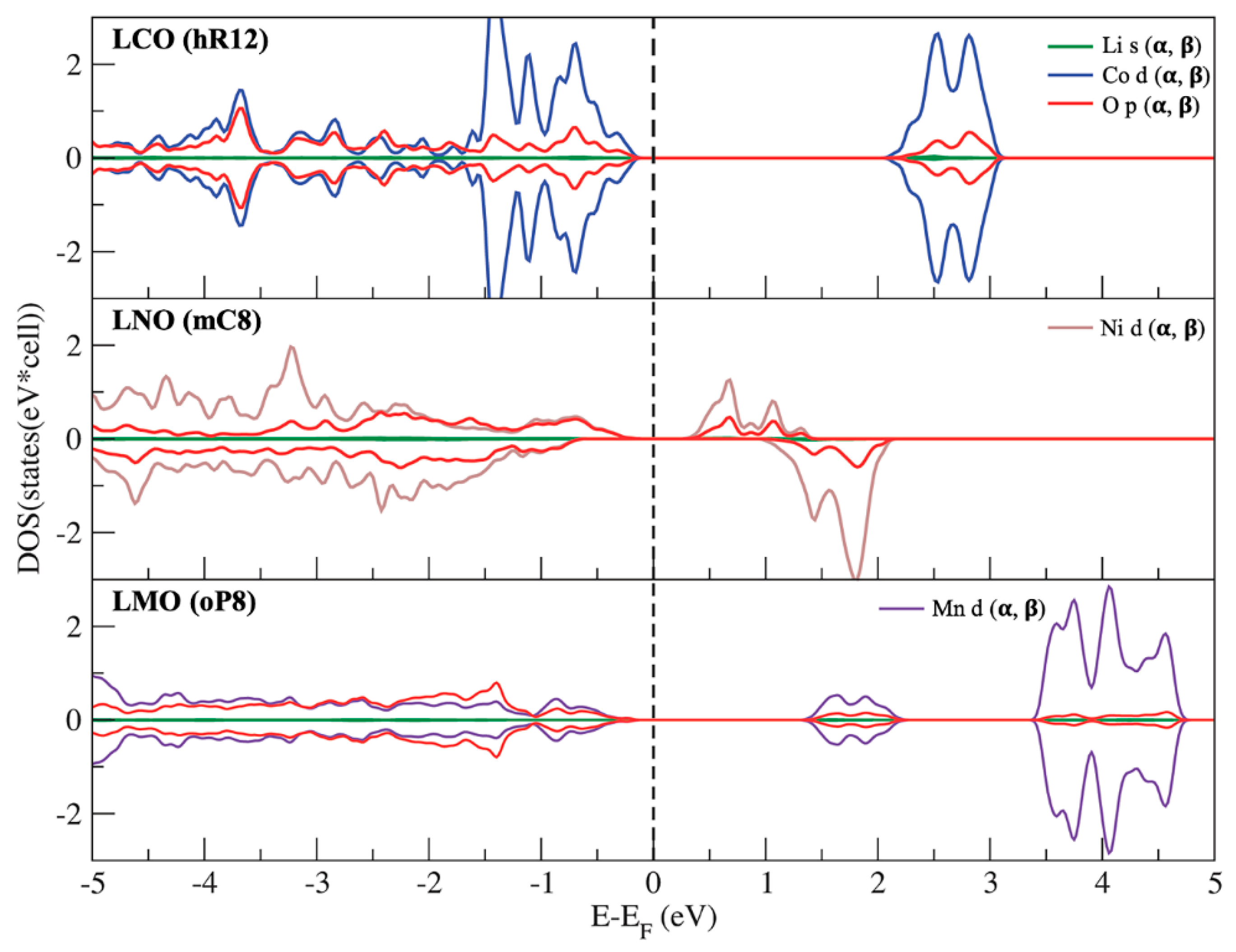
3.3. Phase Stability and Electronic Structures of the Ground States
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Zhang, H.; Li, C.; Eshetu, G.G.; Laruelle, S.; Grugeon, S.; Zaghib, K.; Julien, C.; Mauger, A.; Guyomard, D.; Rojo, T.; et al. From Solid-Solution Electrodes and the Rocking-Chair Concept to Today’s Batteries. Angew. Chem. Int. Ed. 2020, 59, 534–538. [Google Scholar] [CrossRef] [PubMed]
- Budde-Meiwes, H.; Drillkens, J.; Lunz, B.; Muennix, J.; Rothgang, S.; Kowal, J.; Sauer, D.U. A review of current automotive battery technology and future prospects. Proc. Inst. Mech. Eng. Part D J. Automob. Eng. 2013, 227, 761–776. [Google Scholar] [CrossRef]
- Majeau-Bettez, G.; Hawkins, T.R.; StrØmman, A.H. Life cycle environmental assessment of lithium-ion and nickel metal hydride batteries for plug-in hybrid and battery electric vehicles. Environ. Sci. Technol. 2011, 45, 4548–4554. [Google Scholar] [CrossRef]
- Scrosati, B.; Garche, J. Lithium batteries: Status, prospects and future. J. Power Sources 2010, 195, 2419–2430. [Google Scholar] [CrossRef]
- Larcher, D.; Tarascon, J.-M. Towards greener and more sustainable batteries for electrical energy storage. Nat. Chem. 2014, 7, 19–29. [Google Scholar] [CrossRef]
- Mizushima, K.; Jones, P.C.; Wiseman, P.J.; Goodenough, J.B. LixCoO2 (0 < x ≤1): A new cathode material for batteries of high energy density. Solid State Ionics 1981, 3, 171–174. [Google Scholar]
- Bianchini, M.; Roca-Ayats, M.; Hartmann, P.; Brezesinski, T.; Janek, J. There and Back Again—The Journey of LiNiO2 as a Cathode Active Material. Angew. Chem. Int. Ed. 2019, 58, 10434–10458. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.R.; Bruce, P.G. Synthesis of layered LiMnO2 as an electrode for rechargeable lithium batteries. Nature 1996, 381, 499–500. [Google Scholar] [CrossRef]
- Li, T.; Yuan, X.-Z.; Zhang, L.; Song, D.; Shi, K.; Bock, C. Degradation Mechanisms and Mitigation Strategies of Nickel-Rich NMC-Based Lithium-Ion Batteries. Electrochem. Energy Rev. 2019, 3, 43–80. [Google Scholar] [CrossRef]
- Lee, W.; Muhammad, S.; Sergey, C.; Lee, H.; Yoon, J.; Kang, Y.; Yoon, W. Advances in the Cathode Materials for Lithium Rechargeable Batteries. Angew. Chem. Int. Ed. 2020, 59, 2578–2605. [Google Scholar] [CrossRef]
