Industrial Applications of Enzymes: Recent Advances, Techniques, and Outlooks



Abstract
:1. Introduction
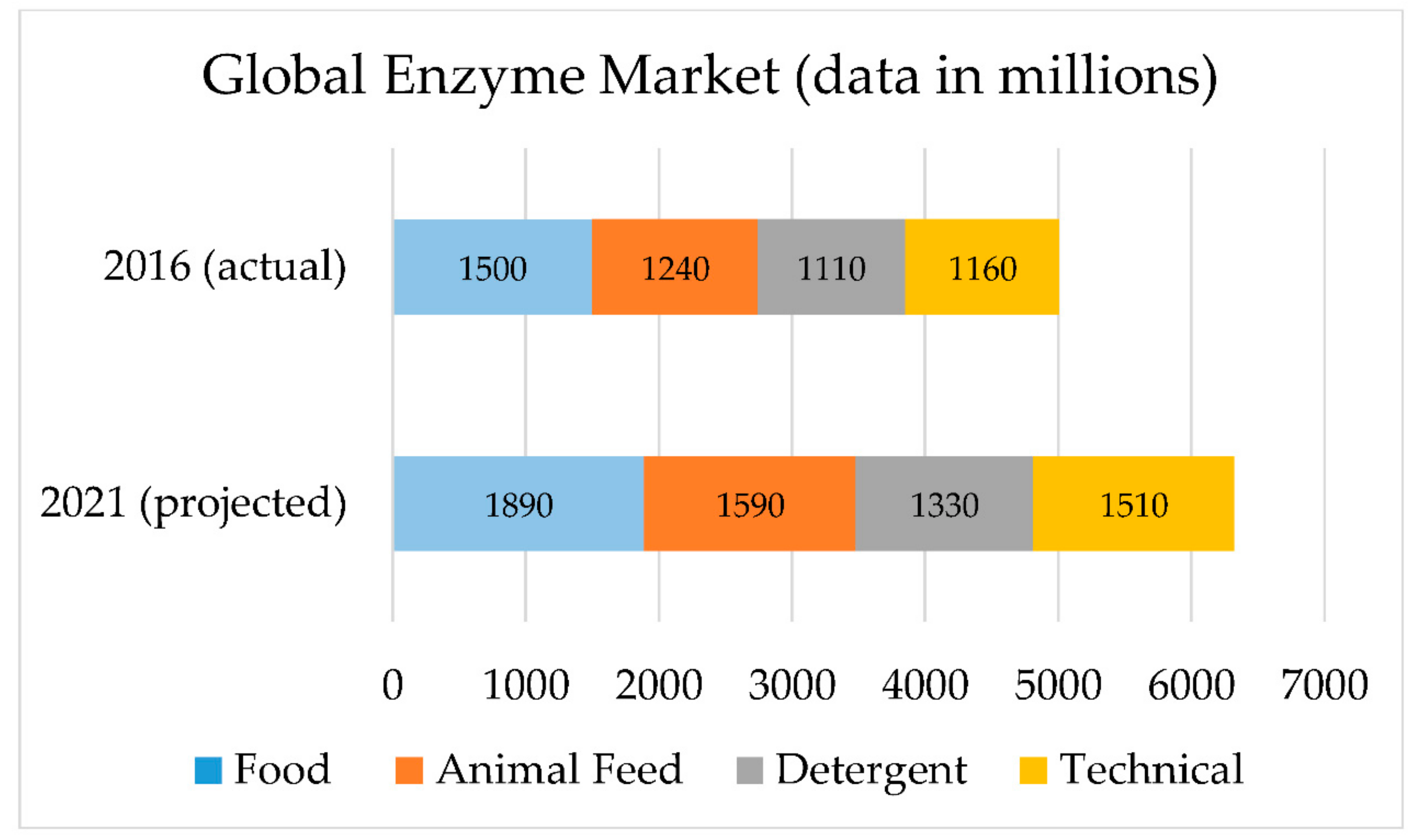
2. Enzyme Implementation: A Societal Need
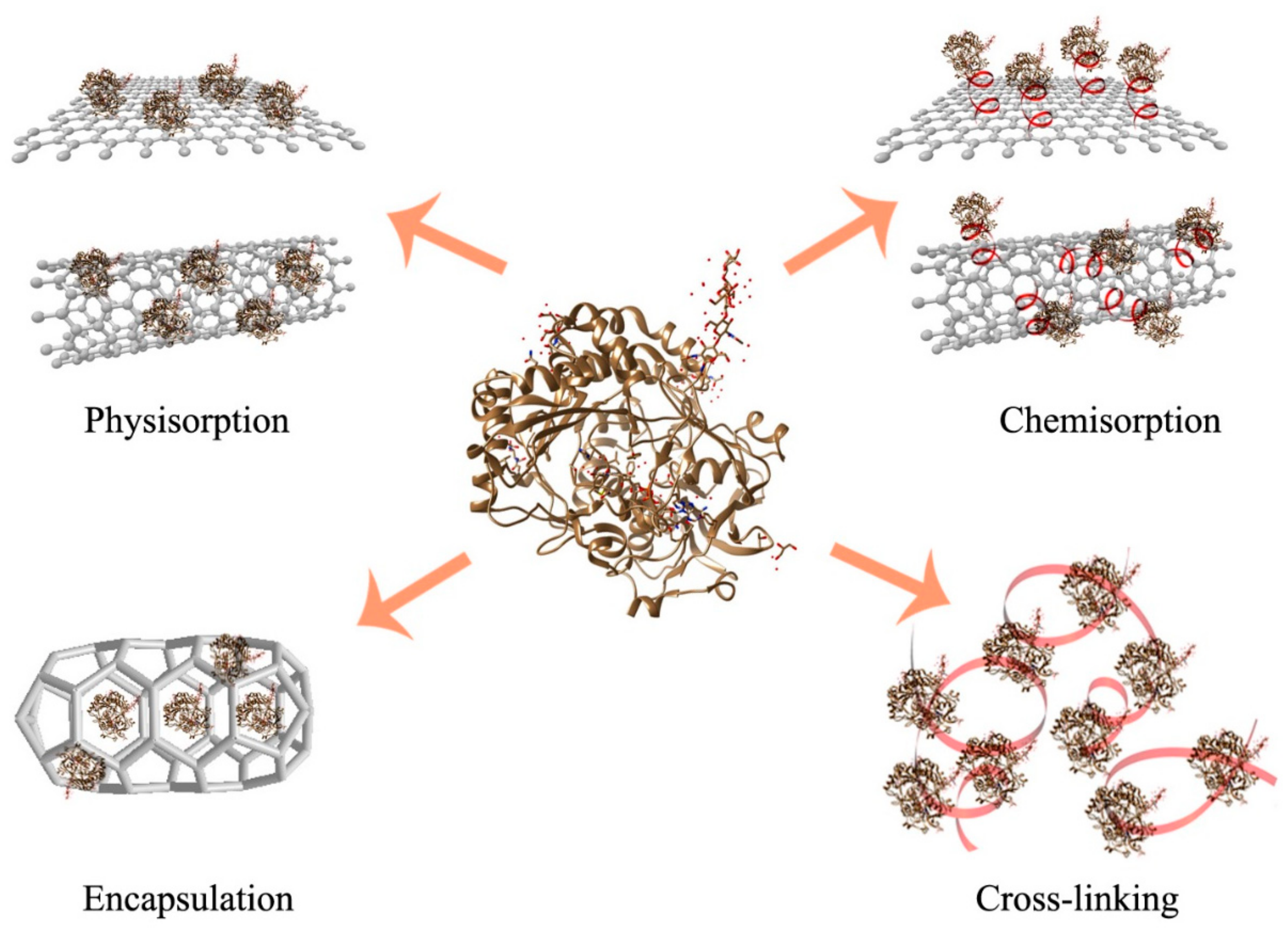
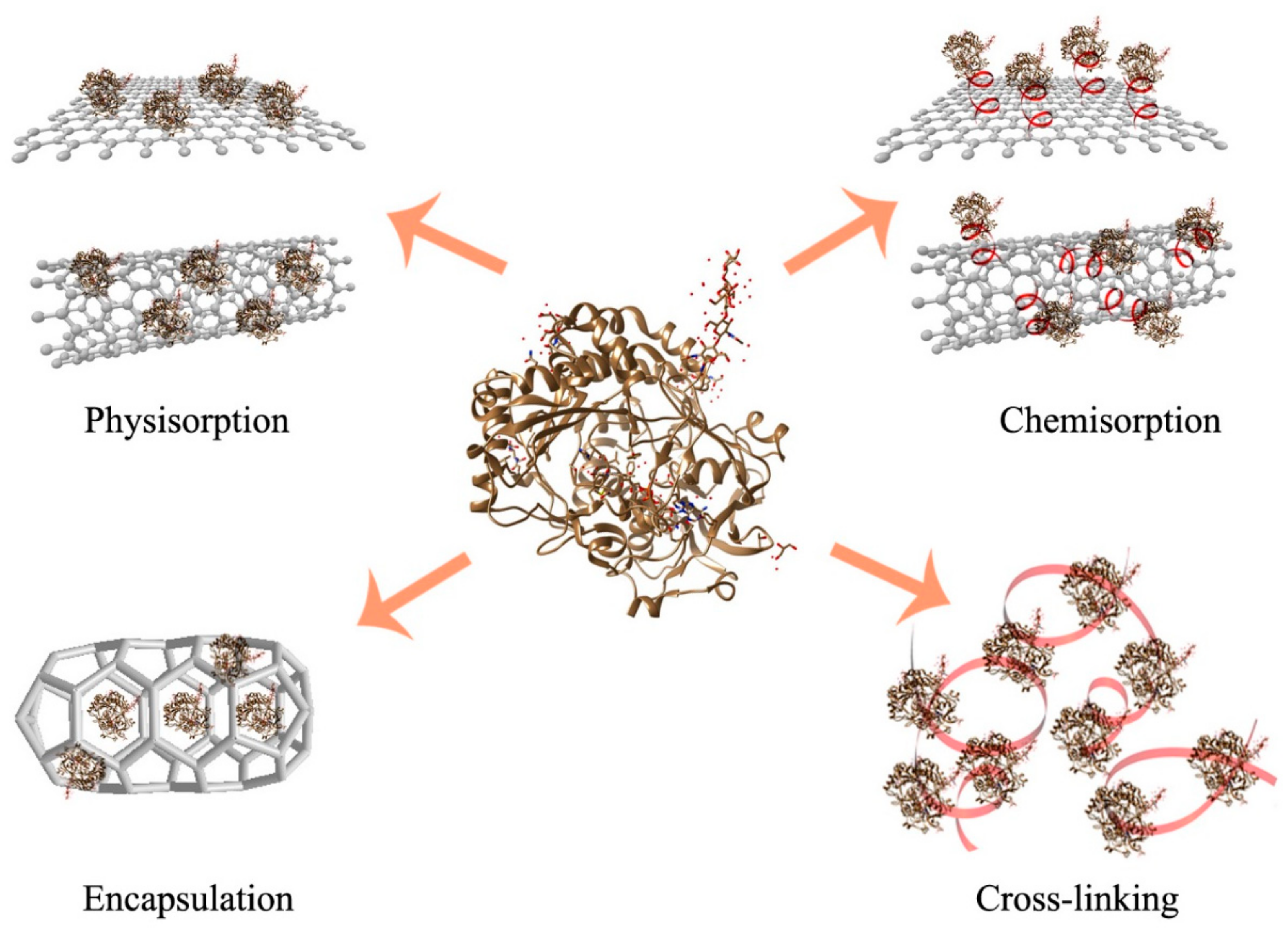
3. Enzyme Immobilization for Expanded Scope of Implementation
3.1. Carrier-Bound Enzyme Immobilization through Both Physical and Chemical Binding
3.2. Enzyme Entrapment
3.3. Cross-Linked Enzyme Aggregates
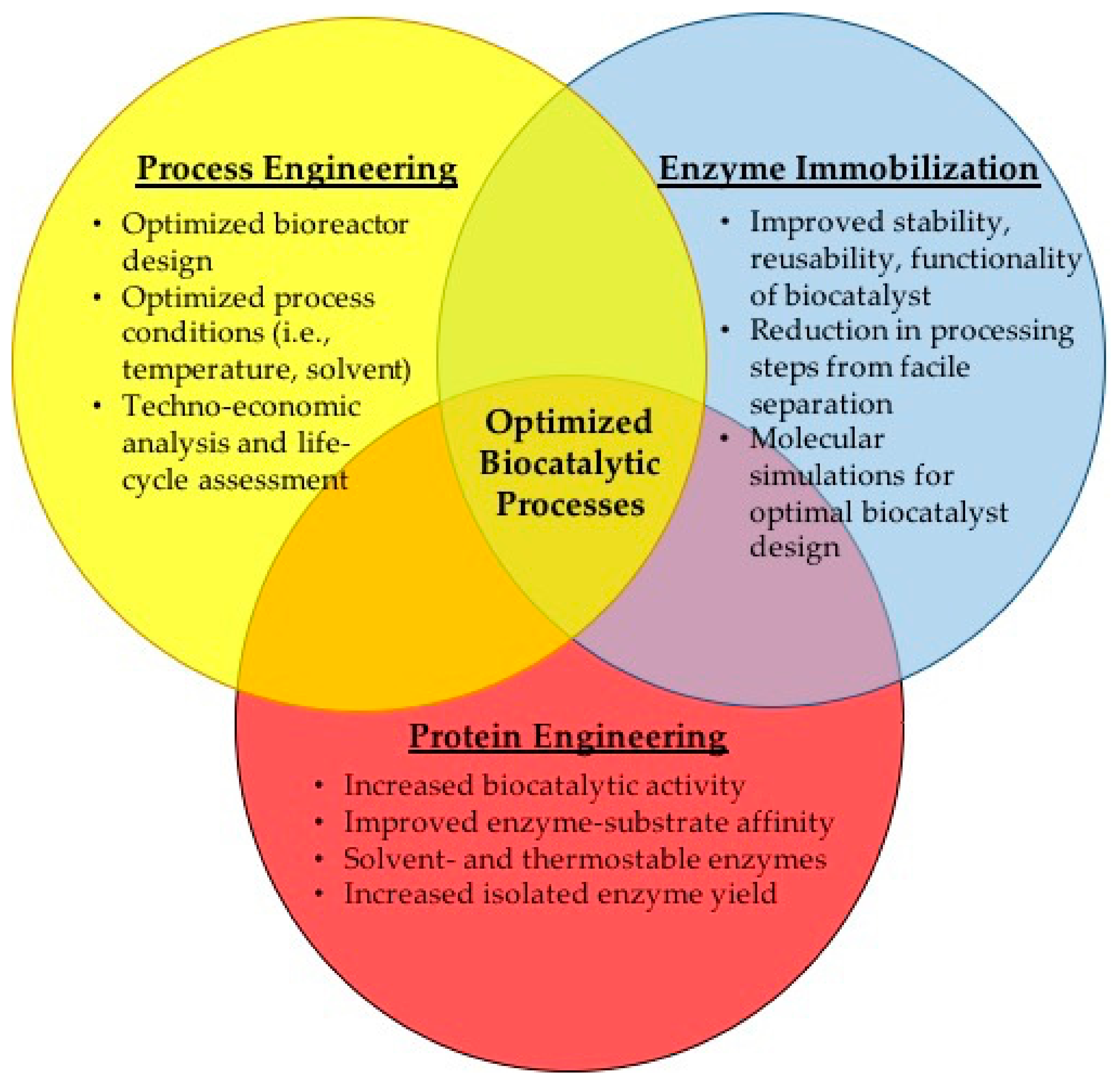
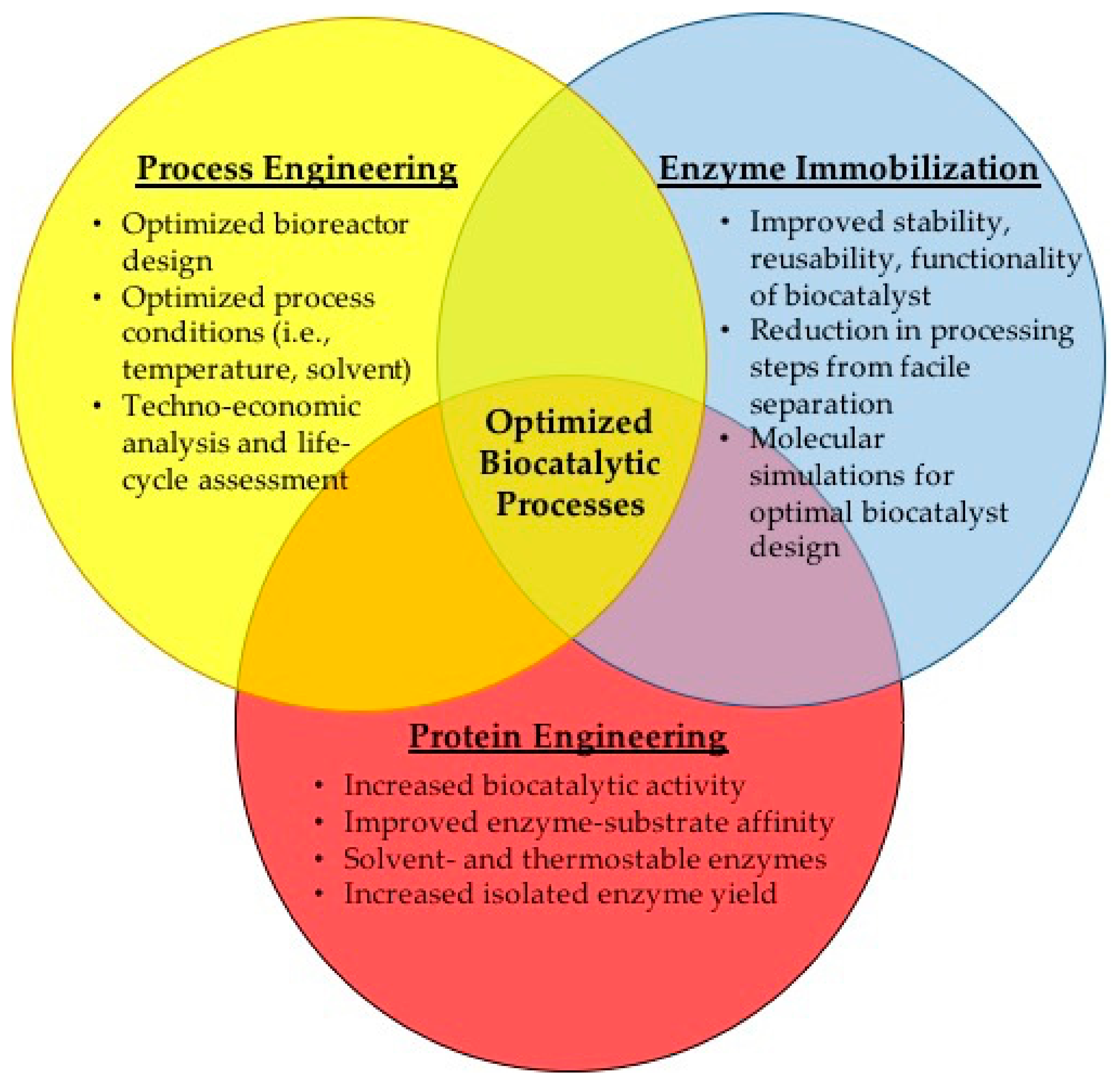
4. Intensified Approach for Designing Improved Biocatalysts
Glucose Isomerase: A Model for Enzyme Immobilization
5. Environmental Impact Assessment and Economic Approaches for Enzyme Implementation in Industrial Catalysis
6. Pertinent Examples of Enzymes Application in Industrial Catalysis
6.1. Pharmaceuticals Industry
6.2. The Food-Water-Fuel Nexus
6.3. Natural Gas Conversion
6.4. Food and Beverage Industry
6.5. Flavors and Aromas Industry
6.6. Detergents Industry
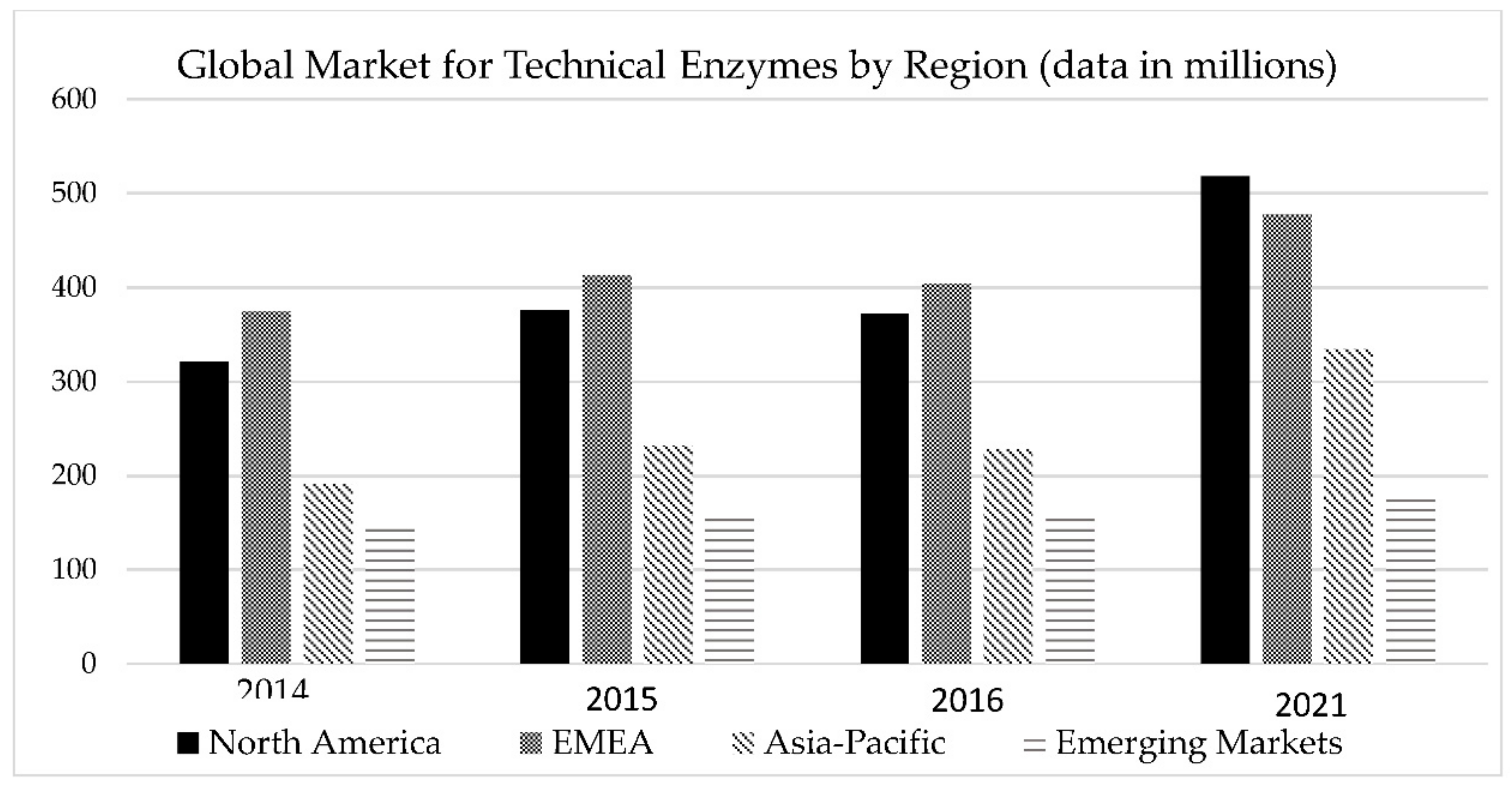
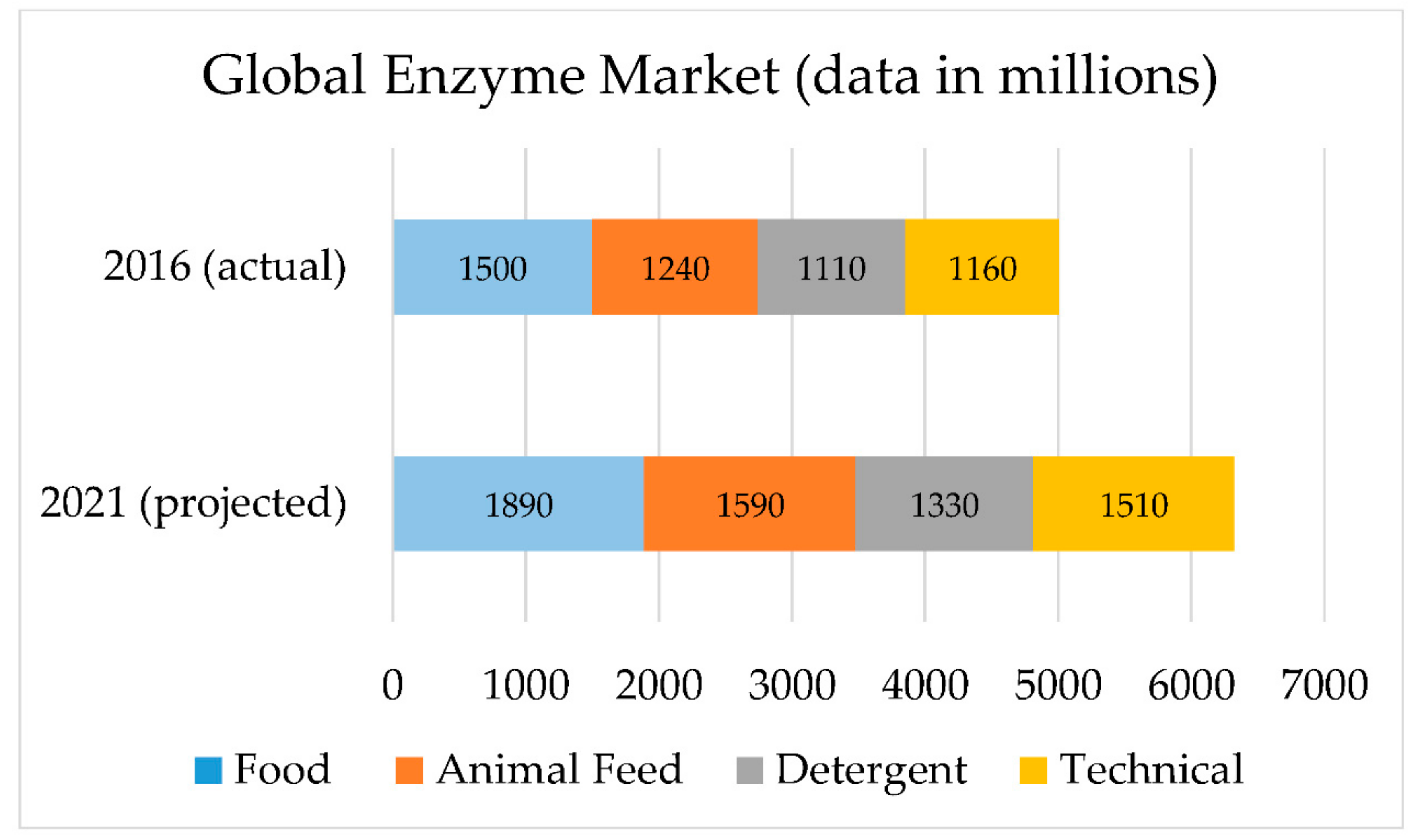
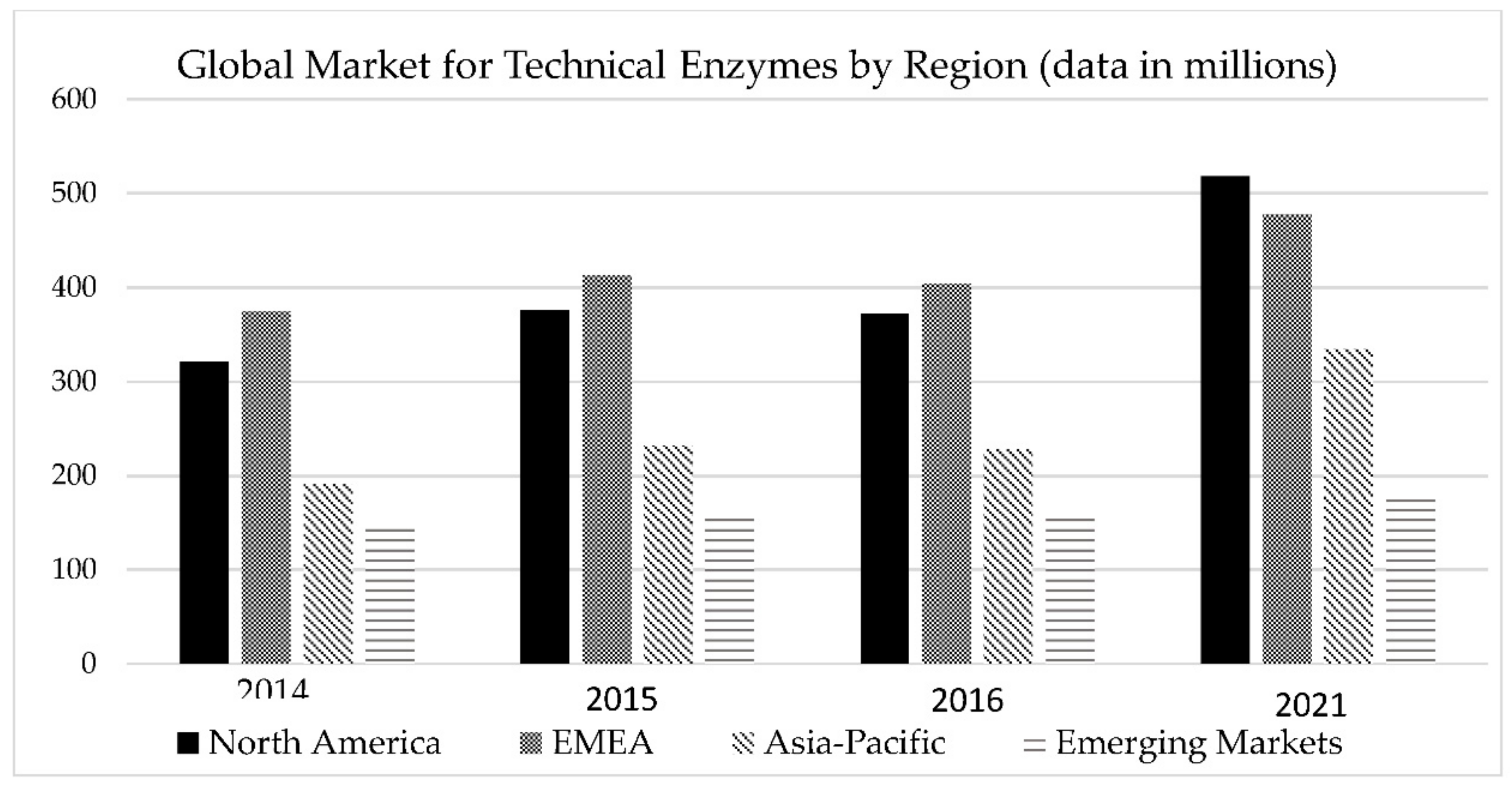
7. Projections of Economic Growth and Implementation Potential
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bommarius, A.S.; Paye, M.F. Stabilizing biocatalysts. Chem. Soc. Rev. 2013, 42, 6534–6565. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-M.; Han, S.-S.; Kim, H.-S. Industrial applications of enzyme biocatalysis: Current status and future aspects. Biotechnol. Adv. 2015, 33, 1443–1454. [Google Scholar] [CrossRef] [PubMed]
- Madhavan, A.; Sindhu, R.; Binod, P.; Sukumaran, R.K.; Pandey, A. Strategies for design of improved biocatalysts for industrial applications. Bioresour. Technol. 2017, 245, 1304–1313. [Google Scholar] [CrossRef] [PubMed]
- Roy, I.; Prasad, S. Converting Enzymes into Tools of Industrial Importance. Recent Pat. Biotechnol. 2017, 12, 33–56. [Google Scholar]
- DiCosimo, R.; McAuliffe, J.; Poulose, A.J.; Bohlmann, G. Industrial use of immobilized enzymes. Chem. Soc. Rev. 2013, 42, 6437–6474. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Zhang, H.; Ang, E.L.; Zhao, H. Biocatalysis for the synthesis of pharmaceuticals and pharmaceutical intermediates. Bioorgan. Med. Chem. 2017, 26, 1275–1284. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.N. Biocatalysis for synthesis of pharmaceuticals. Bioorganic Med. Chem. 2017, 26, 1252–1274. [Google Scholar] [CrossRef] [PubMed]
- Huisman, G.W.; Collier, S.J. On the development of new biocatalytic processes for practical pharmaceutical synthesis. Curr. Opin. Chem. Biol. 2013, 17, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Panesar, P.S.; Kumari, S.; Panesar, R. Biotechnological approaches for the production of prebiotics and their potential applications. Crit. Rev. Biotechnol. 2013, 33, 345–364. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, P. Enzymes in Food Processing: A Condensed Overview on Strategies for Better Biocatalysts. Enzyme Res. 2010. [Google Scholar] [CrossRef] [PubMed]
- Akoh, C.C.; Chang, S.-W.; Lee, G.-C.; Shaw, J.-F. Biocatalysis for the Production of Industrial Products and Functional Foods from Rice and Other Agricultural Produce. J. Agric. Food Chem. 2008, 56, 10445–10451. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, S.; Rafiq, A.; Sharma, S. Protein engineering and its applications in food industry. Crit. Rev. Food Sci. Nutr. 2017, 57, 2321–2329. [Google Scholar] [CrossRef] [PubMed]
- Pellis, A.; Cantone, S.; Ebert, C.; Gardossi, L. Evolving biocatalysis to meet bioeconomy challenges and opportunities. New Biotechnol. 2018, 40, 154–169. [Google Scholar] [CrossRef] [PubMed]
- Strong, P.J.; Kalyuzhnaya, M.; Silverman, J.; Clarke, W.P. A methanotroph-based biorefinery: Potential scenarios for generating multiple products from a single fermentation. Bioresour. Technol. 2016, 215, 314–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fei, Q.; Guarnieri, M.T.; Tao, L.; Laurens, L.M.L.; Dowe, N.; Pienkos, P.T. Bioconversion of natural gas to liquid fuel: Opportunities and challenges. Biotechnol. Adv. 2014, 32, 596–614. [Google Scholar] [CrossRef] [PubMed]
