1. Introduction
The transport sector is responsible for a large fraction of CO2 emissions. To mitigate global warming, in addition to the development of electric vehicles, other solutions, still based on the internal combustion engine (ICE), have been proposed, such as the use of hydrogen as fuel.
Hydrogen is a carbon-free energy carrier and a virtually emission-free fuel, thus representing a good alternative to electric and fuel cell-powered vehicles [
1].
Emissions from a H
2-fueled engine can be reduced to a minimum by employing a suitably designed combustion process in lean-burn operation with exhaust gas recirculation [
2]. However, a completely nitrogen-oxide-free combustion cannot be ensured at all operating points. Under certain conditions, nitrogen oxides can be emitted, mainly driven by the thermal formation mechanism occurring when the combustion temperature exceeds 2000 K, and for this reason, a deNO
x unit is required.
The use of a catalyzed exhaust after-treatment system using H
2 as a reducing agent would have the advantage that only one fuel needs to be carried on board both for propulsion and NO
x reduction, thus avoiding the devices for storage and injection of the urea solution, required for the traditional NH
3-SCR for diesel engines. Moreover, compared to other reducing agents, hydrogen requires the lowest reaction temperature [
3].
The NO
x reduction by H
2 produces, in theory, only elemental nitrogen and water [
4].
Nevertheless, the competitive reaction of hydrogen with the excess oxygen contained in the lean exhaust gas to directly produce water and the potential formation of N
2O, a strong greenhouse gas, can adversely affect the selectivity of the process [
3].
Great efforts have been made recently to understand the relationship between the structure and reactivity of H
2-SCR catalysts, the most active being those based on noble metals such as platinum [
3,
4,
5].
Noble metals-based catalysts are traditionally used for H
2-SCR due to the ability to decompose NO into adsorbed N and O and to generate active nitrate and NH
x+ species from H
2 that is dissociatively adsorbed [
3,
4,
5]. In fact, ammonium species are considered key reaction intermediates [
6].
The use of a proper support for the active metal can largely affect the catalytic performance [
7,
8]. It has been reported that zeolite-supported Pt catalysts exhibited higher N
2 selectivity than metal oxide-supported counterparts; in particular, Pt/MFI provided the highest N
2 selectivity among zeolite-based catalysts [
9,
10]. The support significantly influences the dispersion and stability of the active element, as well as the catalyst’s acidity [
3,
4,
5]. Specifically, it was reported that Pt/ZSM-5 showed a larger amount of strong acid sites compared to Pt/SiO
2 or Pt/Al
2O
3, which, in turn, enhanced the specific catalytic activity [
9]. Moreover, the location of Pt in the zeolite plays a crucial role: Pt
0 on the outer surface activates H
2, whereas Pt
2+ within the zeolitic framework can adsorb NO
x as nitrite/nitrate species [
6].
A further important drawback of Pt-based catalysts for the deNO
x process under excess O
2 is related to the production of significant amounts of NO
2 via the oxidation of NO that increases along with the reaction temperature, resulting in a relatively narrow operating window of Pt/zeolite catalysts [
11,
12]. Therefore, it is required to control and tune the oxidation ability of Pt to limit the NO
2 emissions. Park et al. [
11] reported that a thermal ageing treatment of Pt/ZSM5 catalyst widened the temperature window of H
2-SCR by depressing the NO oxidation to NO
2, whereas Machida and Watanabe [
13] reported enhancements of N
2 selectivity by adding various amounts of sodium to a 1 wt% Pt/ZSM5 catalysts.
In this work, we set out to investigate the H2-SCR reaction of NO over a series of Pt/ZSM5 catalysts with limited metal loadings (0.1–1 wt%) and doped with Na (5–10 wt%) that was added to inhibit the NO oxidation activity. The effects of sodium addition on the main features of the original catalyst, such as the surface area and acidity, were studied to identify its inhibiting mechanism and the optimal loading that preserves the catalytic properties in the H2-SCR. Eventually, an in situ DRIFTS study was performed to investigate the main surface species formed during the reaction both on the unpromoted and the Na-promoted catalysts and to assess their possible role in the reaction mechanism.
