Infrared Characterization of the Bidirectional Oxygen-Sensitive [NiFe]-Hydrogenase from E. coli



Abstract
1. Introduction
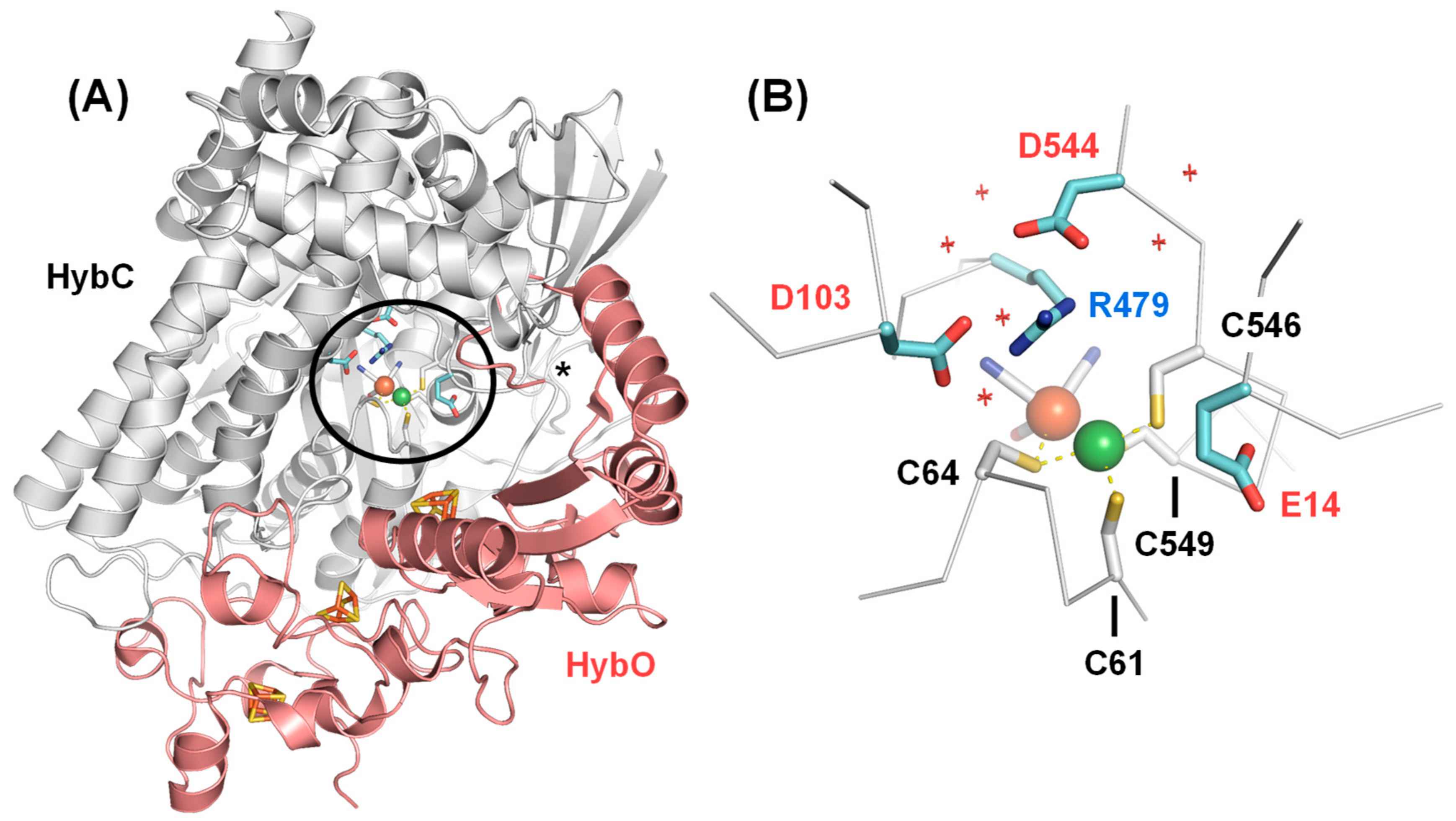
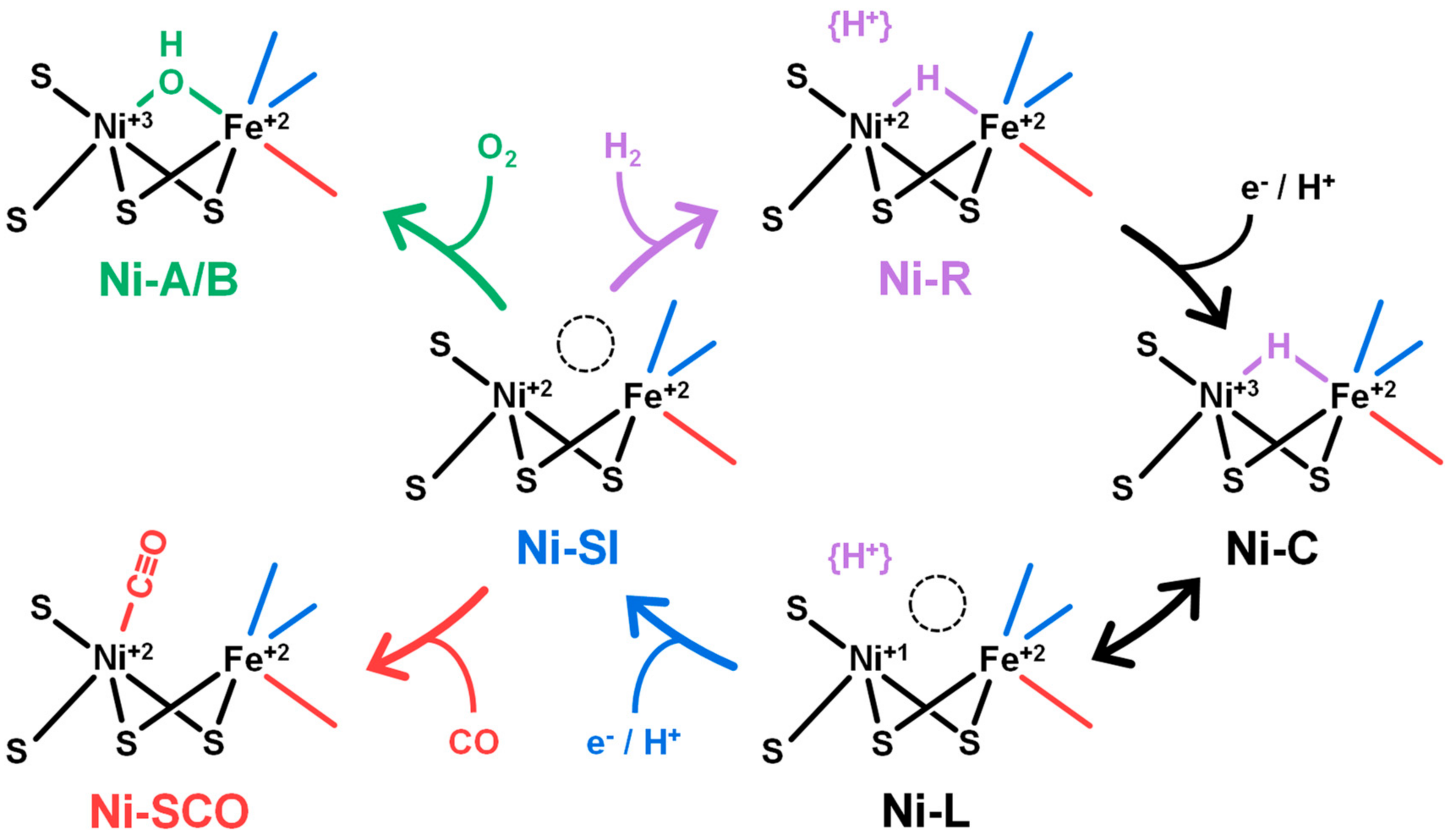
2. Results and Discussion
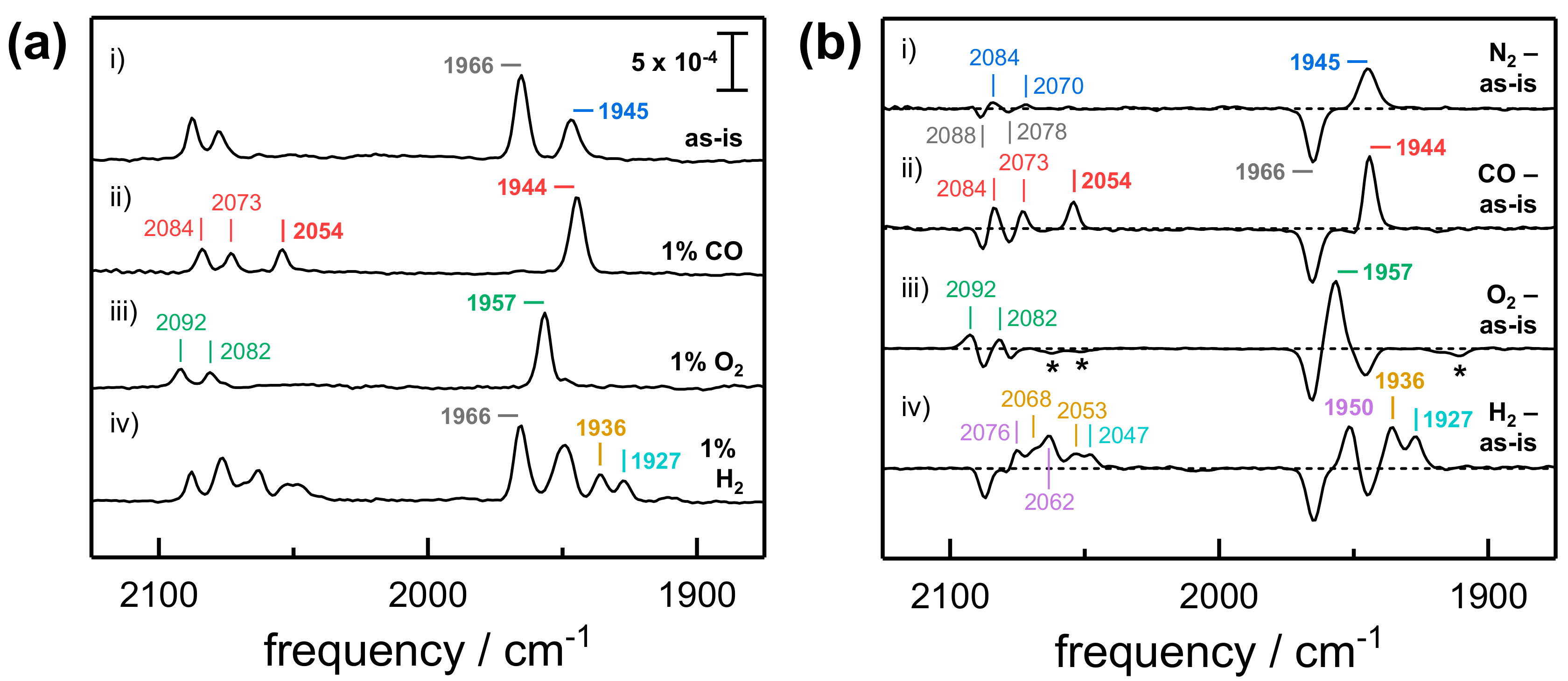
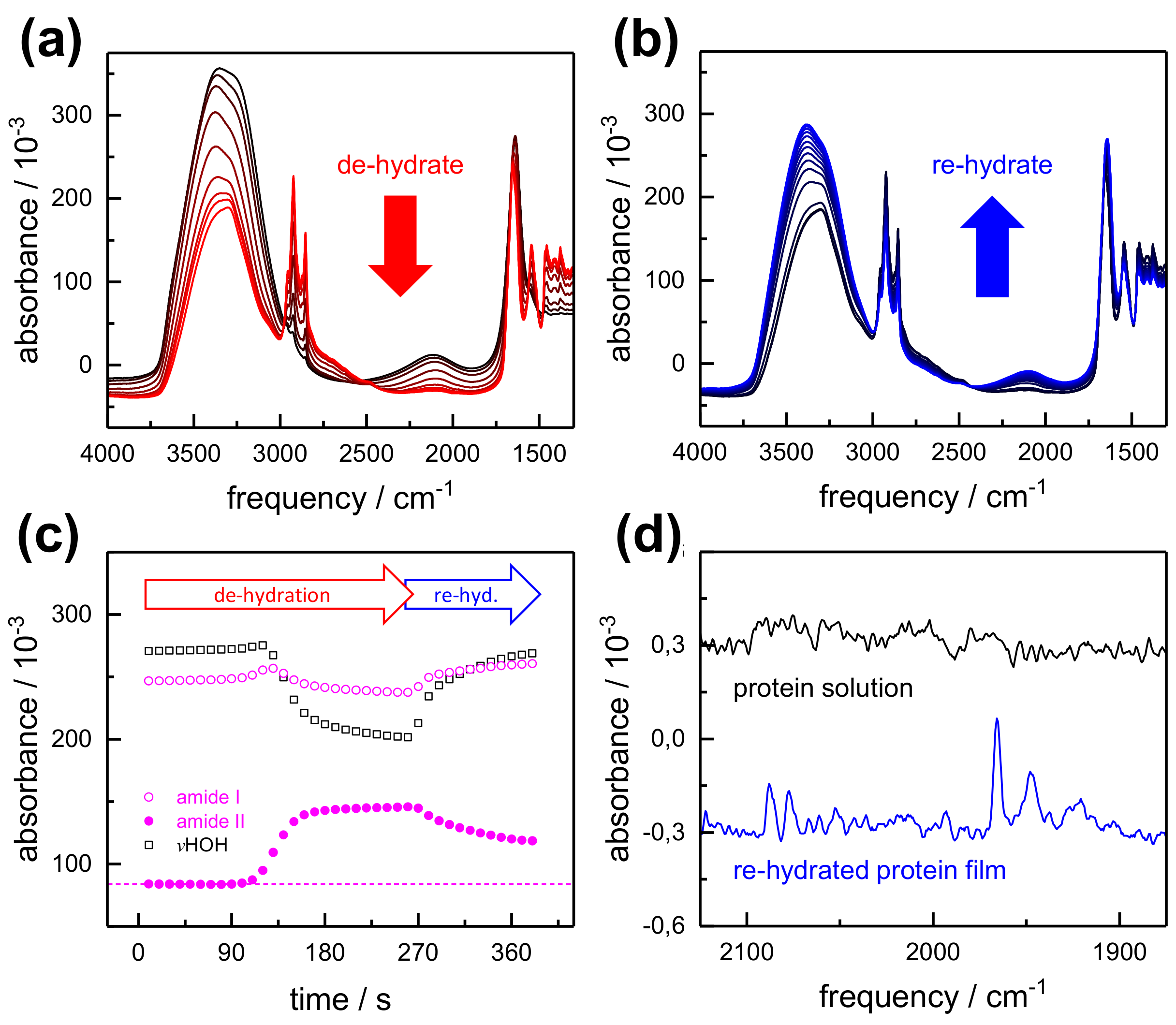
2.1. FTIR Steady-State and In Situ Difference Spectra
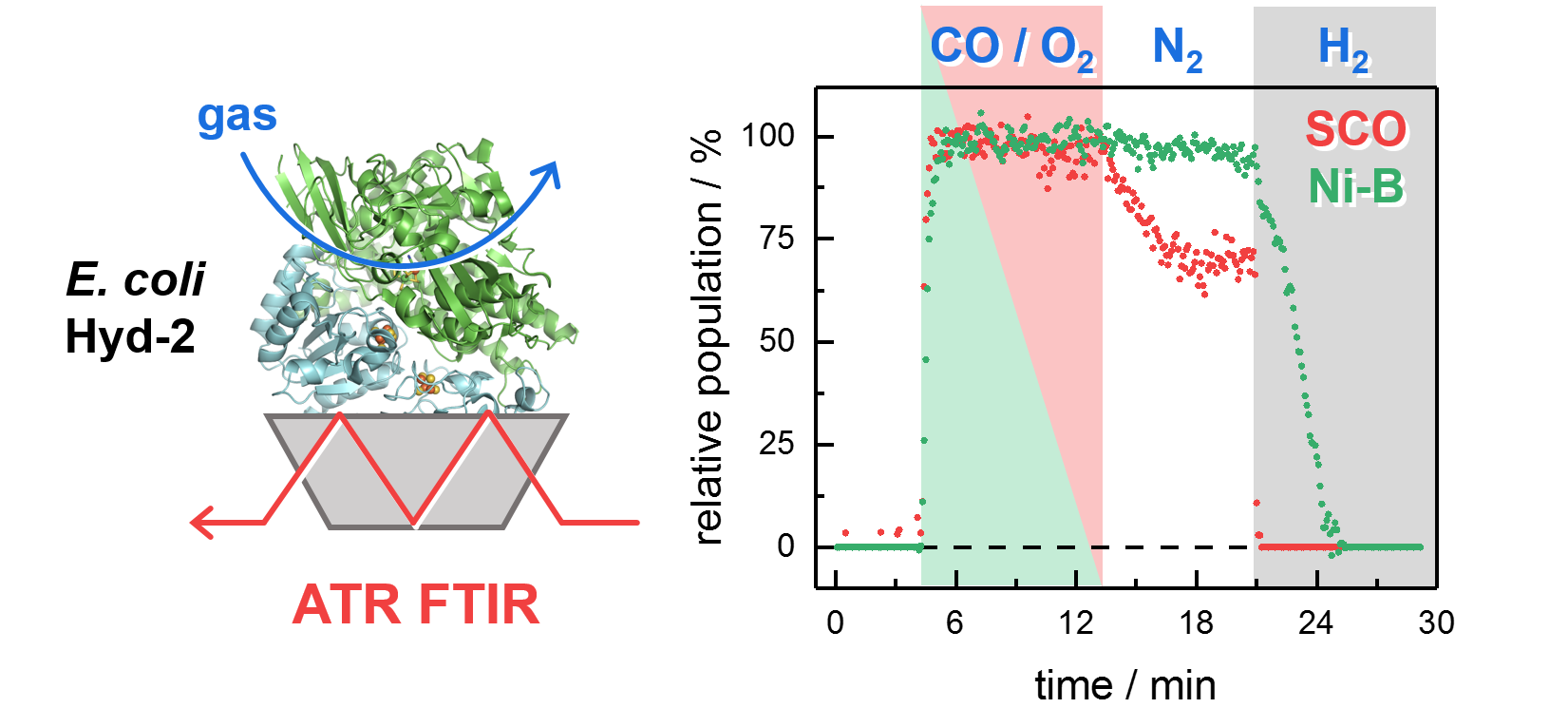
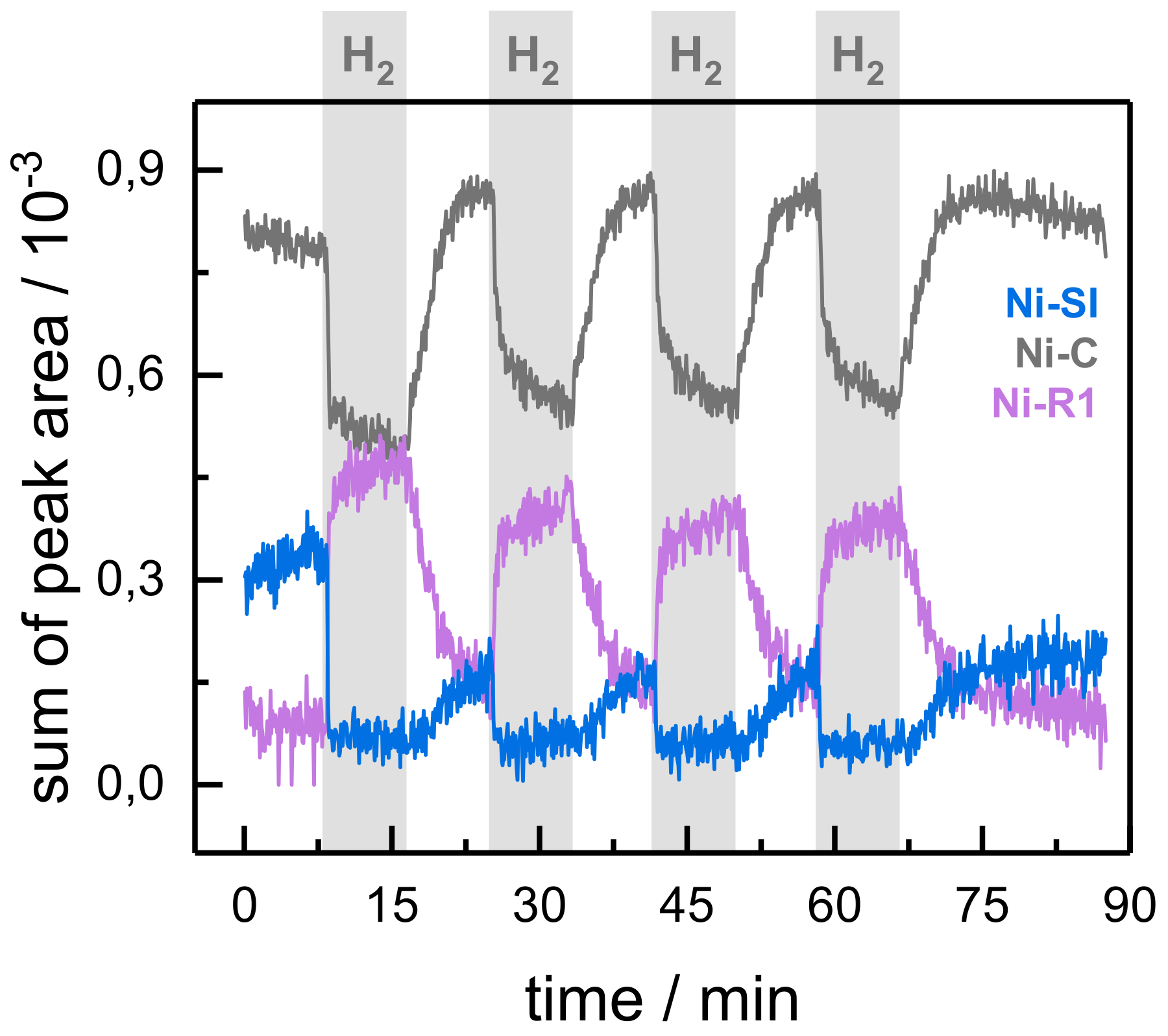
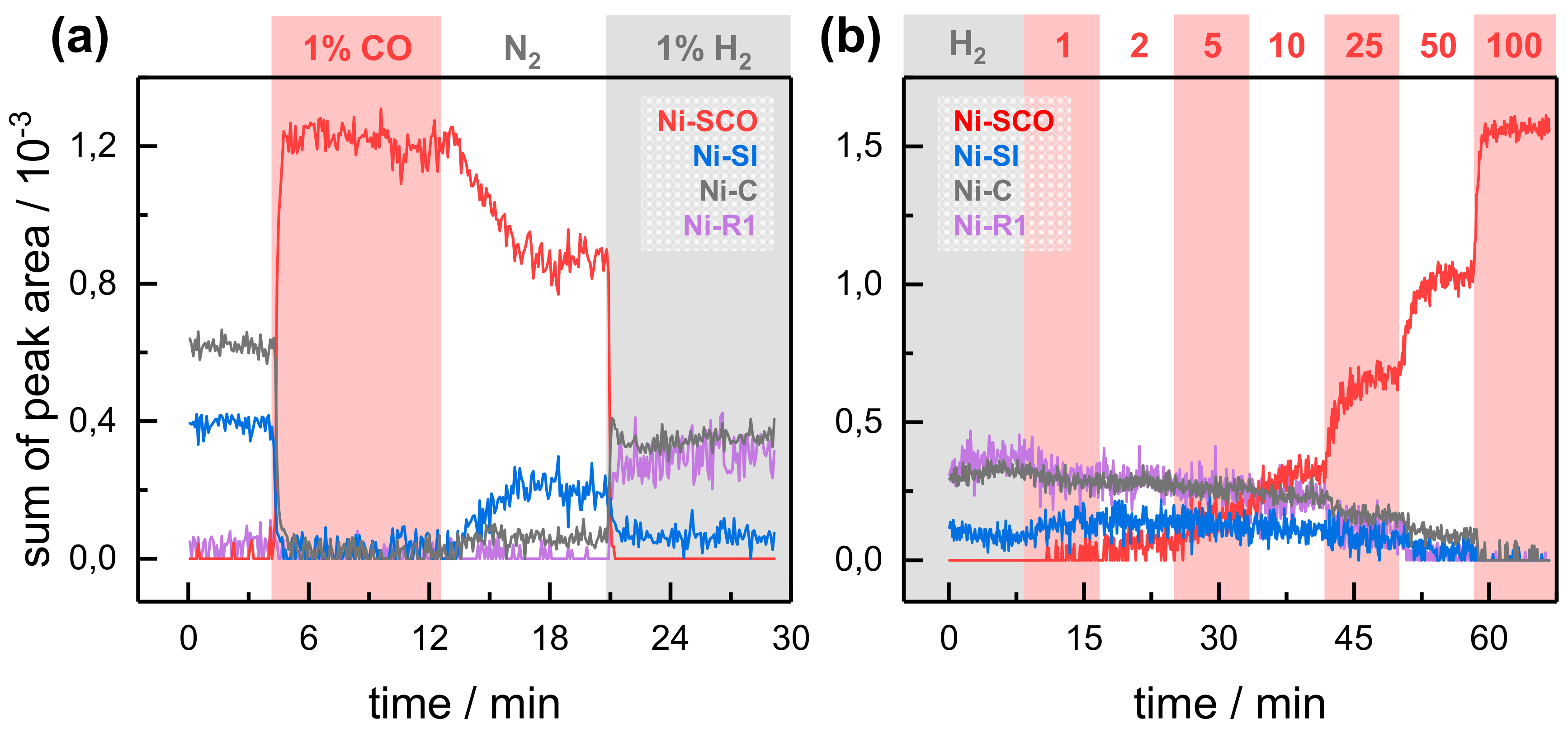
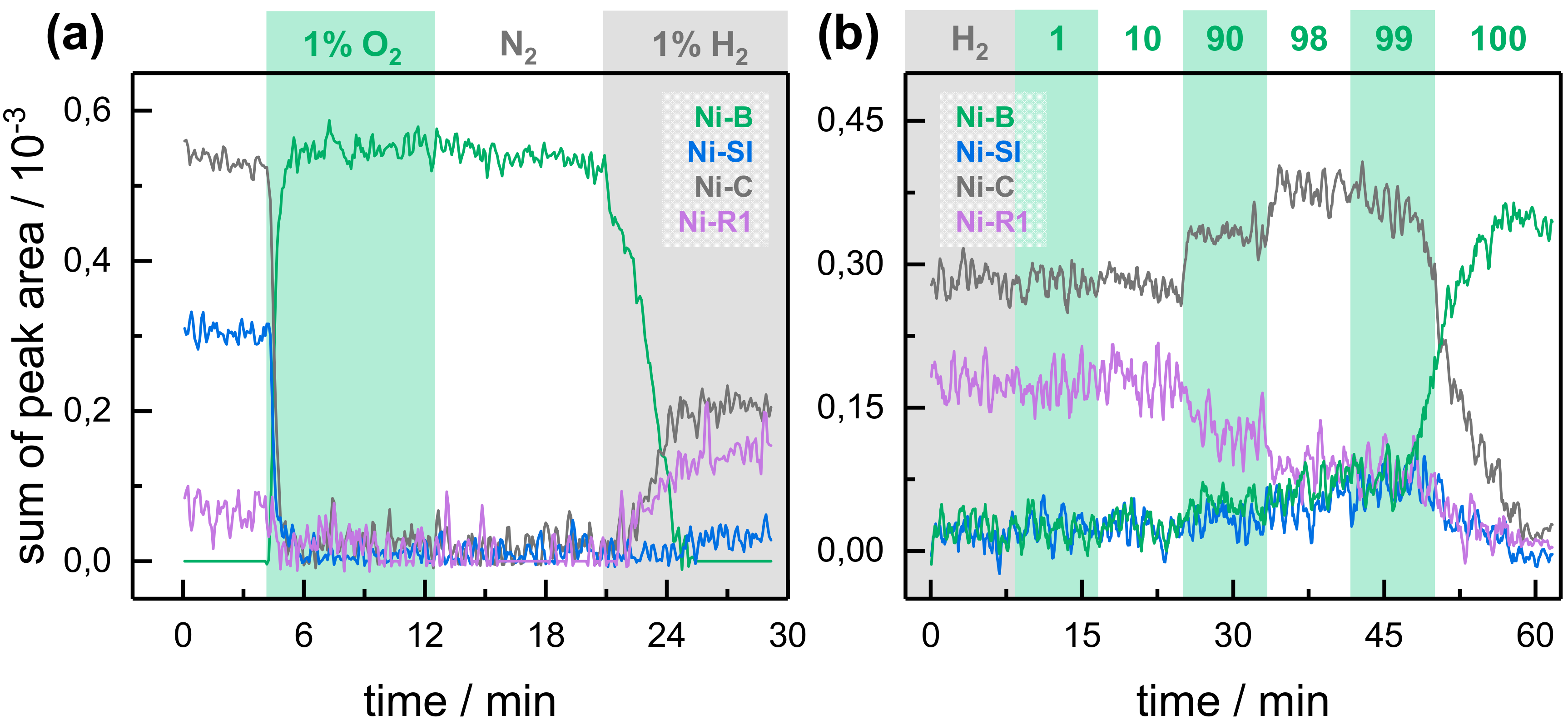
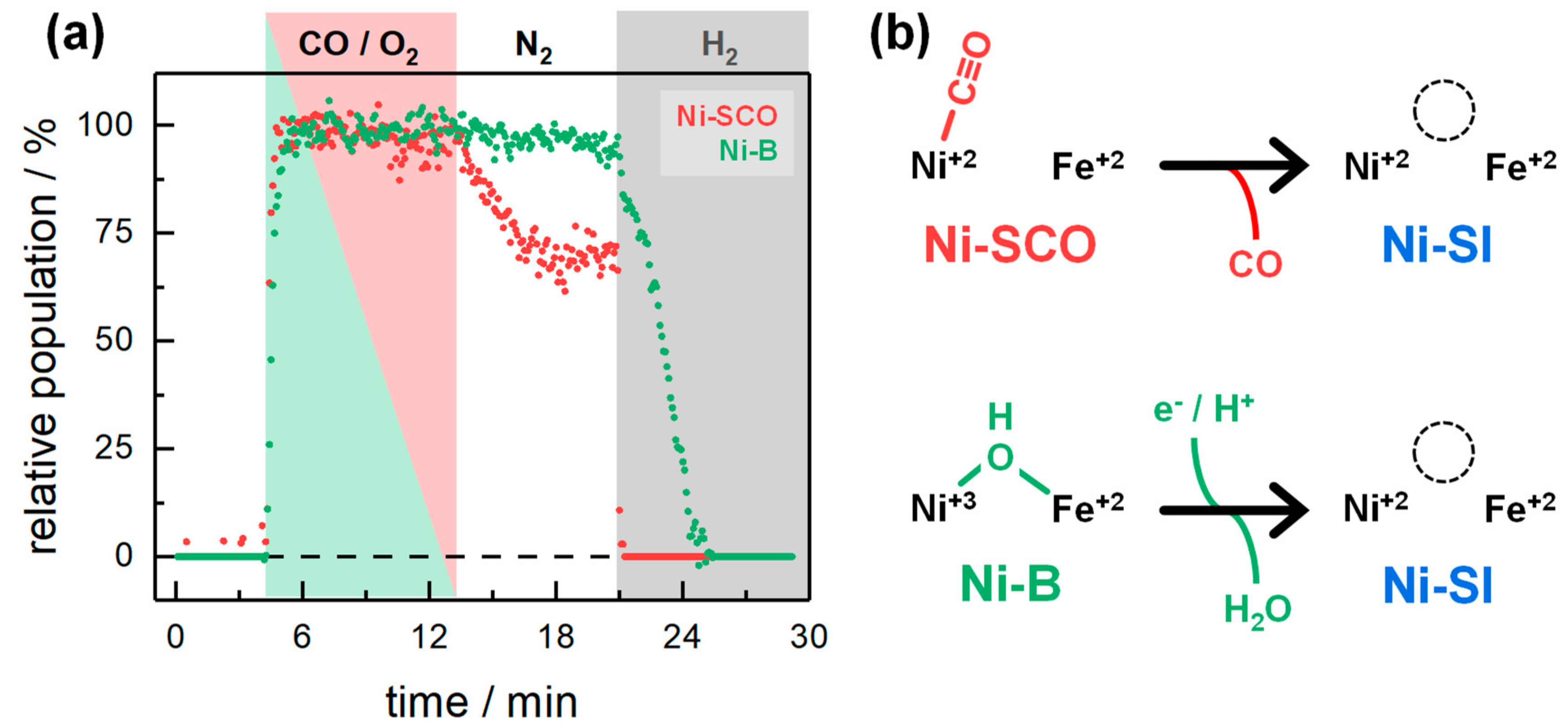
2.2. Kinetic Traces for the Reaction with H2, CO, and O2
3. Materials and Methods
3.1. Synthesis and Isolation of HYD-2 from E. coli
3.2. Infrared Spectroscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Appendix A


References
- Lubitz, W.; Ogata, H.; Ru, O.; Reijerse, E. Hydrogenases. Chem. Rev. 2014, 114, 4081–4148. [Google Scholar] [CrossRef] [PubMed]
- Shima, S.; Thauer, R.K. A third type of hydrogenase catalyzing H2 activation. Chem. Rec. 2007, 7, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Ogata, H.; Lubitz, W.; Higuchi, Y. Structure and function of [NiFe] hydrogenases. J. Biochem. 2016, 160, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Stripp, S.T.; Happe, T. How Algae Produce Hydrogen—News from the Photosynthetic Hydrogenase. Dalton Trans. 2009, 45, 9960–9969. [Google Scholar] [CrossRef] [PubMed]
- Vignais, P.M.; Billoud, B. Occurrence, classification, and biological function of hydrogenases: An overview. Chem. Rev. 2007, 107, 4206–4272. [Google Scholar] [CrossRef] [PubMed]
- Tard, C.; Pickett, C.J. Structural and functional analogues of the active sites of the [Fe]-, [NiFe]-, and [FeFe]-hydrogenases. Chem. Rev. 2009, 109, 2245–2274. [Google Scholar] [CrossRef] [PubMed]
- Simmons, T.R.; Berggren, G.; Bacchi, M.; Fontecave, M.; Artero, V. Mimicking hydrogenases: From biomimetics to artificial enzymes. Coord. Chem. Rev. 2014, 270–271, 127–150. [Google Scholar] [CrossRef]