- Noraini, M.Y.; Ong, H.C.; Badrul, M.J.; Chong, W.T. A review on potential enzymatic reation for biodiesel production from algae. Renew. Sustain. Energy Rev. 2014, 39, 24–34. [Google Scholar] [CrossRef]
- Asgher, M.; Shahid, M.; Kamal, S.; Iqbal, H.M.N. Recent trends and valorization of immobilization strategies and ligninolytic enzymes by industrial biotechnology. J. Mol. Catal. B Enzym. 2014, 101, 56–66. [Google Scholar] [CrossRef]
- Liew, W.H.; Hassim, M.H.; Ng, D.K.S. Review of evolution, technology and sustainability assessments of biofuel production. J. Clean. Prod. 2014, 71, 11–29. [Google Scholar] [CrossRef]
- Cardona, C.A.; Quintero, J.A.; Paz, I.C. Production of bioethanol from sugarcane bagasse: Status and perspectives. Bioresour. Technol. 2010, 101, 4745–4766. [Google Scholar] [CrossRef] [PubMed]
- Grigoras, A.G. Catalase immobilization—A review. Biochem. Eng. J. 2017, 117, 1–20. [Google Scholar] [CrossRef]
- Cao, S.; Xu, P.; Ma, Y.; Yao, X.; Yao, Y.; Zong, M.; Li, X.; Lou, W. Recent advances in immobilized enzymes on nanocarriers. Chin. J. Catal. 2016, 37, 1814–1823. [Google Scholar] [CrossRef]
- Ansari, S.A.; Husain, Q. Potential applications of enzymes immobilized on/in nano materials: A review. Biotechnol. Adv. 2012, 30, 512–523. [Google Scholar] [CrossRef] [PubMed]
- Misson, M.; Zhang, H.; Jin, B. Nanobiocatalyst advancements and bioprocessing applications. Interface 2015, 12, 20140891. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A. Enzyme Immobilization: The Quest for Optimum Performance. Adv. Synth. Catal. 2007, 349, 1289–1307. [Google Scholar] [CrossRef]
- Mehta, J.; Bhardwaj, N.; Bhardwaj, S.K.; Kim, K.-H.; Deep, A. Recent advances in enzyme immobilization techniques: Metal-organic frameworks as novel substrates. Coord. Chem. Rev. 2016, 322, 30–40. [Google Scholar] [CrossRef]
- Cipolatti, E.P.; Valerio, A.; Henriques, R.O.; Moritz, D.E.; Ninow, J.L.; Freire, D.M.G.; Manoel, E.A.; Fernandez-Lafuente, R.; Oliveira, D.D. Nanomaterials for biocatalyst immobilization—State of the art and future trends. RSC Adv. 2016, 6, 104675–104692. [Google Scholar] [CrossRef]
- Bezerra, C.S.; Lemos, C.M.G.D.F.; Sousa, M.D.; Goncalves, L.R.B. Enzyme immobilization onto renewable polymeric matrixes: Past, present and future trends. J. Appl. Polym. Sci. 2015, 132. [Google Scholar] [CrossRef]
- Tracewell, C.A.; Arnold, F.H. Directed enzyme evolution: Climbing fitness peaks on amino acid at a time. Curr. Opin. Chem. Biol. 2009, 13, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Chao, F.-A.; Morelli, A.J.C.H., III; Churchfield, L.; Hagmann, L.N.; Shi, L.; Masterson, L.R.; Sarangi, R.; Veglia, G.; Seelig, B. Structure and dynamics of a primordial catalytic fold generated by in vitro evolution. Nat. Chem. Biol. 2013, 9, 81–83. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Liang, J.; Ambrogelly, A.; Brennan, T.; Gloor, G.; Huisman, G.; Lalonde, J.; Lekhal, A.; Mijts, B.; Muley, S.; et al. Efficient, chemoenzymatic process for manufacture of the boceprevir bicyclic [3.1.0]proline intermediate based on amine oxidase-catalyzed desymmetrization. J. Am. Chem. Soc. 2012, 134, 6467–6472. [Google Scholar] [CrossRef] [PubMed]
- Desai, A.A. Sitagliptin manufacture: A compelling tale of green chemistry, process intensification, and industrial asymmetric catalysis. Angew. Chem. Int. Ed. 2011, 50, 1974–1976. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, A.; Almeida, L.; Silva, A.P.; Fontes-Ribeiro, C.; Ambrosio, A.F.; Cristovao, A.; Fernandes, R. The dipeptidyl peptidase-4 (dpp-4) inhibitor sitagliptin ameliorates retinal endothelial cell dysfunction triggered by inflammation. Biomed. Pharmacother. 2018, 102, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Savile, C.K. Biocatalytic asymmetric synthesis of chiral amines from ketones applied to sitagliptin manufacture. Science 2010, 329, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Kjellin, M.; Wesslen, T.; Lofblad, E.; Lennerstrand, J.; Lannergard, A. The effect of the first-generation hcv-protease inhibitors boceprevir and telaprevir and the relation to baseline ns3 resistance mutations in genotype 1: Experience from a small swedish cohort. Upsala J. Med. Sci. 2018, 123, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Krell, H.V.; Leuchter, A.F.; Cook, I.A.; Abrams, M. Evaluation of reboxetine, a noradrenergic antidepressant, for the treatment of fibromyalgia and chronic low back pain. Psychosomatics 2005, 46, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Hayes, S.T.; Assaf, G.; Checksfield, G.; Cheung, C.; Critcher, D.; Harris, L.; Howard, R.; Mathew, S.; Regius, C.; Scotney, G.; et al. Commercial synthesis of (s,s0-reboxetine succinate: A journey to find the cheapest commercial chemistry for manufacture. Org. Process Res. Dev. 2011, 15, 1305–1314. [Google Scholar] [CrossRef]
