
Frequency-Resolved Modulation Excitation Spectroscopy Methodology for Identifying Surface Reaction Species in Ethanol Oxidation on Gold Catalysts



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
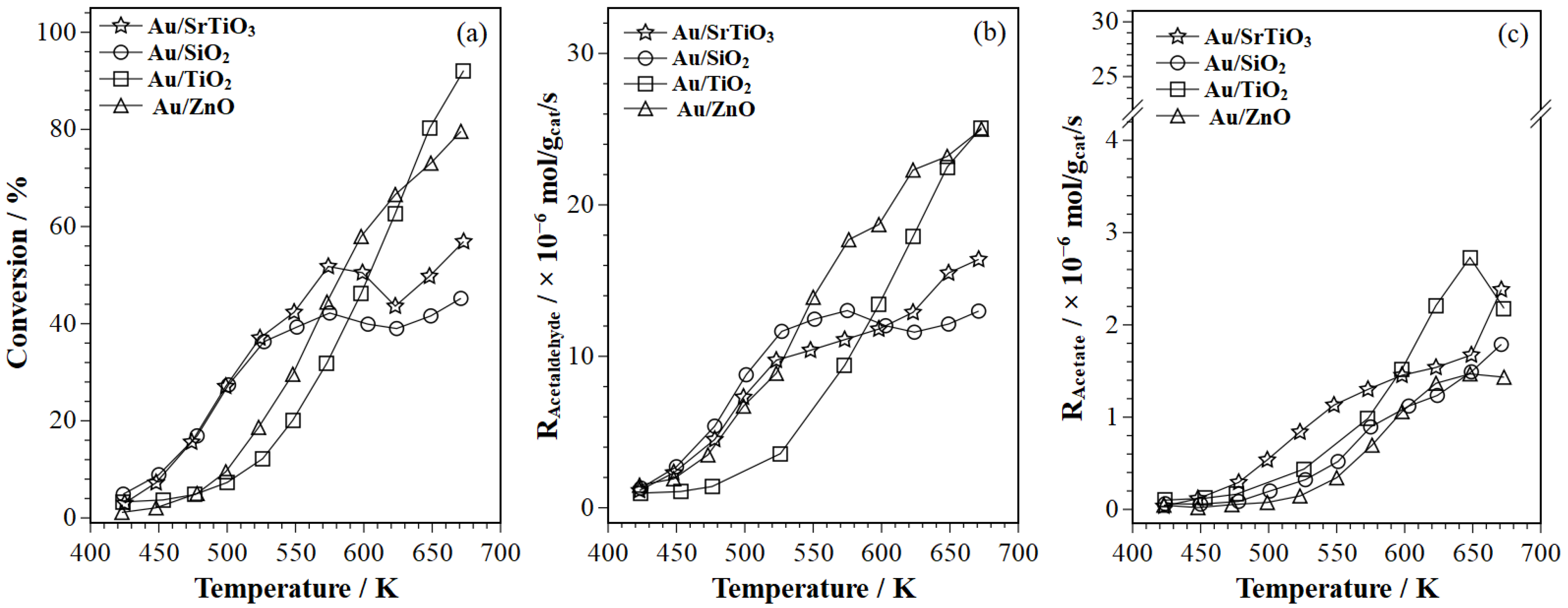