- Schilter, D.; Camara, J.M.; Huynh, M.T.; Hammes-Schiffer, S.; Rauchfuss, T.B. Hydrogenase Enzymes and Their Synthetic Models: The Role of Metal Hydrides. Chem. Rev. 2016, 116, 8693–8749. [Google Scholar] [CrossRef] [PubMed]
- Sargent, F. The Model [NiFe]-Hydrogenases of Escherichia coli, 1st ed.; Elsevier Ltd.: Amsterdam, The Netherlands, 2016; Volume 68, ISBN 9780128048238. [Google Scholar]
- Laurinavichene, T.V.; Tsygankov, A.A. H2 consumption by Escherichia coli coupled via hydrogenase 1 or hydrogenase 2 to different terminal electron acceptors. FEMS Microbiol. Lett. 2001, 202, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Vincent, K.A.; Parkin, A.; Lenz, O.; Albracht, S.P.J.; Fontecilla-Camps, J.C.; Cammack, R.; Friedrich, B.; Armstrong, F.A. Electrochemical definitions of O2 sensitivity and oxidative inactivation in hydrogenases. J. Am. Chem. Soc. 2005, 127, 18179–18189. [Google Scholar] [CrossRef] [PubMed]
- Lukey, M.J.; Roessler, M.M.; Parkin, A.; Evans, R.M.; Davies, R.A.; Lenz, O.; Friedrich, B.; Sargent, F.; Armstrong, F.A. Oxygen-Tolerant [NiFe]-Hydrogenases: The Individual and Collective Importance of Supernumerary Cysteines at the Proximal Fe-S Cluster. J. Am. Chem. Soc. 2011, 133, 16881–16892. [Google Scholar] [CrossRef] [PubMed]
- Beaton, S.E.; Evans, R.M.; Finney, A.J.; Lamont, C.M.; Armstrong, F.A.; Sargent, F.; Carr, S.B. The Structure of Hydrogenase-2 from Escherichia coli: Implications for H2-Driven Proton Pumping. Biochem. J. 2018, 2, BCJ20180053. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, J.; Lenz, O.; Friedrich, B. Structure, function and biosynthesis of O2-tolerant hydrogenases. Nat. Rev. 2013, 11, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, J.; Scheerer, P.; Frielingsdorf, S.; Kroschinsky, S.; Friedrich, B.; Lenz, O.; Spahn, C.M.T. The crystal structure of an oxygen-tolerant hydrogenase uncovers a novel iron-sulphur centre. Nature 2011, 479, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.; Olsen, S.; MacFarlane, D.R.; Sun, C. The oxygen reduction reaction on [NiFe] hydrogenases. Phys. Chem. Chem. Phys. 2018, 20, 23528–23534. [Google Scholar] [CrossRef] [PubMed]