- Martinez, C.A.; Hu, S.; Dumond, Y.; Tao, J.; Kelleher, P.; Tully, L. Development of a Chemoenzymatic Manufacturing Process for Pregabalin. Org. Process Res. Dev. 2008, 12, 392–398. [Google Scholar] [CrossRef]
- Marouf, H.M. Effect of Pregabalin Premedication on Emergence Agitation in Children after Sevoflurane Anesthesia: A Randomized Controlled Study. Anesth. Essays Res. 2018, 12, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Elavarasan, K.; Shamasundar, B.A. Effect of oven drying and freeze drying on the antioxidant and functional properties of protein hydrosylates derived from freshwater fish (Cirrhinus mrigala) using papain enzyme. J. Food Sci. Technol. 2016, 53, 1303–1311. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.-C.; Yang, S.-J.; Hong, D.; Yang, J.-P.; Liu, M.; Lin, Y.; Huang, C.-H.; Wang, C.-J. A simple and convenient method for the preparation of antioxidant peptides from walnut (Juglans regia L.) protein hydrosylates. Chem. Cent. J. 2016, 10, 39. [Google Scholar] [CrossRef] [PubMed]
- Vosmann, K.; Wiege, B.; Weitkamp, P.; Weber, N. Preparation of lipophilic alkyl (hydroxy)benzoates by solvent-free lipase-catalyzed esterification and transesterification. Appl. Microbiol. Biotechnol. 2008, 80, 29–36. [Google Scholar] [CrossRef] [PubMed]
- SA, A.G.A.; Meneses, A.C.D.; Araujo, P.H.H.D.; Oliveira, D.D. A review on enzymatic synthesis of aromatic esters used as flavor ingredients for food, cosmetics an pharmaceutical industries. Trends Food Sci. Technol. 2017, 69, 95–105. [Google Scholar] [CrossRef]
- Badgujar, K.C.; Pai, P.A.; Bhanage, B.M. Enhanced biocatalytic activity of immobilized Pseudomonas cepacia lipase under sonicated condition. Bioprocess Biosyst. Eng. 2016, 39, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Dewan, S.S. Global Markets for Enzymes in Industrial Applications; BCC Research: Wellesly, MA, USA, 2017. [Google Scholar]
- Kumar, D.; Savitri; Thakur, N.; Verma, R.; Bhalla, T.C. Microbial Proteases and Application as Laundry Detergent Additive. Res. J. Microbiol. 2008, 3, 661–672. [Google Scholar] [CrossRef]
- Alfa, M.J.; Jackson, M. A new hydrogen peroxide-based medical-device detergent with germicidal properties: Comparison with enzymatic cleaners. AJIC 2001, 29, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Kumar, M.; Mittal, A.; Mehta, P.K. Microbial enzymes: Industrial progress in 21st century. 3 Biotech 2016, 6, 174. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Yang, X.; Yang, S.; Zhu, M.; Wang, X. Technology Prospecting on Enzymes: Application, Marketing and Engineering. Comput. Struct. Biotechnol. J. 2012, 2, e201209017. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Weng, Y.; Xu, H.; Mao, Z. Enzyme immobilization for biodiesel production. Appl. Microbial. Biotechnol. 2012, 93, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Harding, K.G.; Dennis, J.S.; Blottnitz, H.V.; Harrison, S.T.L. A life-cycle comparison between inorganic and biological catalysis for the production of biodiesel. J. Clean. Prod. 2007, 16, 1368–1378. [Google Scholar] [CrossRef]
- Raman, J.K.; Ting, V.F.W.; Pogaku, R. Life cycle assessment of biodiesel production using alkali, soluble and immobilized enzyme catalyst processes. Biomass Bioenergy 2011, 35, 4221–4229. [Google Scholar] [CrossRef]
- Olofsson, J.; Barta, Z.; Borjesson, P.; Wallberg, O. Integrating enzyme fermentation in lignocellulosic ethanol production: Life-cycle assessment and techno-economic analysis. Biotechnol. Biofuels 2017, 10, 51. [Google Scholar] [CrossRef] [PubMed]