2.1. Ethanol Oxidation Catalytic Activity Results
2.2. Operando Modulation Excitation–Diffuse Reflectance Infrared Spectroscopy–Phase Sensitive Detection (ME–PSD–DRIFTS)
2.2.1. Time-Domain Spectra
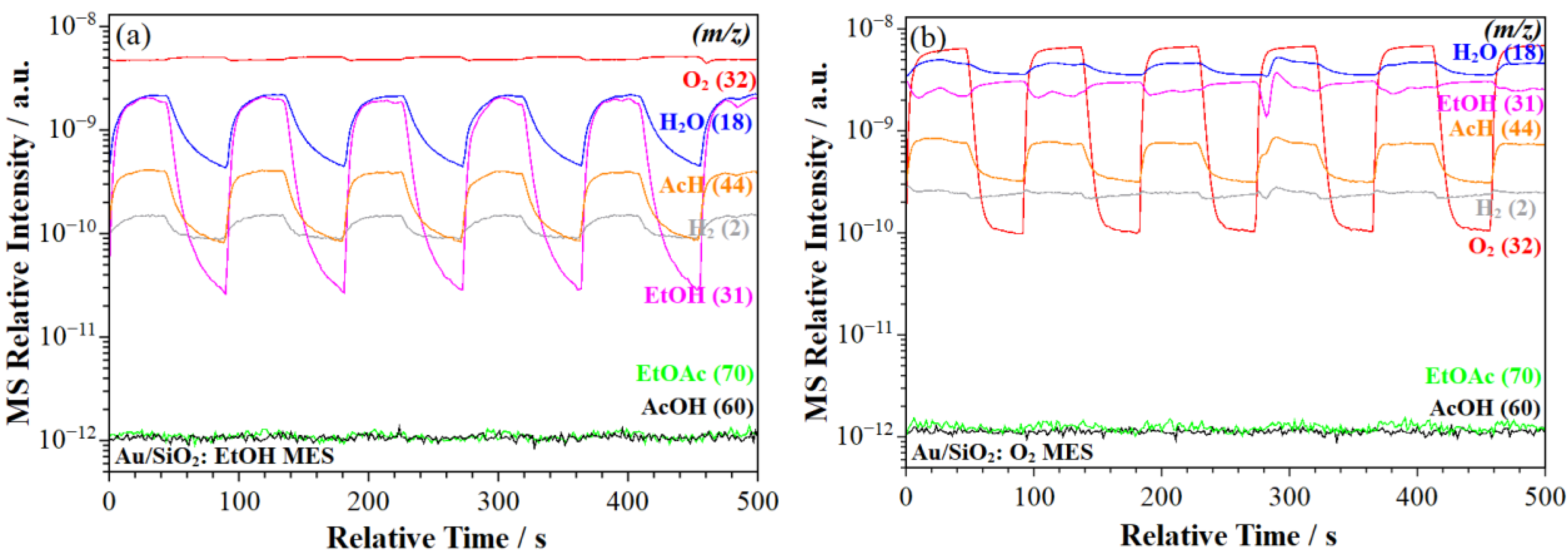
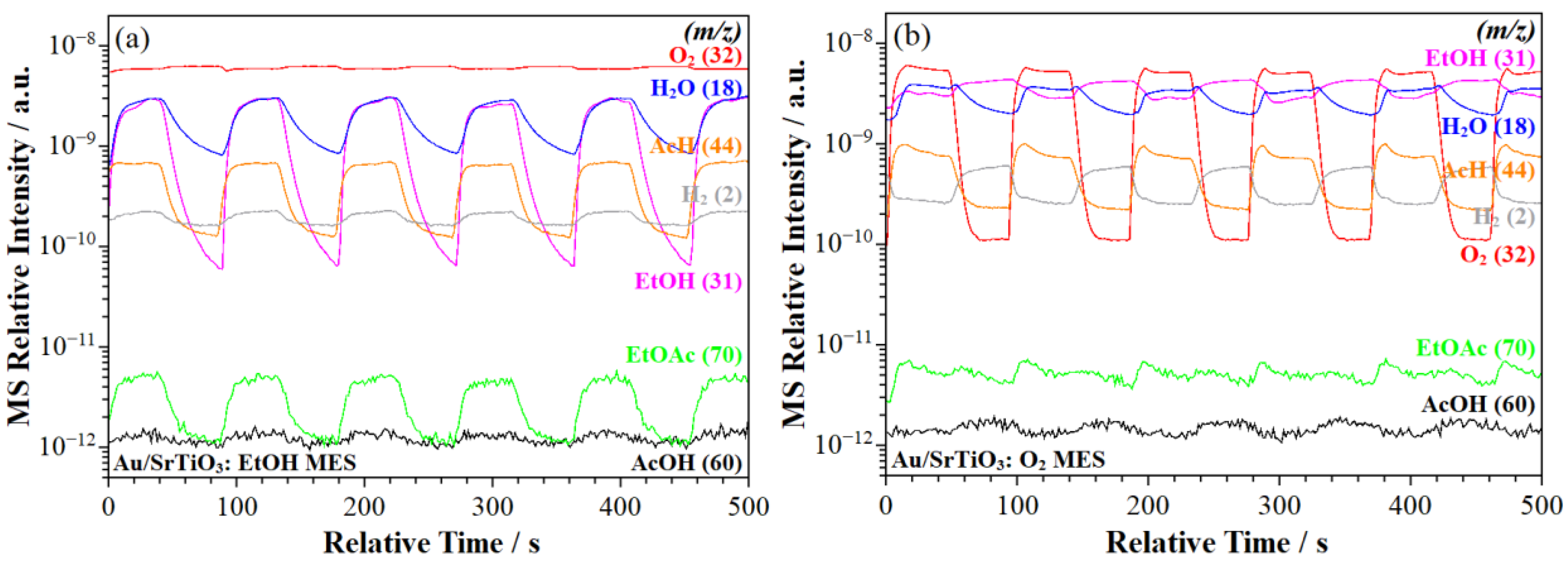