- Volbeda, A.; Amara, P.; Darnault, C.; Mouesca, J.-M.; Parkin, A.; Roessler, M.M.; Armstrong, F.A.; Fontecilla-Camps, J.C. X-ray crystallographic and computational studies of the O2-tolerant [NiFe]-hydrogenase 1 from Escherichia coli. Proc. Natl. Acad. Sci. USA 2012, 109, 5305–5310. [Google Scholar] [CrossRef] [PubMed]
- Volbeda, A.; Darnault, C.; Parkin, A.; Sargent, F.; Armstrong, F.A.; Fontecilla-Camps, J.C. Crystal structure of the O2-tolerant membrane-bound hydrogenase 1 from Escherichia coli in complex with its cognate cytochrome b. Structure 2013, 21, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Pierik, A.J.; Roseboom, W.; Happe, R.P.; Bagley, K.A.; Albracht, S.P.J. Carbon monoxide and cyanide as intrinsic ligands to iron in the active site of [NiFe]-hydrogenases. J. Biol. Chem. 1999, 274, 3331–3337. [Google Scholar] [CrossRef] [PubMed]
- Ash, P.A.; Hidalgo, R.; Vincent, K.A. Proton Transfer in the Catalytic Cycle of [NiFe] Hydrogenases: Insight from Vibrational Spectroscopy. ACS Catal. 2017, 7, 2471–2485. [Google Scholar] [CrossRef] [PubMed]
- Tai, H.; Higuchi, Y.; Hirota, S. Comprehensive reaction mechanisms at and near the Ni-Fe active sites of [NiFe] hydrogenases. Dalton Trans. 2018, 47, 4408–4423. [Google Scholar] [CrossRef] [PubMed]
- Pandelia, M.-E.; Ogata, H.; Lubitz, W. Intermediates in the catalytic cycle of [NiFe] hydrogenase: Functional spectroscopy of the active site. ChemPhysChem 2010, 11, 1127–1140. [Google Scholar] [CrossRef] [PubMed]
- De Lacey, A.L.; Pardo, A.; Fernández, V.M.; Dementin, S.; Adryanczyk-Perrier, G.; Hatchikian, E.; Rousset, M. FTIR spectroelectrochemical study of the activation and inactivation processes of [NiFe] hydrogenases: Effects of solvent isotope replacement and site-directed mutagenesis. J. Biol. Chem. 2004, 9, 636–642. [Google Scholar] [CrossRef] [PubMed]