- Yue, F.; Jian-xin, J.; Li-wei, Z. Recent developments in activities, utilization and sources of cellulase. For. Stud. China 2009, 11, 202–207. [Google Scholar]
- Khorshidi, K.J.; Lenjannezhadian, H.; Jamalan, M.; Zeinali, M. Preparation and characterization of nanomagnetic cross-linked cellulase aggregates for cellulose bioconversion. J. Chem. Technol. Biotechnol. 2016, 91, 539–546. [Google Scholar] [CrossRef]
- Taherzadeh, M.J.; Karimi, K. Enzyme-based hydrolysis processes for ethanol from lignocellulosic materials: A review. BioResources 2007, 2, 707–738. [Google Scholar]
- Lima, J.S.; Araujo, P.H.H.; Sayer, C.; Souza, A.A.U.; Viegas, A.C.; Oliveira, D.D. Cellulase immobilization on magnetic nanoparticles encapsulated in polymer nanospheres. Bioprocess Biosyst. Eng. 2017, 40, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.; Lu, J.; Nie, K.; Deng, L.; Wang, F. Biodiesel production with immobilized lipase: A review. Biotechnol. Adv. 2010, 28, 628–634. [Google Scholar] [CrossRef] [PubMed]
- Bhat, M.K. Cellulases and related enzymes in biotechnology. Biotechnol. Adv. 2000, 18, 355–383. [Google Scholar] [CrossRef] [Green Version]
- Kuhad, R.C.; Gupta, R.; Singh, A. Microbial Cellulases and Their Industrial Applications. Enzyme Res. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Hasan, F.; Shah, A.A.; Hameed, A. Industrial applications of microbial lipases. Enzyme Microb. Technol. 2006, 39, 235–251. [Google Scholar] [CrossRef]
- Bajpai, P. Application of Enzymes in the Pulp and Paper Industry. Biotechnol. Prog. 1999, 15, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A. Characteristic features and biotechnological applications of cross-linked enzyme aggregates (cleas). Appl. Microbial. Biotechnol. 2011, 92, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Jesionowski, T.; Zdarta, J.; Krajewska, B. Enzyme immobilization by adsorption: A review. Adsorption 2014, 20, 801–821. [Google Scholar] [CrossRef]
- Betancor, L.; Luckarift, H.R. Bioinspired enzyme encapsulation for biocatalysis. Trends Biotechnol. 2008, 26, 566–572. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Ji, P. Enzymes immobilized on carbon nanotubes. Biotechnol. Adv. 2011, 29, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, G.S. Evaluation of nanogels as supports for enzyme immobilization. Polym. Int. 2014, 63, 1889–1894. [Google Scholar] [CrossRef]
- Kommoju, P.R.; Chen, Z.W.; Bruckner, R.C.; Mathews, F.S.; Jorns, M.S. Probing oxygen activation sites in two flavoprotein oxidases using chloride as an oxygen surrogate. Biochemistry 2011, 50, 5521–5534. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Quan, J.; Branford-White, C.; Williams, G.R.; Wu, J.-X.; Zhu, L.-M. Electrospun polyacrylonitrile-glycopolymer nanofibrous membranes for enzyme immobilization. J. Mol. Catal. B Enzym. 2012, 76, 15–22. [Google Scholar] [CrossRef]
- Kumar, D.; Nagar, S.; Bhushan, I.; Kumar, L.; Parshad, R. Covalent immobilization of organic solvent tolerant lipase on aluminum oxide pellets and its potential application in esterification reaction. J. Mol. Catal. B Enzym. 2013, 87, 51–61. [Google Scholar] [CrossRef]
- Tran, D.-T.; Chen, C.-L.; Chang, J.-S. Immobilization of Burkholderia sp. lipase on ferric silica nanocomposite for biodiesel production. J. Biotechnol. 2012, 158, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Premaratne, G.; Nerimetla, R.; Matlock, R.; Sunday, L.; Koralege, R.S.H.; Ramsey, J.D.; Krishnan, S. Stability, scalability, and reusability of a volume efficient biocatalytic system constructed on magnetic nanoparticles. Catal. Sci. Technol. 2016, 6, 2361–2369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Luo, S.; Chen, W. Activity of catalase adsorbed to carbon nanotubes: Effects of carbon nanotube surface properties. Talanta 2013, 113, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.V.; Prevost, N.T.; Condon, B.; French, A.; Wu, Q. Immobilization of lysozyme-cellulose amide-linked conjugates on cellulose I and II cotton nanocrystalline preparations. Cellulose 2012, 19, 495–506. [Google Scholar] [CrossRef]
- Hong, J.; Xu, D.; Gong, P.; Yu, J.; Ma, H.; Yao, S. Covalent-bonded immobilization of enzyme on hydrophilic polymer covering magnetic nanogels. Microporous Mesoporous Mater. 2008, 109, 470–477. [Google Scholar] [CrossRef]