2. Results and Discussion
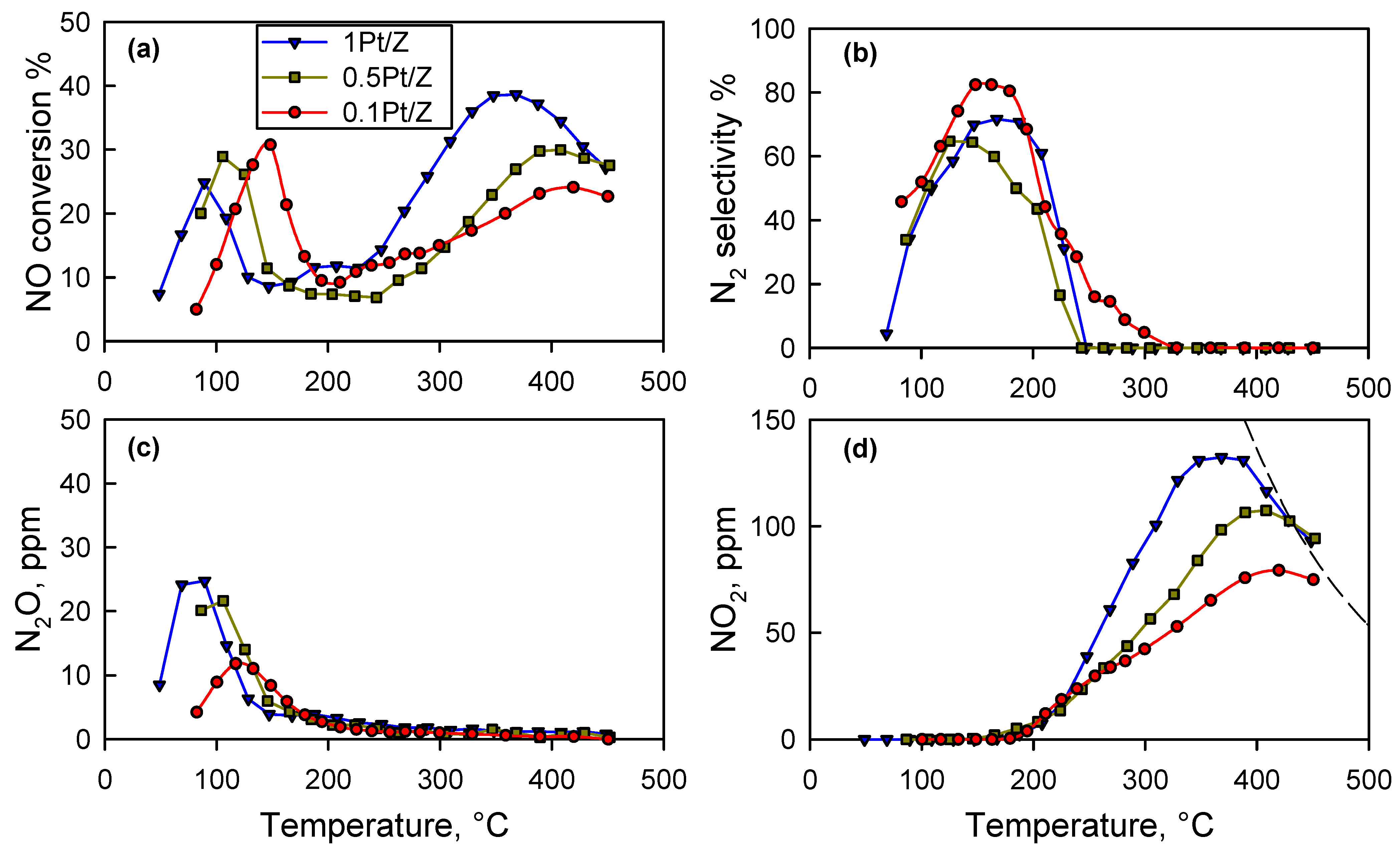
The parent commercial ZSM-5 zeolite did not show any activity for the H
2-SCR reaction of NO, confirming the results reported by others [
14,
15,
16]. The addition of even a low amount of Pt (i.e., 0.1 wt%) activated the NO molecule, which was consumed in two different temperature ranges: from ca 80 to 200 °C and from 220 up to 450 °C (
Figure 1). However, it must be noticed that for T > 200 °C, a NO
2 production was detected that increased along with the temperature and peaked at 380–410 °C depending on the precious metal loading. As a consequence, only in the lower temperature range, the NO conversion was related to the occurrence of reaction (1) with high selectivity to N
2 (
Figure 1b), which inevitably dropped to zero as soon as all of the H
2 in the feed was consumed (oxidized to H
2O).
The maximum NO conversion in the low temperature range, was observed with 0.1Pt/Z catalyst at 144 °C and corresponded to 31%. This value is lower than others reported in the literature for zeolite-supported platinum catalysts (summarized in
Table 1) even if a direct comparison is somehow precluded due to significant differences in the experimental conditions (particularly feed compositions and WHSV). The undesired formation of some N
2O was observed, as also reported by others [
9]: it was higher for larger platinum loadings, and it peaked at the same temperature of the NO conversion with a direct impact on the process selectivity to N
2 (
Figure 1b,c). Burch et al. [
7,
17] stated that N
2 and N
2O are produced by a parallel process involving a pre-adsorbed NO intermediate that can react with another adsorbed NO species to produce N
2O or with an adsorbed N atom to give N
2. The ratio between adsorbed hydrogen and adsorbed NO is also considered a key parameter in determining the N
2 selectivity with respect to N
2O [
18]. According to these hypotheses, it can be argued that larger platinum clusters/nanoparticles can promote the adsorption of more NO intermediates close to each other, thus favouring their reaction to form N
2O.
As already mentioned, for T > 200 °C, the conversion of NO was mostly associated with its oxidation with O
2, producing NO
2. This agrees well with data reported in [
13] showing that NO conversion to NO
2 increased between 150 and 300 °C with increasing concentrations of O
2 up to 10% in the feed. Hong et al. [
14] investigated the NO oxidation activity of Pt/ZSM5 by feeding a mixture of NO and O
2 without H
2. They observed an increasing NO
2 formation from 100 to about 250 °C catalyzed by platinum. As shown in
Figure 1d, the oxidation of NO to NO
2 significantly increased along with the Pt content in the catalysts and became limited by the approach to the thermodynamic equilibrium [
11,
14] above 380–420 °C.