- Saubanere, M.; McCalla, E.; Tarascon, J.-M.; Doublet, M.-L. The intriguing question of anionic redox in high-energy density cathodes for Li-ion batteries. Energy Environ. Sci. 2016, 9, 984–991. [Google Scholar] [CrossRef]
- Du, T.; Xu, B.; Wu, M.; Liu, G.; Ouyang, C. Insight into the Vibrational and Thermodynamic Properties of Layered Lithium Transition-Metal Oxides LiMO 2 (M = Co, Ni, Mn): A First-Principles Study. J. Phys. Chem. C 2016, 120, 5876–5882. [Google Scholar] [CrossRef]
- Wang, M.; Navrotsky, A. LiMO2 (M = Mn, Fe, and Co): Energetics, polymorphism and phase transformation. J. Solid State Chem. 2005, 178, 1230–1240. [Google Scholar] [CrossRef]
- Yu, Z.; Shang, S.-L.; Gordin, M.L.; Mousharraf, A.; Liu, Z.-K.; Wang, D. Ti-substituted Li[Li 0.26 Mn 0.6-x Ti x Ni 0.07 Co 0.07 ]O 2 layered cathode material with improved structural stability and suppressed voltage fading. J. Mater. Chem. A 2015, 3, 17376–17384. [Google Scholar] [CrossRef]
- Koyama, Y.; Arai, H.; Tanaka, I.; Uchimoto, Y.; Ogumi, Z. High temperature defect chemistry in layered lithium transition-metal oxides based on first-principles calculations. J. Power Sources 2013, 244, 592–596. [Google Scholar] [CrossRef]
- Brutti, S.; Balducci, G.; Ciccioli, A.; Gigli, G. Thermodynamic assessment of the Yb-Si system. Calphad Comput. Coupling Phase Diagr. Thermochem. 2005, 29, 254–261. [Google Scholar] [CrossRef]
- Lalena, J.N.; Cleary, D.A. Principles of Inorganic Materials Design, 2nd ed.; John Wiley and Sons: Hoboken, NJ, USA, 2010; ISBN 9780470404034. [Google Scholar]
- Luo, J. Developing Interfacial Phase Diagrams for Applications in Activated Sintering and Beyond: Current Status and Future Directions. J. Am. Ceram. Soc. 2012, 95, 2358–2371. [Google Scholar] [CrossRef]
- Jin, L.; Wang, J.; Rousselot, S.; Dollé, M.; Chartrand, P. Experimental and thermodynamic study of Li-O and Li 2O-P 2 O5 systems. Can. J. Chem. Eng. 2019, 97, 2234–2241. [Google Scholar] [CrossRef]
- Phan, A.T.; Gheribi, A.E.; Chartrand, P. Modelling of phase equilibria of LiFePO4 -FePO4 olivine join for cathode material. Can. J. Chem. Eng. 2019, 97, 2224–2233. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Eyert, V.; Christensen, M.; Wolf, W.; Reith, D.; Mavromaras, A.; Freeman, C.; Wimmer, E. Unravelling the Potential of Density Functional Theory through Integrated Computational Environments: Recent Applications of the Vienna Ab Initio Simulation Package in the MedeA® Software. Computation 2018, 6, 63. [Google Scholar] [CrossRef]