- Zhu, J.; Sun, G. Lipase immobilization on glutaraldehyde-activated nanofibrous membranes for improved enzyme stabilities and activities. React. Funct. Polym. 2012, 72, 839–845. [Google Scholar] [CrossRef]
- Ottone, C.; Bernal, C.; Serna, N.; Illanes, A.; Wilson, L. Enhanced long-chain fatty alcohol oxidation by immobilization of alcohol dehydrogenase from S. cerevisiae. Appl. Microbiol. Biotechnol. 2017, 102, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.-H.; Chen, G.-J.; Chen, C.-I.; Liu, Y.-C.; Shieh, C.-J. Kinetics and optimization of lipase-catalyzed synthesis of rose fragrance 2-phenylethyl acetate through transesterification. Process Biochem. 2014, 49, 437–444. [Google Scholar] [CrossRef]
- Tomke, P.D.; Rathod, V.K. Ultrasound assisted lipase catalyzed synthesis of cinnamyl acetate via transesterification reaction in a solvent free medium. Ultrason. Sonochemistry 2015, 27, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, A.M.; Pedroni, V.; Volpe, M.; Ferreira, M.L. Immobilization of catalase from Aspergillus niger on inorganic and biopolymeric supports for H2O2 decomposition. Appl. Catal. B Environ. 2004, 47, 153–163. [Google Scholar] [CrossRef]
- Homaei, A.A.; Sajedi, R.H.; Sariri, R.; Seyfzadeh, S.; Stevanato, R. Cysteine enhances activity and stability of immobilized papain. Amino Acids 2010, 38, 937–942. [Google Scholar] [CrossRef] [PubMed]
- Dincer, A.; Telefoncu, A. Improving the stability of cellulase by immobilization on modified polyvinyl alcohol coated chitosan beads. J. Mol. Catal. B Enzym. 2007, 45, 10–14. [Google Scholar] [CrossRef]
- Lopez-Gallego, F.; Betancor, L.; Hidalgo, A.; Dellamora-Ortiz, G.; Mateo, C.; Fernandez-Lafuente, R.; Guisan, J.M. Stabilization of different alcohol oxidases via immobilization and post immobilization techniques. Enzyme Microb. Technol. 2007, 40, 278–284. [Google Scholar] [CrossRef]
- Falus, P.; Cerioli, L.; Bajnoczi, G.; Boros, Z.; Weiser, D.; Nagy, J.; Tessaro, D.; Servi, S.; Poppe, L. A Continuous-Flow Cascade Reactor System for Subtilisin A-Catalyzed Dynamic Kinetic Resolution of N-tert-butyloxy-carbonylphenylalanine Ethyl Thioester with Benzylamine. Adv. Synth. Catal. 2016, 358, 1608–1617. [Google Scholar] [CrossRef]
- Knezevic, Z.; Milosavic, N.; Bezbradica, D.; Jakovljevic, Z.; Prodanovic, R. Immobilization of lipase from Candida rugosa on Eupergit C supports by covalent attachment. Biochem. Eng. J. 2006, 30, 269–278. [Google Scholar] [CrossRef]
- Bolivar, J.M.; Nidetzky, B. Positively Charged Mini-Protein Zbasic2 as a Highly Efficient Silica Binding Module: Opportunities for Enzyme Immobilization on Unmodified Silica Supports. Langmuir 2012, 28, 10040–10049. [Google Scholar] [CrossRef] [PubMed]
- Bolivar, J.M.; Nidetzky, B. Smart enzyme immobilization in microstructured reactors. Microreactors 2013, 31, 50–54. [Google Scholar]
- Zhao, X.; Qi, F.; Yuan, C.; Du, W.; Liu, D. Lipase-catalyzed process for biodiesel production: Enzyme immobilization, process simulation and optimization. Renew. Sustain. Energy Rev. 2015, 44, 182–197. [Google Scholar] [CrossRef]
- Mendes, A.A.; Freitas, L.; Carvalho, A.K.F.D.; Oliveira, P.C.D.; Castro, H.F.D. Immobilization of a Commercial Lipase from Penicillium camembertii (Lipase G) by Different Strategies. Enzyme Res. 2011. [Google Scholar] [CrossRef] [PubMed]
- Petri, A.; Marconcini, P.; Salvadori, P. Efficient immobilization of epoxide hydrolase onto silica gel and use in the enantioselective hydrolysis of racemic para-nitrostyrene oxide. J. Mol. Catal. B Enzym. 2005, 32, 219–224. [Google Scholar] [CrossRef]
- Alptekin, O.; Tukel, S.S.; Yildirim, D.; Alagoz, D. Immobilization of catalase onto Eupergit C and its characterization. J. Mol. Catal. B Enzym. 2010, 64, 177–183. [Google Scholar] [CrossRef]
- Ferraz, L.I.R.; Possebom, G.; Alvez, E.V.; Cansian, R.L.; Paroul, N.; Oliveira, D.D.; Treichel, H. Application of home-made lipase in the production of geranyl propionate by esterification of geraniol and propionic acid in solvent-free system. Biocatal. Agric. Biotechnol. 2015, 4, 44–48. [Google Scholar] [CrossRef]