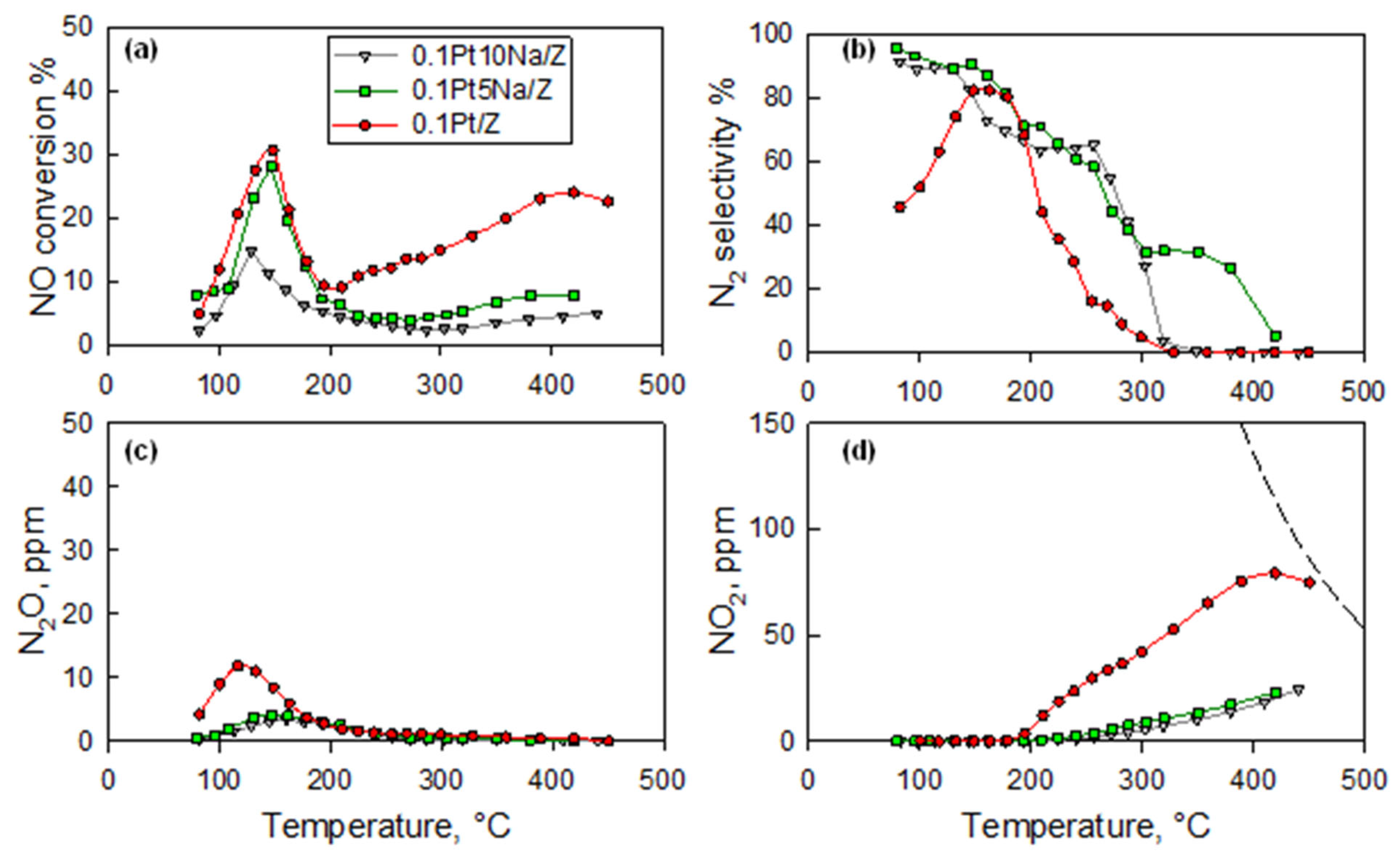
Figure 2 reports the effect of the addition of 5 or 10% Na to the 0.1Pt/Z catalyst on the NO conversion as well as the undesired production of NO
2 and N
2O. The addition of 10% Na almost completely suppressed the NO
2 formation at high temperatures, but, unfortunately, it also strongly inhibited the overall NO conversion rate. On the other hand, the addition of 5% Na proved effective in depressing the undesired NO
2 formation at high temperatures as well as the N
2O formation (reaction (2)), which took place simultaneously with reaction (1) in the low-temperature range while preserving the activity towards the H
2-SCR. Such results align with those reported by Machida and Watanabe on similar catalysts [
13], who found that Na doping could promote both NO conversion and N
2 selectivity. Burch and Coleman [
17] observed a similar qualitative effect of Na doping on Pt/Al
2O
3 catalyst for H
2-SCR under lean conditions, although they did not detect the inhibition of N
2O formation, which was observed for the ZSM5-supported catalyst.
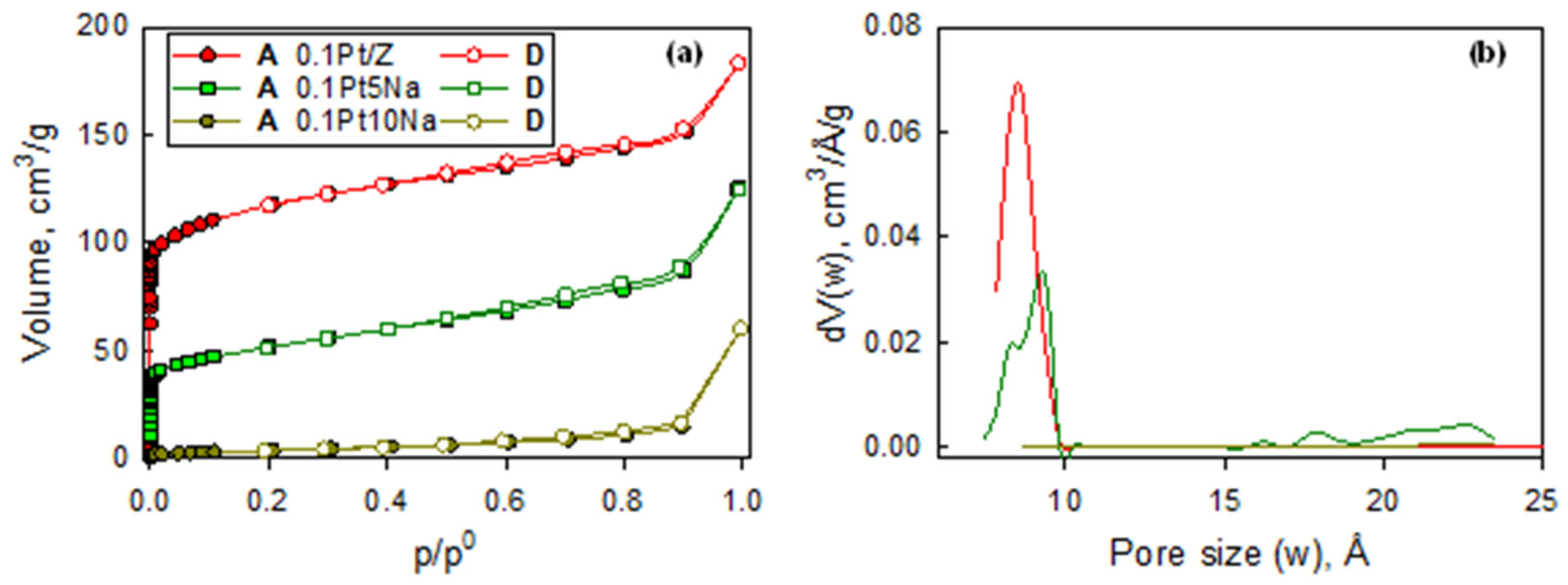
Figure 3 presents the N
2 isotherms at 77 K for 0.1Pt/Z, 0.1Pt5Na/Z, and 0.1Pt10Na/Z catalysts. It can be observed that while 0.1Pt/Z and 0.1Pt5Na/Z preserved their typical shape characteristic of type I microporous materials (such as their parent ZSM5 support), the addition of 10% Na caused the almost complete loss of microporosity. The progressive reduction in the volume of micropores is clearly recognized in
Figure 3b, where the area of the contribution of pores with a size <10 Å is roughly halved upon the addition of 5% Na, and it is basically zero upon the addition of 10% Na (
Table 2).