- Greene, B.L.; Wu, C.; Vansuch, G.E.; Adams, M.W.W.; Dyer, R.B. Proton Inventory and Dynamics in the Nia-S to Nia-C Transition of a [NiFe] Hydrogenase. Biochemistry 2016, 55, 1813–1825. [Google Scholar] [CrossRef] [PubMed]
- Ogata, H.; Nishikawa, K.; Lubitz, W. Hydrogens detected by subatomic resolution protein crystallography in a [NiFe] hydrogenase. Nature 2015, 520, 571–574. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.M.; Brooke, E.J.; Wehlin, S.A.M.; Nomerotskaia, E.; Sargent, F.; Carr, S.B.; Phillips, S.E.V.; Armstrong, F.A. Mechanism of hydrogen activation by [NiFe] hydrogenases. Nat. Chem. Biol. 2016, 12, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Lubitz, W.; Reijerse, E.; van Gastel, M. [NiFe] and [FeFe] hydrogenases studied by advanced magnetic resonance techniques. Chem. Rev. 2007, 107, 4331–4365. [Google Scholar] [CrossRef] [PubMed]
- Vincent, K.A.; Parkin, A.; Armstrong, F.A. Investigating and exploiting the electrocatalytic properties of hydrogenases. Chem. Rev. 2007, 107, 4366–4413. [Google Scholar] [CrossRef] [PubMed]
- Fontecilla-Camps, J.C.; Volbeda, A.; Cavazza, C.; Nicolet, Y. Structure/function relationships of [NiFe]- and [FeFe]-hydrogenases. Chem. Rev. 2007, 107, 4273–4303. [Google Scholar] [CrossRef] [PubMed]
- Van der Spek, T.M.; Arendsen, A.F.; Happe, R.P.; Yun, S.; Bagley, K.A.; Stufkens, D.J.; Hagen, W.R.; Albracht, S.P. Similarities in the architecture of the active sites of Ni-hydrogenases and Fe-hydrogenases detected by means of infrared spectroscopy. Eur. J. Biochem. 1996, 237, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Brecht, M.; Van Gastel, M.; Buhrke, T.; Friedrich, B.; Lubitz, W. Direct Detection of a Hydrogen Ligand in the [NiFe] Center of the Regulatory H2-Sensing Hydrogenase from Ralstonia eutropha in Its Reduced State by HYSCORE and ENDOR Spectroscopy. J. Am. Chem. Soc. 2003, 125, 13075–13083. [Google Scholar] [CrossRef] [PubMed]
- De Lacey, A.L.; Fernandez, V.M.; Rousset, M.; Cammack, R. Activation and inactivation of hydrogenase function and the catalytic cycle: Spectroelectrochemical studies. Chem. Rev. 2007, 107, 4304–4330. [Google Scholar] [CrossRef] [PubMed]