- Risso, F.V.A.; Mazutti, M.A.; Treichel, H.; Costa, F.; Maugeri, F.; Rodrigues, M.I. Effect of Organic Solvent on the Characteristics of Free and Immobilized Inulinase from Kluyveromyces marxianus ATCC 16045. Food Technol. Biotechnol. 2010, 48, 143–150. [Google Scholar]
- Arica, M.Y.; Oktem, H.A.; Oktem, Z.; Tuncel, S.A. Immobilization of catalase in poly(isopropylacrylamide-co-hydroxythylmethacrylate) thermally reversible hydrogels. Polym. Int. 1999, 48, 879–884. [Google Scholar] [CrossRef]
- Singh, H.P.; Gupta, N.; Sharma, R.K. Hollow Silica Nanoparticles as Support for Catalase Enzyme Immobilization. Catal. Lett. 2013, 143, 1304–1311. [Google Scholar] [CrossRef]
- Yan, M.; Liu, Z.; Lu, D.; Liu, Z. Fabrication of Single Carbonic Anhydrase Nanogel against Denaturation and Aggregation at High Temperature. Biomacromolecules 2007, 8, 560–565. [Google Scholar] [CrossRef] [PubMed]
- Prakasham, R.S.; Devi, G.S.; Rao, C.S.; Sivakumar, V.S.S.; Sathish, T.; Sarma, P.N. Nickel-Impregnated Silica Nanoparticle Synthesis and Their Evaluation for Biocatalyst Immobilization. Appl. Biochem. Biotechnol. 2010, 160, 1888–1895. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Ge, J.; Yang, C.; Hou, M.; Liu, Z. Facile synthesis of multiple enzyme-containing metal-organic frameworks in a biomolecule friendly environment. Chem. Commun. 2015, 51, 13408–13411. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Xiao, Y.; Wang, L.; Yin, Y.; Zheng, J.; Yang, H.; Chen, G. Facile synthesis of enzyme-inorganic hybrid nanoflowers and their application as an immobilized trypsin reactor for highly efficient protein digestion. RSC Adv. 2014, 4, 13888–13891. [Google Scholar] [CrossRef]
- Cao, L.; Rantwijk, F.V.; Sheldon, R.A. Cross-Linked Enzyme Aggregates: A Simple and Effective Method for the Immobilization of Penicillin Acylase. Org. Lett. 2000, 2, 1361–1364. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A. Cross-Linked Enzyme Aggregates as Industrial Biocatalysts. Org. Process Res. Dev. 2011, 15, 213–223. [Google Scholar] [CrossRef]
- Illanes, A.; Wilson, L.; Altamirano, C.; Cabrera, Z.; Alvarez, L.; Aguirre, C. Production of cephalexin in organic medium at high substrate concentrations with CLEA of penicillin acylase and PGA-450. Enzyme Microb. Technol. 2007, 40, 195–203. [Google Scholar] [CrossRef]
- Vafiadi, C.; Topakas, E.; Christakopoulos, P. Preparation of multipurpose cross-linked enzyme aggregates and their application to production of alkyl ferulates. J. Mol. Catal. B Enzym. 2008, 54, 35–41. [Google Scholar] [CrossRef]
- Zhao, L.; Zheng, L.; Gao, G.; Jia, F.; Cao, S. Resolution of N-(2-ethyl-6-methyl-phenyl) alanine via cross-linked aggregates of Pseudomonas sp. Lipase. J. Mol. Catal. B Enzym. 2008, 54, 7–12. [Google Scholar] [CrossRef]
- Martins, S.L.; Albuquerque, B.F.; Nunes, M.A.P.; Ribeiro, M.H.L. Exploring magnetic and imprinted cross-linked enzyme aggregates of rhamnopyranosidase in microbioreactors. Bioresour. Technol. 2018, 249, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Yang, K.-L. Combined cross-linked enzyme aggregates of horseradish peroxidase and glucose oxidase for catalyzing cascade chemical reactions. Enzyme Microb. Technol. 2017, 100, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.-Q.; Hu, Z.-L.; Sheldon, R.A.; Yang, Z. Catalytic performance of cross-linked enzyme aggregates of Penicillium expansum lipase and their use as a catalyst for biodiesel production. Process Biochem. 2012, 47, 2058–2063. [Google Scholar] [CrossRef]
- Nuijens, T.; Cusan, C.; Schepers, A.C.H.M.; Kruijtzer, J.A.W.; Rijkers, D.T.S.; Liskamp, R.M.J.; Quaedflieg, P.J.L.M. Enzymatic synthesis of activated esters and their subsequent use in enzyme-based peptide synthesis. J. Mol. Catal. B Enzym. 2011, 71, 79–84. [Google Scholar] [CrossRef]
- Cui, J.-D.; Zhang, S.; Sun, L.-M. Cross-Linked Enzyme Aggregates of Phenylalanine Ammonia Lyase: Novel Biocatalysts for Synthesis of L-Phenylalanine. Appl. Biochem. Biotechnol. 2012, 167, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Taboada-Puig, R.; Junghanns, C.; Demarche, P.; Moreira, M.T.; Feijoo, G.; Lema, J.M.; Agathos, S.N. Combined cross-linked enzyme aggregates from versatile peroxidase and glucose oxidase: Production, partial characterization and application for the elimination of endocrine disruptors. Bioresour. Technol. 2011, 102, 6593–6599. [Google Scholar] [CrossRef] [PubMed]