- Pavone, M.; Ritzmann, A.M.; Carter, E.A. Quantum-mechanics-based design principles for solid oxide fuel cell cathode materials. Energy Environ. Sci. 2011, 4, 4933–4937. [Google Scholar] [CrossRef]
- Dudarev, S.; Botton, G. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys. Rev. B Condens. Matter Mater. Phys. 1998, 57, 1505–1509. [Google Scholar] [CrossRef]
- Muñoz-García, A.B.; Pavone, M. Structure and energy level alignment at the dye-electrode interface in p-type DSSCs: New hints on the role of anchoring modes from ab initio calculations. Phys. Chem. Chem. Phys. 2015, 17, 12238–12246. [Google Scholar] [CrossRef]
- Muñoz-García, A.B.; Sannino, F.; Vitiello, G.; Pirozzi, D.; Minieri, L.; Aronne, A.; Pernice, P.; Pavone, M.; D’Errico, G. Origin and Electronic Features of Reactive Oxygen Species at Hybrid Zirconia-Acetylacetonate Interfaces. ACS Appl. Mater. Interfaces 2015, 7, 21662–21667. [Google Scholar] [CrossRef]
- Muñoz-García, A.B.; Tuccillo, M.; Pavone, M. Computational design of cobalt-free mixed proton-electron conductors for solid oxide electrochemical cells. J. Mater. Chem. A 2017, 5, 11825–11833. [Google Scholar] [CrossRef]
- Wang, L.; Maxisch, T.; Ceder, G. Oxidation energies of transition metal oxides within the GGA+ U framework. Phys. Rev. B 2006, 73, 195107. [Google Scholar] [CrossRef]
- Jain, A.; Hautier, G.; Ping Ong, S.; Moore, C.J.; Fischer, C.C.; Persson, K.A.; Ceder, G. Formation enthalpies by mixing GGA and GGA + U calculations. Phys. Rev. B 2011, 84, 45115. [Google Scholar] [CrossRef]
- Greedan, J.E.; Raju, N.P.; Davidson, I.J. Long Range and Short Range Magnetic Order in Orthorhombic LiMnO2. J. Solid State Chem. 1997, 128, 209–214. [Google Scholar] [CrossRef]
- Singh, D.J. Magnetic and electronic properties of LiMnO2. Phys. Rev. B Condens. Matter Mater. Phys. 1997, 55, 309–312. [Google Scholar] [CrossRef]
- Thomas, M.G.S.R.; Bruce, P.G.; Goodenough, J.B. Lithium mobility in the layered oxide Li1-xCoO2. Solid State Ion. 1985, 17, 13–19. [Google Scholar] [CrossRef]
- Yamada, S.; Fujiwara, M.; Kanda, M. Synthesis and properties of LiNiO2 as cathode material for secondary batteries. J. Power Sources 1995, 54, 209–213. [Google Scholar] [CrossRef]