- Albracht, S.P.J. Nickel hydrogenases: In search of the active site. BBA Bioenerg. 1994, 1188, 167–204. [Google Scholar] [CrossRef]
- Fichtner, C.; van Gastel, M.; Lubitz, W. Wavelength dependence of the photo-induced conversion of the Ni-C to the Ni-L redox state in the [NiFe] hydrogenase of Desulfovibrio vulgaris Miyazaki F. Phys. Chem. Chem. Phys. 2003, 5, 5507–5513. [Google Scholar] [CrossRef]
- Schröder, O.; Bleijlevens, B.; De Jongh, T.E.; Chen, Z.; Li, T.; Fischer, J.; Förster, J.; Friedrich, C.G.; Bagley, K.A.; Albracht, S.P.J.; et al. Characterization of a cyanobacterial-like uptake [NiFe] hydrogenase: EPR and FTIR spectroscopic studies of the enzyme from Acidithiobacillus ferrooxidans. J. Biol. Inorg. Chem. 2007, 12, 212–233. [Google Scholar] [CrossRef] [PubMed]
- Pandelia, M.E.; Infossi, P.; Stein, M.; Giudici-Orticoni, M.T.; Lubitz, W. Spectroscopic characterization of the key catalytic intermediate Ni-C in the O2-tolerant [NiFe] hydrogenase i from Aquifex aeolicus: Evidence of a weakly bound hydride. Chem. Commun. 2012, 48, 823–825. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, R.; Ash, P.A.; Healy, A.J.; Vincent, K.A. Infrared spectroscopy during electrocatalytic turnover reveals the Ni-L active site state during H2 oxidation by a NiFe hydrogenase. Angew. Chem. Int. Ed. 2015, 54, 7110–7113. [Google Scholar] [CrossRef] [PubMed]
- Murphy, B.J.; Hidalgo, R.; Roessler, M.M.; Evans, R.M.; Ash, P.A.; Myers, W.K.; Vincent, K.A.; Armstrong, F.A. Discovery of Dark pH-Dependent H+ Migration in a [NiFe]-Hydrogenase and Its Mechanistic Relevance: Mobilizing the Hydrido Ligand of the Ni-C Intermediate. J. Am. Chem. Soc. 2015, 137, 8484–8489. [Google Scholar] [CrossRef] [PubMed]
- Tai, H.; Nishikawa, K.; Inoue, S.; Higuchi, Y.; Hirota, S. FT-IR Characterization of the Light-Induced Ni-L2 and Ni-L3 States of [NiFe] Hydrogenase from Desulfovibrio vulgaris Miyazaki F. J. Phys. Chem. B 2015, 150430101225004. [Google Scholar] [CrossRef] [PubMed]