The parent zeolite, after thermal treatment at 550 °C, required to decompose the ammonium ion and obtain the zeolite in its H-form, showed a BET area of 430 m
2 g
−1 [
19]. The addition of 0.1% Pt did not change the textural properties or the crystalline structure (PDF n. 04-13-2411), as confirmed by the comparison of the relevant XRD patterns shown in
Figure 4. However, doping with 5% or 10% Na reduced the BET surface area of the samples down to 184 and to only 14 m
2 g
−1, respectively (
Table 2). XRD analysis indicated the loss of surface area and porosity (particularly evident for the 0.1Pt10Na/Z catalyst) was not accompanied by any major modification of the crystalline structure of the parent zeolite, but rather it was due to pore blocking caused by the accumulation of Na-nitrate species giving a characteristic peak at 29.3° (PDF n. 079-2056) as shown in
Figure 4.
Accordingly, SEM analysis performed on the 0.1Pt5Na/Z catalyst (
Figure 5) revealed the presence of needle-like crystals with a variable dimension (length 5–20 µm, width 1–5 µm) dispersed among irregular particles. The corresponding EDX mapping of Si, Al, and Na indicated that these crystals are mostly composed of sodium, thus corresponding to the NaNO
3 phase detected by XRD. On the other hand, the irregular particles mainly consisted of silicon and aluminum, clearly suggesting the assignment to the zeolite phase.
A 49% platinum dispersion and an average particle size of 2.3 nm was evaluated for the 1Pt/Z catalyst using the H2 chemisorption technique, whereas a correct estimation of dispersion was not possible for those materials with 0.1% Pt content due to the overall small quantities of H2 chemisorbed. Therefore, a semi-quantitative analysis of platinum dispersion was performed by DRIFTS measurement of CO adsorption.
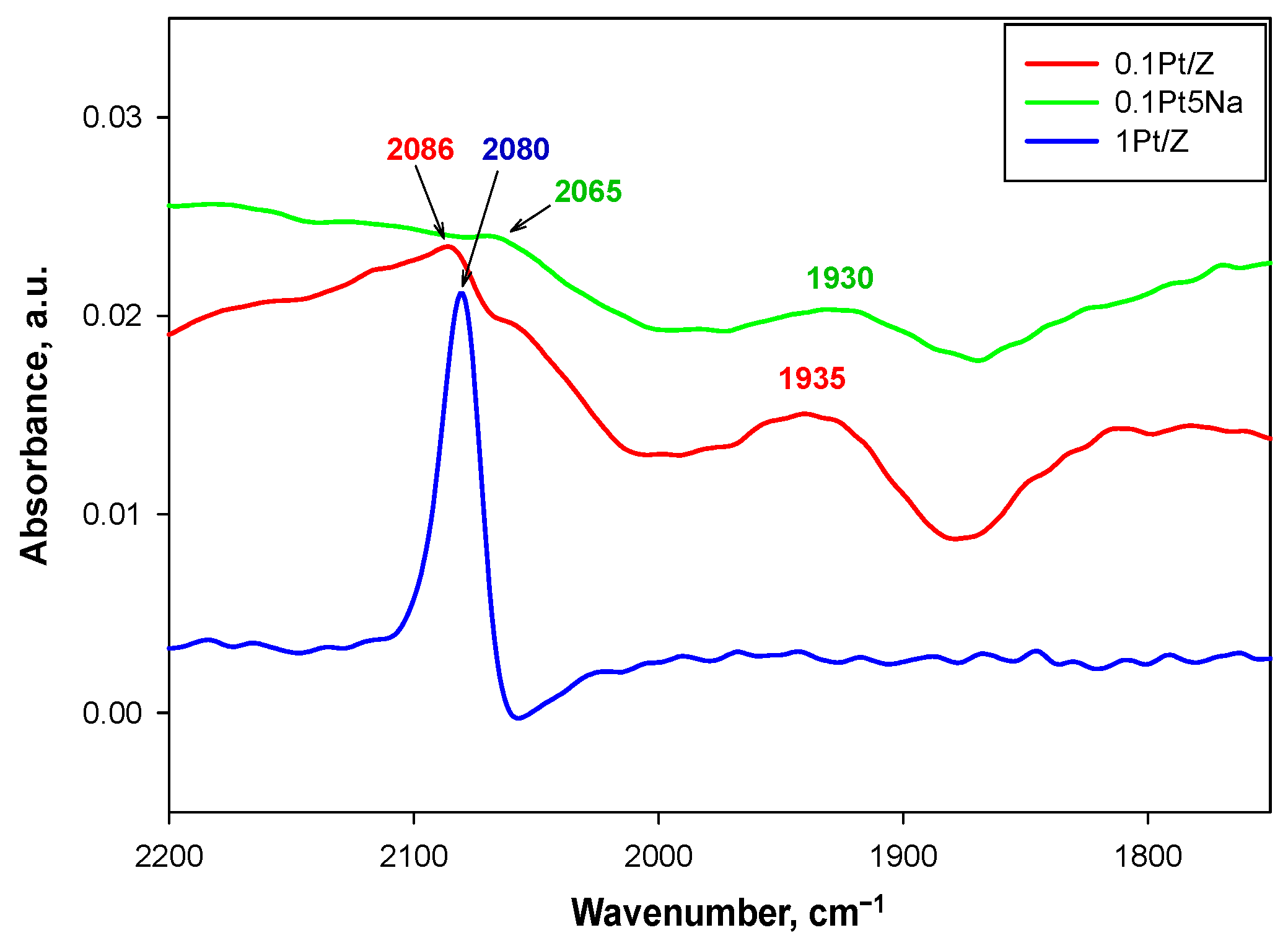
In
Figure 6, the DRIFT spectra relevant to CO adsorption on the 0.1Pt/Z, 0.1Pt5Na/Z, and 1Pt/Z catalysts are presented. A single large and sharp band was detected at 2080 cm
−1 for the 1Pt/Z sample, whereas very small bands were detected in the same region for the two catalysts with 0.1%Pt loading. The 0.1Pt/Z catalyst showed a broad band at 1935 cm
−1 in addition to a small band at 2086 cm
−1. The band at 2080–2060 cm
−1 was almost absent for the 0.1Pt5Na/Z sample, and one at 1930 cm
−1 was significantly reduced compared to the Na-free counterpart.
A band at 2075–2085 cm
−1 has been commonly reported for CO adsorbed on Pt supported on zeolites and also other oxide supports [
15,
20,
21,
22,
23,
24].
For ν(CO) < 2100 cm
−1, it is assumed that CO is linearly adsorbed on Pt
0, whereas for ν(CO) > 2100 cm
−1, platinum is partially oxidized (Pt
δ+) [
22,
24]. Therefore, it can be argued that platinum was completely reduced in all three catalysts after the H
2 pre-treatment. Nevertheless, the shift in the carbonyl band from 2086 to 2065 cm
−1 for 0.1Pt/Z in the presence of sodium could indicate a lower oxidation state of platinum, as also suggested by Zhang et al. [