- Ohzuku, T. Electrochemistry and Structural Chemistry of LiNiO2 (R3m) for 4 Volt Secondary Lithium Cells. J. Electrochem. Soc. 1993, 140, 1862. [Google Scholar] [CrossRef]
- Chen, H.; Freeman, C.L.; Harding, J.H. Charge disproportionation and Jahn-Teller distortion in LiNiO2 and NaNiO2: A density functional theory study. Phys. Rev. B Condens. Matter Mater. Phys. 2011, 84, 085108. [Google Scholar] [CrossRef]
- Rougier, A.; Delmas, C.; Chadwick, A.V. Non-cooperative Jahn-Teller effect in LiNiO2: An EXAFS study. Solid State Commun. 1995, 94, 123–127. [Google Scholar] [CrossRef]
- Chung, J.H.; Proffen, T.; Shamoto, S.; Ghorayeb, A.M.; Croguennec, L.; Tian, W.; Sales, B.C.; Jin, R.; Mandrus, D.; Egami, T. Local structure of LiNiO 2 studied by neutron diffraction. Phys. Rev. B Condens. Matter Mater. Phys. 2005, 71, 064410. [Google Scholar] [CrossRef]
- Nakai, I.; Takahashi, K.; Shiraishi, Y.; Nakagome, T.; Nishikawa, F. Study of the Jahn-Teller Distortion in LiNiO 2, a Cathode Material in a Rechargeable Lithium Battery, byin SituX-Ray Absorption Fine Structure Analysis. J. Solid State Chem. 1998, 140, 145–148. [Google Scholar] [CrossRef]
- Dittrich, G.; Hoppe, R. Zur Kristallstruktur von LiMnO2. Zeitschrift fur Anorganische und Allgemeine Chemie 1969, 368, 262–270. [Google Scholar] [CrossRef]
- Hoppe, R.; Brachtel, G.; Jansen, M. Zur Kenntnis der Oxomanganate(III): LiMnO2 und a-NaMnO2. Zeitschrift fur Anorganische und Allgemeine Chemie 1975, 417, 1–10. [Google Scholar] [CrossRef]
- Minenkov, Y.; Singstad, Å.; Occhipinti, G.; Jensen, V.R. The accuracy of DFT-optimized geometries of functional transition metal compounds: A validation study of catalysts for olefin metathesis and other reactions in the homogeneous phase. Dalton Trans. 2012, 41, 5526–5541. [Google Scholar] [CrossRef]
- Ceder, G.; Mishra, S.K. Stability of orthorhombic and monoclinic-layered LiMnO2. Electrochem. Solid State Lett. 1999, 2, 550–552. [Google Scholar] [CrossRef]
- Brutti, S.A.; Nguyen-Manh, D.B.; Pettifor, D.G.C. Lattice stability of Ca, Sr and Yb disilicides. Intermetallics 2006, 14, 1472–1486. [Google Scholar] [CrossRef]
- Hu, Y.Q.; Xiong, L.; Liu, X.Q.; Zhao, H.Y.; Liu, G.T.; Bai, L.G.; Cui, W.R.; Gong, Y.; Li, X.D. Equation of state of LiCoO2 under 30 GPa pressure. Chin. Phys. B 2019, 28, 2–6. [Google Scholar] [CrossRef]