- Ogata, H.; Mizoguchi, Y.; Mizuno, N.; Miki, K.; Adachi, S.; Yasuoka, N.; Yagi, T.; Yamauchi, O.; Hirota, S.; Higuchi, Y. Structural studies of the carbon monoxide complex of [NiFe]hydrogenase from Desulfovibrio vulgaris Miyazaki F: Suggestion for the initial activation site for dihydrogen. J. Am. Chem. Soc. 2002, 124, 11628–11635. [Google Scholar] [CrossRef] [PubMed]
- Pandelia, M.E.; Ogata, H.; Currell, L.J.; Flores, M.; Lubitz, W. Inhibition of the [NiFe] hydrogenase from Desulfovibrio vulgaris Miyazaki F by carbon monoxide: An FTIR and EPR spectroscopic study. Biochim. Biophys. Acta Bioenerg. 2010, 1797, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Lamle, S.E.; Albracht, S.P.J.; Armstrong, F.A. Electrochemical potential-step investigations of the aerobic interconversions of [NiFe]-hydrogenase from allochromatium vinosum: Insights into the puzzling difference between unready and ready oxidized inactive states. J. Am. Chem. Soc. 2004, 126, 14899–14909. [Google Scholar] [CrossRef] [PubMed]
- Bleijlevens, B.; van Broekhuizen, F.A.; De Lacey, A.L.; Roseboom, W.; Fernandez, V.M.; Albracht, S.P.J. The activation of the [NiFe]-hydrogenase from Allochromatium vinosum. An infrared spectro-electrochemical study. J. Biol. Inorg. Chem. 2004, 9, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Volbeda, A.; Martin, L.; Cavazza, C.; Matho, M.; Faber, B.W.; Roseboom, W.; Albracht, S.P.J.; Garcin, E.; Rousset, M.; Fontecilla-Camps, J.C. Structural differences between the ready and unready oxidized states of [NiFe] hydrogenases. J. Biol. Inorg. Chem. 2005, 10, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Ogata, H.; Hirota, S.; Nakahara, A.; Komori, H.; Shibata, N.; Kato, T.; Kano, K.; Higuchi, Y. Activation process of [NiFe] hydrogenase elucidated by high-resolution X-ray analyses: Conversion of the ready to the unready state. Structure 2005, 13, 1635–1642. [Google Scholar] [CrossRef] [PubMed]