15] for W-promoted Pt/ZSM5 catalysts. In good agreement with our results, Rivallan et al. [
21] assumed for 2%Pt/ZSM5 that the band they detected at 2095 cm
−1 was associated with CO on large Pt
0 particles with an average dimension of 6 nm, whereas a shift towards 2085 cm
−1 indicated CO adsorption on smaller (about 2.5 nm) Pt
0 particles. Similarly, Han et al. [
20] attributed the band at 2075 cm
−1, detected for platinum dispersed on L zeolite (load ranging from about 0.5 to 0.7%), to CO on Pt small clusters into zeolite pores.
The band at lower frequencies (1930–1935 cm
−1) is less commonly reported in the literature. Han et al. [
20] reported that signals in the region 1900–1700 cm
−1 can be assigned to bridged-bonded carbonyl species; in particular, they assigned a 1935–1920 cm
−1 band to CO species between zeolite framework and Pt atoms. Consistently, Bazin et al. [
22] reported that this low-frequency band was due to CO on very low coordinated Pt atoms strongly interacting with the support. Based on these assignments, it can be argued that at very low loadings (0.1% wt%), all the Pt strongly interacted with the ZSM5 support being atomically dispersed. However, the accessibility of these Pt sites was somewhat limited by the further addition of sodium. At variance, when the metal loading was 1 wt%, the nature of the noble metal sites changed significantly due to the strong prevalence of small clusters with a characteristic size of ca 2–3 nm.
Figure 7 presents the NH
3-TPD profiles following ammonia saturation at room temperature. The characteristic traces for the parent zeolite and the two Pt/Z supported catalysts were almost overlapped regardless of the different Pt loadings, thus suggesting that the acid sites typical of the H-ZSM5 were preserved after the dispersion of the noble metal. Specifically, three main desorption events were detected, peaking at 110–120 °C, 215 °C and 440 °C. Hong et al. [
14] reported that all Pt/ZSM-5 samples prepared by different methods showed two distinct NH
3 desorption peaks (below 300 °C and up to 600 °C) associated, respectively, to weakly acidic silanol groups and weak Lewis (or Brønsted) acid sites, and to the stronger acid sites. They also observed that the peaks at high temperatures became less intense after the introduction of platinum, indicating that some of the available strong acid sites were blocked by platinum. The same effect was reported by Yu et al. [
25] for Pt/ZSM5 and Pt/ZSM35 but it was rather limited for our catalysts.
As expected, the addition of sodium strongly changed the acid properties of the catalyst: the most evident feature, common to both Na loadings, was the disappearance of the peak in the range 300–500 °C, representing the strong acid centres. However, for the 0.1Pt5Na/Z catalyst, most of the acid sites desorbing ammonia up to 300 °C were preserved, whereas they were totally neutralized at 10%Na load. For the 0.1Pt5Na/Z catalyst, new additional acid centres at 250–350 °C were formed whose nature was not defined.