- Kugel, K.I.; Khomskii, D.I. The jahn-teller effect and magnetism: Transition metal compounds. Sov. Phys. Uspekhi 1982, 25, 621–641. [Google Scholar] [CrossRef]
- Longuet-Higgins, H.C.; Öpik, U.; Lecorney Pryce, M.H.; Sack, R.A. Studies of the Jahn-Teller effect II. The dynamical problem. Proc. R. Soc. Lond. Ser. A Math. Phys. Sci. 1958, 244, 1–16. [Google Scholar]
- Mishra, S.K.; Ceder, G. Structural stability of lithium manganese oxides. Phys. Rev. B Condens. Matter Mater. Phys. 1999, 59, 6120–6130. [Google Scholar] [CrossRef]
- Rao, C.N.R.; Raveau, B. Transition Metal Oxides: Structure, Properties, and Synthesis of Ceramic Oxides; Wiley-VCH: Weinheim, Germany, 1998; ISBN 9780471189718. [Google Scholar]
- Li, W.; Reimers, J.N.; Dahn, J.R. Crystal structure of LixNi2-xO2 and a lattice-gas model for the order-disorder transition. Phys. Rev. B 1992, 46, 3236–3246. [Google Scholar] [CrossRef]
- Chevrier, V.L.; Ong, S.P.; Armiento, R.; Chan, M.K.Y.; Ceder, G. Hybrid density functional calculations of redox potentials and formation energies of transition metal compounds. Phys. Rev. B Condens. Matter Mater. Phys. 2010, 82, 075122. [Google Scholar] [CrossRef]
- Van Elp, J.; Wieland, J.L.; Eskes, H.; Kuiper, P.; Sawatzky, G.A.; De Groot, F.M.F.; Turner, T.S. Electronic structure of CoO, Li-doped CoO, and LiCoO2. Phys. Rev. B 1991, 44, 6090–6103. [Google Scholar] [CrossRef]
- Kong, F.; Longo, R.C.; Park, M.S.; Yoon, J.; Yeon, D.H.; Park, J.H.; Wang, W.H.; Kc, S.; Doo, S.G.; Cho, K. Ab initio study of doping effects on LiMnO2 and Li2MnO3 cathode materials for Li-ion batteries. J. Mater. Chem. A 2015, 3, 8489–8500. [Google Scholar] [CrossRef]
- Nath Shukla, N.; Prasad, R. Electronic structure of LiMnO2: A comparative study of the LSDA and LSDA + U methods. J. Phys. Chem. Solids 2006, 67, 1731–1740. [Google Scholar] [CrossRef][Green Version]
- Cupid, D.M.; Li, D.; Gebert, C.; Reif, A.; Flandorfer, H.; Seifert, H.J. Enthalpy of formation and heat capacity of Li 2 MnO 3. J. Ceram. Soc. Jpn. 2016, 124, 1072–1082. [Google Scholar] [CrossRef]
- Chase, M.W., Jr.; Davies, C.A.; Downey, J.R., Jr.; Frurip, D.J.; McDonald, R.A.; Syverud, A.N. NIST-JANAF Thermochemical Tables; Standard Reference Data Program National Institute of Standards and Technology: Gaithersburg, MD, USA, 1985.