- Barilone, J.L.; Ogata, H.; Lubitz, W.; Van Gastel, M. Structural differences between the active sites of the Ni-A and Ni-B states of the [NiFe] hydrogenase: An approach by quantum chemistry and single crystal ENDOR spectroscopy. Phys. Chem. Chem. Phys. 2015, 17, 16204–16212. [Google Scholar] [CrossRef] [PubMed]
- Hexter, S.V.; Chung, M.-W.; Vincent, K.A.; Armstrong, F.A. Unusual Reaction of [NiFe]-Hydrogenases with Cyanide. J. Am. Chem. Soc. 2014, 136, 10470–10477. [Google Scholar] [CrossRef] [PubMed]
- Senger, M.; Stripp, S.T.; Soboh, B. Proteolytic cleavage orchestrates cofactor insertion and protein assembly in [NiFe]-hydrogenase biosynthesis. J. Biol. Chem. 2017, 292, 11670–11681. [Google Scholar] [CrossRef] [PubMed]
- Senger, M.; Mebs, S.; Duan, J.; Wittkamp, F.; Apfel, U.-P.; Heberle, J.; Haumann, M.; Stripp, S.T. Stepwise isotope editing of [FeFe]-hydrogenases exposes cofactor dynamics. Proc. Natl. Acad. Sci. USA 2016, 113, 8454–8459. [Google Scholar] [CrossRef] [PubMed]
- Senger, M.; Mebs, S.; Duan, J.; Shulenina, O.; Laun, K.; Kertess, L.; Wittkamp, F.; Apfel, U.-P.; Happe, T.; Winkler, M.; et al. Protonation/reduction dynamics at the [4Fe–4S] cluster of the hydrogen-forming cofactor in [FeFe]-hydrogenases. Phys. Chem. Chem. Phys. 2018, 20, 3128–3140. [Google Scholar] [CrossRef] [PubMed]
- Haumann, M.; Stripp, S.T. The Molecular Proceedings of Biological Hydrogen Turnover. Acc. Chem. Res. 2018, 51, 1755–1763. [Google Scholar] [CrossRef] [PubMed]
- Lukey, M.J.; Parkin, A.; Roessler, M.M.; Murphy, B.J.; Harmer, J.; Palmer, T.; Sargent, F.; Armstrong, F.A. How Escherichia coli is equipped to oxidize hydrogen under different redox conditions. J. Biol. Chem. 2010, 285, 3928–3938. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, Y.; Ogata, H.; Miki, K.; Yasuoka, N.; Yagi, T. Removal of the bridging ligand atom at the Ni-Fe active site of [NiFe] hydrogenase upon reduction with H2, as revealed by X-ray structure analysis at 1.4 Å resolution. Structure 1999, 7, 549–556. [Google Scholar] [CrossRef]