Therefore, it seems that the target acid sites upon sodium addition are mainly those releasing ammonia in the 350–500 °C temperature range, as also observed by Hong et al. [
14] for higher Pt loads. Zhang et al. [
15] also observed a slight reduction in acid sites in the same temperature range upon tungsten addition to a 1Pt/ZSM5 catalyst. They associated this reduction with that of the silanols and Brønsted bands detected by FTIR, concluding that both platinum and tungsten occupied these positions. These certainly are the main targets of sodium in our catalysts, although other acid sites cannot be totally excluded. However, at 10 wt% of Na, medium acid strength sites were also neutralized with an evident negative impact on the SCR activity (see
Figure 2a).
Hong et al. [
14] found a rough correspondence between the total amount of acid sites and the NO conversion at 90 °C, corresponding to the maximum selectivity to N
2. Yu et al. [
25] reported that at high Pt load, when the large platinum particles blocked acid sites, the H
2-SCR dropped. Our results seem to suggest that weak and medium-strength acid sites (responsible for the first two partially overlapped peaks in NH
3-TPDs) can be involved in the H
2-SCR, whereas strong acid sites could promote the NO oxidation to NO
2.
Figure 8 presents the in situ DRIFTS spectra recorded over the 0.1Pt/Z, 1Pt/Z, and 0.1Pt5Na/Z catalysts at 30 °C under a NO/O
2/Ar mixture and after H
2 was added to the gaseous feed at increasing temperatures up to 230 °C. These temperature levels were selected in order to investigate a range (60–130 °C) where reaction (1) mainly occurred and a temperature (230 °C) where NO oxidation to NO
2 took place.
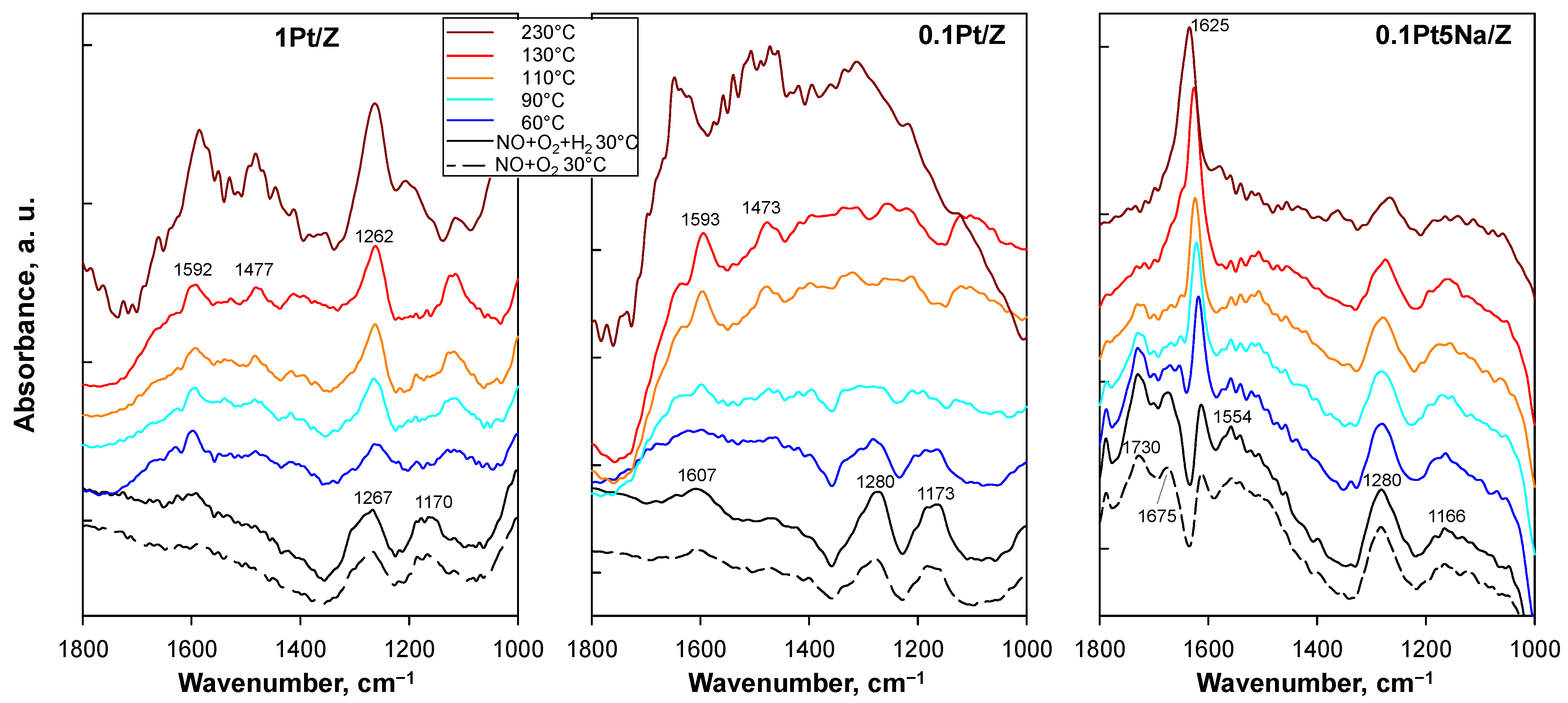
Two bands at 1267–1280 cm−1 and at 1170–1173 cm−1 were present in the spectra of both 0.1Pt/Z and 1Pt/Z, along with a very small signal at ca 1600 cm−1 after the adsorption of NO+O2 at 30 °C (dashed lines). The intensity of these signals did not change significantly regardless of the Pt loading, and they were also observed in the parent H-ZSM5, suggesting they can be assigned to NO on the zeolite rather than on platinum. The simultaneous presence of H2 at this temperature increased the intensity of all these bands. New bands at 1593 cm−1, with a shoulder at 1625 cm−1 more evident for 0.1Pt/Z, and 1473 cm−1 appeared by increasing the temperature up to 130 °C. Interestingly, the intensity of the band at 1267–1280 cm−1 increased along with the temperature for the 1Pt/Z catalyst, whilst this feature disappeared for the sample with a low platinum content. Moreover, the signal at 1170 cm−1 shifted towards lower wavenumbers for 1Pt/Z.