- Thirkettle, J. SB-253514 and Analogues; Novel Inhibitors of Lipoprotein Associated Phospholipase A2 Produced by Pseudomonas fluorescens DSM 11579. J. Antibiot. 2000, 53, 733–735. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, P.J.; Harms, C.T.; Allen, J.R.F. Metolachlor, S-metolachlor and their role within sustainable weed management. Crop Prot. 1998, 17, 207–212. [Google Scholar] [CrossRef]
- Dinu, C.Z.; Borkar, I.V.; Bale, S.S.; Campbell, A.S.; Kane, R.S.; Dordick, J.S. Perhydrolase-nanotube-paint sporicidal composites stabilized by intramolecular crosslinking. J. Mol. Catal. B Enzym. 2012, 75, 20–26. [Google Scholar] [CrossRef]
- Ren, T.; Mao, Z.; Moya, S.E.; Gao, C. Immobilization of Enzymes on 2-Hydroxyethylm Methacrylate and Glycidyl Methacrylate Copolymer Brushes. Chem. Asian. J. 2014, 9, 2132–2139. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, M.; Segura, R.L.; Abian, O.; Betancor, L.; Hidalgo, A.; Mateo, C.; Fernandez-Lafuente, R.; Guisan, J.M. Determination of protein-protein interactions through aldehyde-dextran intermolecular cross-linking. Proteomics 2004, 4, 2602–2607. [Google Scholar] [CrossRef] [PubMed]
- Liese, A.; Hilterhaus, L. Evaluation of immobilized enzymes for industrial applications. Chem. Soc. Rev. 2013, 42, 6236–6249. [Google Scholar] [CrossRef] [PubMed]
- Dach, R.; Song, J.J.; Roschangar, F.; Samstag, W.; Senanayake, C.H. The Eight Criteria Defining a Good Chemical Manufacturing Process. Org. Process Res. Dev. 2012, 16, 1697–1706. [Google Scholar] [CrossRef]
- Zhou, H.; Qu, Y.; Chunlei; Kong; Li, D.; Shen, E.; Ma, Q.; Zhang, X.; Wang, J.; Zhou, J. Catalytic performance and molecular dynamic simulation of immobilized C-C bond hydrolase based on carbon nanotube matrix. Colloids Surfaces B Biointerfaces 2014, 116, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Basso, A.; Spizzo, P.; Ferrario, V.; Knapic, L.; Savko, N.; Braiuca, P.; Ebert, C.; Ricca, E.; Calabro, V.; Gardossi, L. Endo- and Exo-Inulinases: Enzyme-Substrate Interaction and Rational Immobilization. Biotechnol. Prog. 2010, 26, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Franca, E.F.; Leite, F.L.; Cunha, R.A.; Oliveira, O.N., Jr.; Freitas, L.C.G. Designing and enzyme-based nanobiosensor using molecular modeling techniques. Phys. Chem. Chem. Phys. 2011, 13, 8894–8899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathpati, A.C.; Vyas, V.K.; Bhanage, B.M. Kinetic resolution of 1,2-diols using immobilized Burkholderia cepacia lipase: A combined experimental and molecular dynamics investigation. J. Biotechnol. 2017, 262, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.P.; Teldesley, D.J. Computer Simulations of Liquids; Oxford University Press: New York, NY, USA, 1987. [Google Scholar]
- Maarel, M.J.E.C.V.D.; Veen, B.V.D.; Uitdehaag, J.C.M.; Leemhuis, H.; Dijkhuizen, L. Properties and applications of starch-converting enzymes of the alpha-amylase family. J. Biotechnol. 2002, 94, 137–155. [Google Scholar] [CrossRef]
- Rasor, J.P.; Voss, E. Enzyme-catalyzed processes in pharmaceutical industry. Appl. Catal. A Gen. 2001, 221, 145–158. [Google Scholar] [CrossRef]
- Petroleum, British. BP Statistical Review of World Energy; Petroleum, British: London, UK, 2017. [Google Scholar]
- Qiu, C.; Colson, G.; Escalante, C.; Wetzstein, M. Considering macroeconomic indicators in the food before fuel nexus. Energy Econ. 2012, 34, 2021–2028. [Google Scholar] [CrossRef]
- Rabideau, B.D.; Ismail, A.E. Mechanisms of hydrogen bond formation between ionic liquids and cellulose and the influence of water content. Phys. Chem. Chem. Phys. 2015, 17, 5767–5775. [Google Scholar] [CrossRef] [PubMed]
- Viell, J.; Inouye, H.; Szekely, N.K.; Frielinghaus, H.; Marks, C.; Wang, Y.; Anders, N.; Spiess, A.C.; Makowski, L. Multi-scale processes of beech wood disintegration and pretreatment with 1-ethyl-3-methylimidazolium acetate/water mixtures. Biotechnol. Biofuels 2016, 9, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinkert, A.; Marsh, K.N.; Pang, S.; Staiger, M.P. Ionic Liquids and Their Interaction with Cellulose. Chem. Rev. 2009, 109, 6712–6728. [Google Scholar] [CrossRef] [PubMed]