- Chang, K.; Hallstedt, B. Thermodynamic assessment of the Li–O system. Calphad 2011, 35, 160–164. [Google Scholar] [CrossRef]
- Robie, R.A.; Hemingway, B.S. Low-temperature molar heat capacities and entropies of MnO2 (pyrolusite), Mn3O4 (hausmanite), and Mn2O3 (bixbyite). J. Chem. Thermodyn. 1985, 17, 165–181. [Google Scholar] [CrossRef]
- Alcock, C.B.; Zador, S. Thermodynamic study of the manganese/ manganous-oxide system by the use of solid oxide electrolytes. Electrochim. Acta 1967, 12, 673–677. [Google Scholar] [CrossRef]
- Fritsch, S.; Navrotsky, A. Thermodynamic Properties of Manganese Oxides. J. Am. Ceram. Soc. 1996, 79, 1761–1768. [Google Scholar] [CrossRef]
- Seltz, H.; DeWitt, B.J.; McDonald, H.J. The Heat Capacity of Nickel Oxide from 68-298°K. and the Thermodynamic Properties of the Oxide. J. Am. Chem. Soc. 1940, 62, 88–89. [Google Scholar] [CrossRef]
- Kawaji, H.; Takematsu, M.; Tojo, T.; Atake, T.; Hirano, A.; Kanno, R. Low temperature heat capacity and thermodynamic functions of LiCoO2. J. Therm. Anal. Calorim. 2002, 68, 833–839. [Google Scholar]
- Wang, M.; Navrotsky, A. Enthalpy of formation of LiNiO2, LiCoO2 and their solid solution, LiNi1-xCoxO2. Solid State Ion. 2004, 166, 167–173. [Google Scholar] [CrossRef]
- Spencer, P.J. Estimation of thermodynamic data for metallurgical applications. Thermochim. Acta 1998, 314, 1–21. [Google Scholar] [CrossRef]
- Leitner, J.; Chuchvalec, P.; Sedmidubský, D.; Strejc, A.; Abrman, P. Estimation of heat capacities of solid mixed oxides. Thermochim. Acta 2002, 395, 27–46. [Google Scholar] [CrossRef]
- Gotcu-Freis, P.; Cupid, D.M.; Rohde, M.; Seifert, H.J. New experimental heat capacity and enthalpy of formation of lithium cobalt oxide. J. Chem. Thermodyn. 2015, 84, 118–127. [Google Scholar] [CrossRef]
- Chang, K.; Hallstedt, B.; Music, D.; Fischer, J.; Ziebert, C.; Ulrich, S.; Seifert, H.J. Thermodynamic description of the layered O3 and O2 structural LiCoO 2-CoO2 pseudo-binary systems. Calphad Comput. Coupling Phase Diagr. Thermochem. 2013, 41, 6–15. [Google Scholar] [CrossRef]
- Jankovský, O.; Kovařík, J.; Leitner, J.; Růžička, K.; Sedmidubský, D. Thermodynamic properties of stoichiometric lithium cobaltite LiCoO2. Thermochim. Acta 2016, 634, 26–30. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LCO | |||
| hR12 | mC8 | oP8 | |
| a (Å) | 2.836 (2.816) | 5.229 | 2.721 |
| b (Å) | 2.763 | 4.411 | |
| c (Å) | 14.111 (14.054) | 5.141 | 5.572 |
| β (°) | 115.412 | ||
| V (Å3 at−1) | 8.191 (8.043) | 8.386 | 8.359 |
| LMO | |||
| hR12 | mC8 | oP8 | |
| a (Å) | 3.002 | 5.480 (5.44) | 2.851 (2.806) |