At 230 °C, when the NO oxidation prevailed on the SCR, different bands were detected on both catalysts: for 0.1Pt/Z at 1650, 1480, and 1312 cm−1 and for 1Pt/Z at 1586 and 1480 cm−1 whereas a shoulder at 1203 cm−1 appeared on the sharp band peaked at 1260 cm−1, already present at lower temperatures.
Na-doping strongly affected the main features of the DRIFT spectra. In addition to the low-frequency bands (at 1280 and 1166 cm
−1) also observed for the unpromoted catalysts, which confirmed the major role of the zeolite support in this spectral region, bands at 1554, 1675, and 1730 cm
−1 were detected, together with a sharp one at 1625 cm
−1 becoming even more intense at higher temperatures. A quite intense band at 1625 cm
−1 was also detected by Zhang et al. [
15] upon NO or NO+O
2 adsorption on Pt/ZSM5 catalysts, either with or without tungsten-doping, whilst its intensity was reduced under the complete NO-H
2-O
2 mixture for the W-containing catalyst. A band at this position was previously reported by Szanyi and Paffett [
26] for NaH-ZSM5, and it was found to increase with the reaction time, thus suggesting it was due to nitrate species bound to Na
+ ions in cationic positions. The possible contribution of bulk sodium nitrate from the crystallites detected by XRD analysis, expected at ca 1270 and 1635 cm
−1 [
27], must be excluded because it was deleted by the background ratioing operation. As a consequence, this feature should represent nitrate species formed upon exposure to the NO-containing mixture and can be definitively associated with the presence of sodium for our catalysts since the same band was very weak for the unpromoted sample.
The presence of H2 in the feed mixture did not significantly modify the main signals for the Na-doped catalysts; however, the temperature increase resulted in the disappearance of the bands at 1675, 1730, and 1554 cm−1, as well as a reduction in the intensity of the signals at 1280 and 1166 cm−1.
The attribution of the main bands of adsorbed NO
x species was reported by Morrow et al. [
28] for Pt/SiO
2. They found that the intensity of bands at 1785 and 1620 cm
−1, assigned to linear and bent or bridged NO on Pt, respectively, decreased upon O
2 introduction, replaced by a band at 1710 cm
−1 and one at 1545 cm
−1 assigned to linear NO and bidentate nitrate species on oxidized Pt, respectively. A band at 1540 cm
−1 was also assigned to adsorbed N
2O
3 dimer on the zeolite, which was detected by Szanyi et al. [
29] on Na-Y upon the addition of O
2 to gaseous NO, suggesting these are oxidized adsorbed species. The low-frequency bands were assigned by the same authors to nitrate species on the support.
On this basis, the formation of a band around 1590 cm−1 by increasing the temperature in the presence of O2 suggests that some platinum could be oxidized under excess O2, even if in the co-presence of H2. Moreover, the reduction in the bands at 1785 and 1675 cm−1, coupled with the growth of the 1625 cm−1 signal, which remained stable upon H2 addition at increasing temperatures, suggests the formation of a species which does not evolve into NO2.
The band at 1473–1477 cm
−1, well detected for both unpromoted catalysts in the presence of H
2 at high temperature, was attributed to NH
4+ (on Brønsted acid sites) produced by the reduction in the nitrous species [
16,
18]. Eventually, the bands identified only for the Na-containing catalyst in the 1800–700 cm
−1 range were assigned to NO dimers [
13,
30].
At 230 °C, a temperature promoting the oxidation of NO to NO2, the main features of the spectrum did not markedly change for the Na-promoted catalyst, whereas bands similar but with a worse signal/noise ratio were detected for both 0.1Pt/Z and 1Pt/Z.
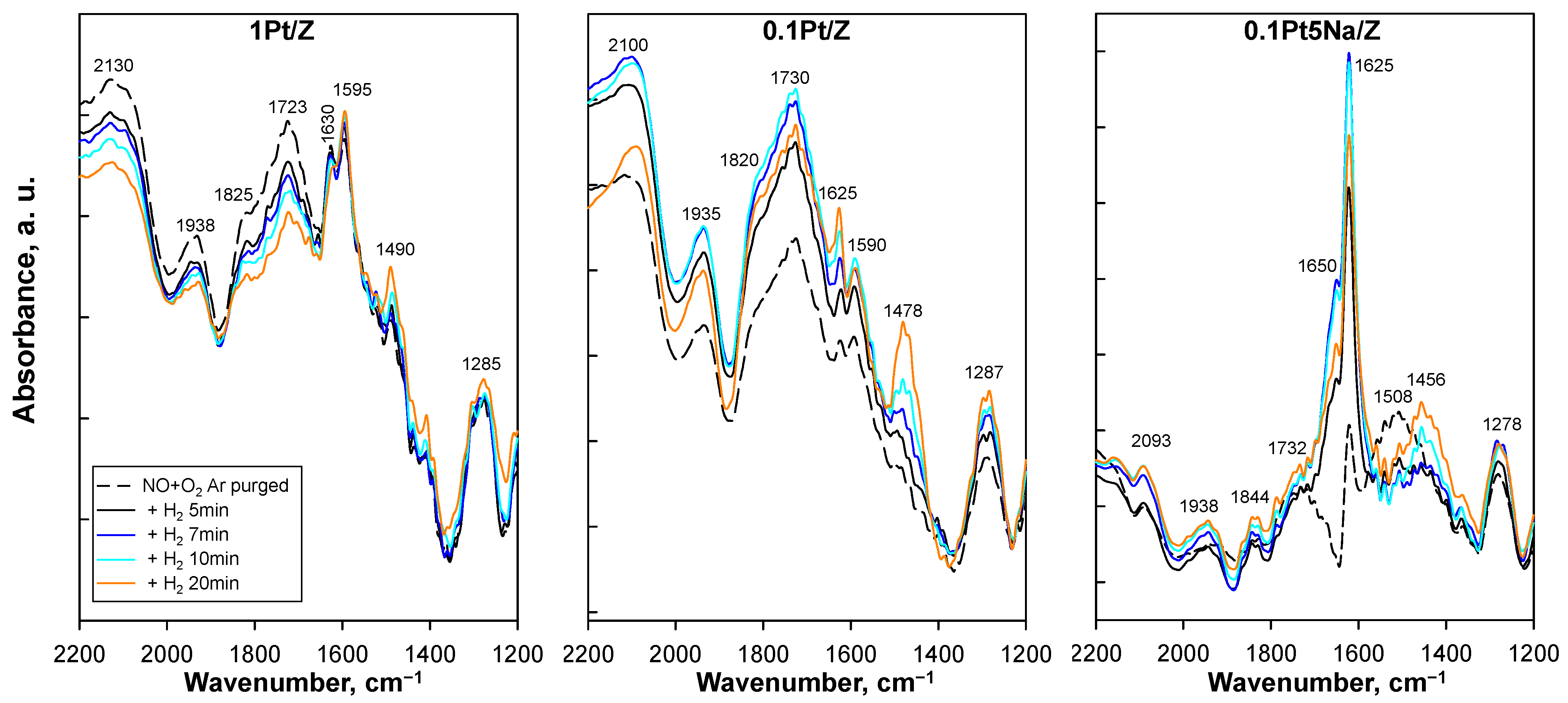
In order to better identify the adsorbed NO
x species involved in the SCR reaction, H
2 was introduced at 100 °C, a temperature where the SCR prevails after NO/O