- Shafaat, H.S.; Rüdiger, O.; Ogata, H.; Lubitz, W. [NiFe] hydrogenases: A common active site for hydrogen metabolism under diverse conditions. Biochim. Biophys. Acta 2013, 1827, 986–1002. [Google Scholar] [CrossRef] [PubMed]
- Soboh, B.; Lindenstrauss, U.; Granich, C.; Javed, M.; Herzberg, M.; Thomas, C.; Stripp, S.T. [NiFe]-hydrogenase maturation in vitro: Analysis of the roles of the HybG and HypD accessory proteins. Biochem. J. 2014, 464, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, S.; Frielingsdorf, S.; Ciaccafava, A.; Lorent, C.; Fritsch, J.; Siebert, E.; Priebe, J.; Haumann, M.; Zebger, I.; Lenz, O. O2-tolerant H2 activation by an isolated large subunit of a [NiFe]-hydrogenase. Biochemistry 2018, 57, 5339–5349. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | vCO | vCN− | vCN− |
|---|---|---|---|
| Ni-B | 1957 | 2082 | 2092 |
| Ni-SI | 1945 | 2070 | 2084 |
| Ni-SCO 1 | 2054/1944 | 2073 | 2084 |
| Ni-C | 1966 | 2078 | 2088 |
| Ni-R1 | 1950 | 2062 | 2076 |
| Ni-R2 | 1936 | 2053 | 2068 |
| Ni-R3 | 1929 | 2047 | 2063 |
| Ni-L | 1911 | 2052 | 2062 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Senger, M.; Laun, K.; Soboh, B.; Stripp, S.T. Infrared Characterization of the Bidirectional Oxygen-Sensitive [NiFe]-Hydrogenase from E. coli. Catalysts 2018, 8, 530. https://doi.org/10.3390/catal8110530
Senger M, Laun K, Soboh B, Stripp ST. Infrared Characterization of the Bidirectional Oxygen-Sensitive [NiFe]-Hydrogenase from E. coli. Catalysts. 2018; 8(11):530. https://doi.org/10.3390/catal8110530
Chicago/Turabian StyleSenger, Moritz, Konstantin Laun, Basem Soboh, and Sven T. Stripp. 2018. "Infrared Characterization of the Bidirectional Oxygen-Sensitive [NiFe]-Hydrogenase from E. coli" Catalysts 8, no. 11: 530. https://doi.org/10.3390/catal8110530
APA StyleSenger, M., Laun, K., Soboh, B., & Stripp, S. T. (2018). Infrared Characterization of the Bidirectional Oxygen-Sensitive [NiFe]-Hydrogenase from E. coli. Catalysts, 8(11), 530. https://doi.org/10.3390/catal8110530