| b (Å) | 2.867 (2.80) | 4.623 (4.550) | |
| c (Å) | 14.550 | 5.387 (5.39) | 5.839 (5.747) |
| β (°) | 115.412 (116) | ||
| V (Å3 at−1) | 9.463 | 9.556 (9.224) | 9.620 (9.172) |
| LNO | |||
| hR12 | mC8 | oP8 | |
| a (Å) | 2.884 (2.879) | 5.288 (4.969) | 2.759 |
| b (Å) | 2.767 (2.877) | 4.473 | |
| c (Å) | 14.271 (14.203) | 5.199 (4.997) | 5.650 |
| β (°) | 115.412 (109.20) | ||
| V (Å3 at−1) | 8.566 (8.460) | 8.589 (8.715) | 8.716 |
| ΔcohE (eV Atom−1) | |||
|---|---|---|---|
| hR12 | mC8 | oP8 | |
| LCO | −22.361 | −22.189 | −21.690 |
| LNO | −20.521 | −20.614 | −20.519 |
| LMO | −26.173 | −26.321 | −26.337 |
| ΔE0 K (eV mol−1) | ||||
|---|---|---|---|---|
| Reaction | hR12 | mC8 | oP8 | |
| ΔE3E | CoO + Li2O → LiCoO2 + Li | 1.092 | 1.264 | 1.763 |
| ΔE4E | NiO + Li2O → LiNiO2 + Li | 2.380 | 2.287 | 2.382 |
| ΔE5E | MnO + Li2O → LiMnO2 + Li | 1.753 | 1.605 | 1.589 |
| ΔE6E | Mn2O3 + Li2O → 2 LiMnO2 | −0.771 | −1.067 | −1.099 |
| ΔfH°298 K (kJ mol−1) | ||||
|---|---|---|---|---|
| Reaction | hR12 | mC8 | oP8 | |
| LiCoO2 | Co(s) + Li(s) + O2(g) → LiCoO2(s) | −734 ± 7 | −717 ± 7 | −669 ± 7 |
| LiNiO2 | Ni(s) + Li(s) + O2(g) → LiNiO2(s) | −610 ± 7 | −619 ± 7 | −609 ± 7 |
| LiMnO2 | Mn(s) + Li(s) + O2(g) → LiMnO2(s) | −820 ± 6 | −834 ± 6 | −836 ± 6 |
| ΔfG°298 K (kJ mol−1) | ||||
|---|---|---|---|---|
| Reaction | hR12 | mC8 | oP8 | |
| LiCoO2 | Co(s) + Li(s) + O2(g) → LiCoO2(s) | −672 ± 8 | −655 ± 8 | −607 ± 8 |
| LiNiO2 | Ni(s) + Li(s) + O2(g) → LiNiO2(s) | −549 ± 8 | −557 ± 8 | −548 ± 8 |
| LiMnO2 | Mn(s) + Li(s) + O2(g) → LiMnO2(s) | −765 ± 10 | −779 ± 10 | −780 ± 10 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tuccillo, M.; Palumbo, O.; Pavone, M.; Muñoz-García, A.B.; Paolone, A.; Brutti, S. Analysis of the Phase Stability of LiMO2 Layered Oxides (M = Co, Mn, Ni). Crystals 2020, 10, 526. https://doi.org/10.3390/cryst10060526
Tuccillo M, Palumbo O, Pavone M, Muñoz-García AB, Paolone A, Brutti S. Analysis of the Phase Stability of LiMO2 Layered Oxides (M = Co, Mn, Ni). Crystals. 2020; 10(6):526. https://doi.org/10.3390/cryst10060526
Chicago/Turabian StyleTuccillo, Mariarosaria, Oriele Palumbo, Michele Pavone, Ana Belen Muñoz-García, Annalisa Paolone, and Sergio Brutti. 2020. "Analysis of the Phase Stability of LiMO2 Layered Oxides (M = Co, Mn, Ni)" Crystals 10, no. 6: 526. https://doi.org/10.3390/cryst10060526
APA StyleTuccillo, M., Palumbo, O., Pavone, M., Muñoz-García, A. B., Paolone, A., & Brutti, S. (2020). Analysis of the Phase Stability of LiMO2 Layered Oxides (M = Co, Mn, Ni). Crystals, 10(6), 526. https://doi.org/10.3390/cryst10060526