2 was pre-adsorbed on the catalyst. In
Figure 9, the corresponding DRIFT spectra are reported for the 0.1Pt/Z, 1Pt/Z, and 0.1Pt5Na/Z catalysts. The same experiments performed over the parent H-ZSM5 revealed low-intensity broad bands in the regions 1800–1600, 1600–1400, and 1400–1200 cm
−1 [
31] that did not change upon H
2 introduction, thus confirming the key role of platinum to activate the H
2-SCR reaction.
As for the spectra recorded under reaction conditions, the features of 0.1PtNa/Z were markedly different from those of the unpromoted catalysts. Following exposure to NO/O
2, both unpromoted samples showed a broad band peaked at 1723–1730 cm
−1 with a shoulder at 1819–1826 cm
−1 that can be attributed to adsorbed nitrate species on Pt [
13] or to NO dimer [
14] also on H-ZSM5 [
29,
30]. These species were not detected when NO, O
2, and H
2 were simultaneously co-fed at a similar temperature level, and this could indicate their steady-state consumption under reaction conditions. Two further bands at 1590–1595 and 1625–1630 cm
−1 were clearly detected over 1Pt/Z as well as 0.1Pt/Z, though with lower intensity. At variance, a large broad band peaked at ca 2100 cm
−1 for 0.1Pt/Z, decreased with platinum loading and even more with Na addition. This feature was assigned to 2O-NO
+ generated by the reaction between N
2O
3 (from NO+NO
2) and protons of H-ZSM5 when it is exposed to NO+O
2 [
30]. The decrease in this band in 1Pt/Z and 0.1Pt5Na/Z indicates the disappearance of acid centres related to the dispersion of metals and can be associated with the loss of weak acid centres detected in TPD experiments.
The addition of H2 at 100 °C did not modify the large band in the range 1700–1850 cm−1 for 0.1Pt/Z, whereas it progressively reduced it along with the time on stream for 1Pt/Z suggesting these species could be involved in the undesired formation of N2O. Nevertheless, it must be reiterated that the 1Pt/Z catalyst activated the SCR reaction at a lower temperature. The other bands did not change significantly with time on stream, excluding the increase in the small band at ca 1490 cm−1 along with the small signal at 1625 cm−1, highlighting a major role of ammonium as a reaction intermediate at low Pt load.
As already observed for the experiments under steady-state reaction conditions, the band at 1625 cm
−1 represented the dominant band in the spectrum of 0.1Pt5Na/Z, and it strongly increased upon H
2 addition with ToS up to 10 min along with a shoulder at 1650 cm
−1, associated with linear NO on Pt [
28]. Furthermore, a band initially present at 1510 cm
−1 in the absence of H
2 was gradually replaced by a new band at 1450 cm
−1, suggesting that bidentate nitrate species on oxidized Pt were transformed into adsorbed ammonium species, which then produced molecular nitrogen according to the reaction:
as proposed by Wang et al. [
6]. The effect of H
2 on all the other signals was negligible for the Na-promoted catalyst.
It must be noticed that the ammonium band was well detectable for the two catalysts providing the best N2 selectivity whereas the formation of ammonium appeared to be inhibited at high Pt loading.
In conclusion, it can be argued that the catalyst with a low Pt content (and high dispersion) activates the reaction path involving the formation of nitrates (likely dimers) on highly dispersed platinum associated with the band in the 1590–1510 cm−1 region, which can easily evolve into ammonium intermediate. Moreover, the neutralization of the strong acid sites provided by Na-doping (at 5 wt%) can suppress the undesired NO oxidation without affecting the Pt activity or the low- and medium-strength acidity, which plays an important role in the activation of the H2-SCR reaction. The selective reaction path is indeed still preserved at a low Na load, as shown by the detection of active nitrate and ammonium-adsorbed species on this promoted catalyst.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}