- Blanchette, C.D.; Knipe, J.M.; Stolaroff, J.K.; DeOtte, J.R.; Oakdale, J.S.; Maiti, A.; Lenhardt, J.; Sirajuddin, S.; Rosenzweig, A.C.; Baker, S.E. Printable enzyme-embedded materials for methane to methanol conversion. Nat. Commun. 2016, 7, 11900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, K.A.; Hilliard, M.V.; He, Q.P.; Wang, J. A mini review on bioreactor configurations and gas transfer enhancements for biochemical methane conversion. Biochem. Eng. J. 2017, 128, 83–92. [Google Scholar] [CrossRef]
- Strong, P.J.; Xie, S.; Clarke, W.P. Methane as a Resource: Can the Methanotrophs Add Value? Environ. Sci. Technol. 2015, 49, 4001–4018. [Google Scholar] [CrossRef] [PubMed]
- Agyei, D.; Danquah, M.K. Industrial-scale manufacturing of pharmaceutical-grade bioactive peptides. Biotechnol. Adv. 2011, 2011, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Lucas, J.; Castaneda, D.; Hormigo, D. New trends for a classical enzyme: Papain, a biotechnological success story in the food industry. Trends Food Sci. Technol. 2017, 68, 91–101. [Google Scholar] [CrossRef]
- Lima-Ramos, J.; Tufvesson, P.; Woodley, J.M. Application of environmental and economic metrics to guide the development of biocatalytic processes. Green Process. Synth. 2014, 3, 195–213. [Google Scholar] [CrossRef]
- Tufvesson, P.; Lima-Ramos, J.; Haque, N.A.; Gernaey, K.V.; Woodly, J.M. Advances in the Process Development of Biocatalytic Processes. Org. Process Res. Dev. 2013, 17, 1233–1238. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Woodley, J.M. Role of Biocatalysis in Sustainable Chemistry. Chem. Rev. 2018, 118, 801–838. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sector | Enzymes | Applications | References |
|---|---|---|---|
| Pharmaceuticals | Nitrile hydratase, transaminase, monoamine oxidase, lipase, penicillin acylase | Synthesis of intermediates for production of active pharmaceutical ingredients | [2,4,30,31,32,33,34,35,36,37,38] |
| Food Processing | Trypsin, amylase, glucose isomerase, papain, pectinase | Conversion of starch to glucose, production of high fructose corn syrup, production of prebiotics, debittering of fruit juice | [2,5,9,10,11,12,39,40,41,42,43] |
| Detergent | Protease, lipase, amylase, cellulase | Stain removal, removal of fats and oils, color retention, | [44,45,46,47,48] |
| Biofuels | Lipase, cellulase, xylanase | Production of fatty acid methyl esters, decomposition of lignocellulotic material for bioethanol production | [18,19,44,49,50,51,52,53,54,55,56,57] |
| Paper and Pulp | Lipase, cellulase, xylanase | Removal of lignin for improved bleaching, improvement in fiber properties | [2,4,44,58,59,60,61] |
| Advantages | Disadvantages |
|---|---|
| Functionality for use in continuous processes | Loss of enzyme activity |
| Improved stability in broader range of operating conditions (e.g., pH, temperature etc.) | Immobilization of enzyme in undesired conformation and subsequent loss of activity |
| Facile separation of enzyme from product | Cost of carrier and additional preparation materials and methods, as well as laborious training strategies |
| Reusability of enzyme | Mass transfer limitations |
| Immobilization in preferred conformation and at preferred location | Laborious and time-consuming immobilization processes |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chapman, J.; Ismail, A.E.; Dinu, C.Z. Industrial Applications of Enzymes: Recent Advances, Techniques, and Outlooks. Catalysts 2018, 8, 238. https://doi.org/10.3390/catal8060238
Chapman J, Ismail AE, Dinu CZ. Industrial Applications of Enzymes: Recent Advances, Techniques, and Outlooks. Catalysts. 2018; 8(6):238. https://doi.org/10.3390/catal8060238
Chicago/Turabian StyleChapman, Jordan, Ahmed E. Ismail, and Cerasela Zoica Dinu. 2018. "Industrial Applications of Enzymes: Recent Advances, Techniques, and Outlooks" Catalysts 8, no. 6: 238. https://doi.org/10.3390/catal8060238
APA StyleChapman, J., Ismail, A. E., & Dinu, C. Z. (2018). Industrial Applications of Enzymes: Recent Advances, Techniques, and Outlooks. Catalysts, 8(6), 238. https://doi.org/10.3390/catal8060238