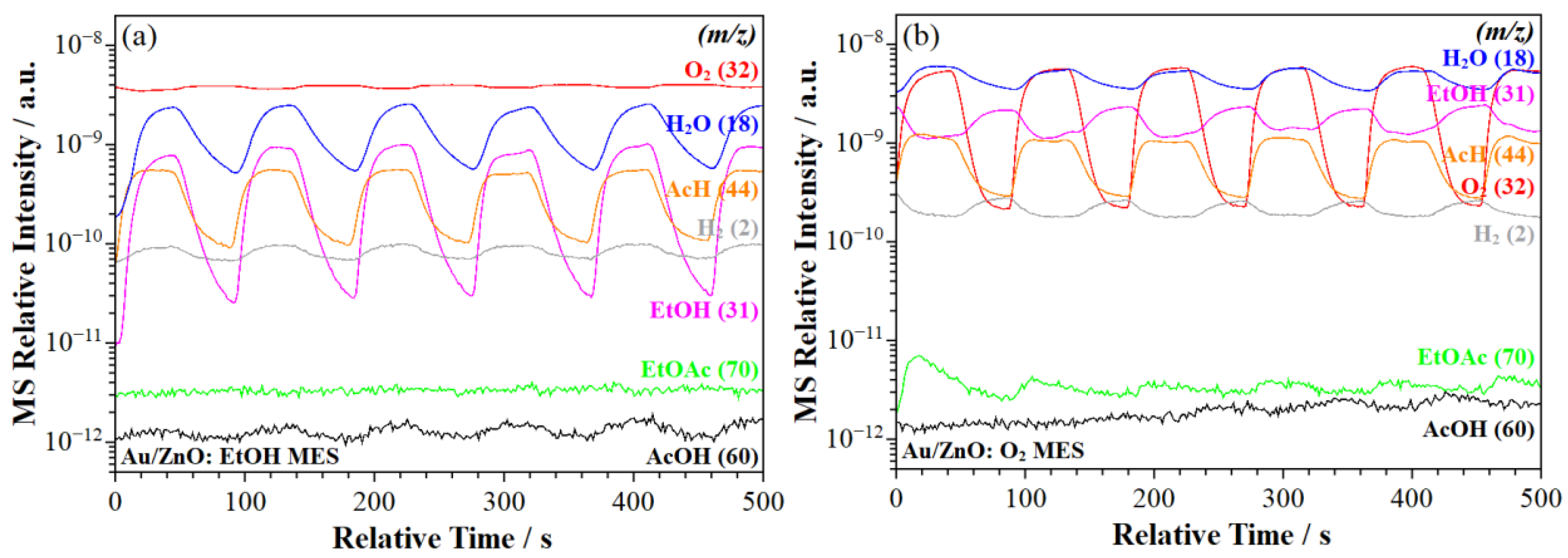
2.2.2. MS Signal Response
2.2.3. Time and Frequency-Domain Absorbance IR Spectra
2.2.4. Phase-Domain Contour Plots During ME–PSD–DRIFTS
2.2.5. Phase–Domain Trace Plots During ME–PSD DRIFTS with PSD at the Fundamental Frequency (1f0)
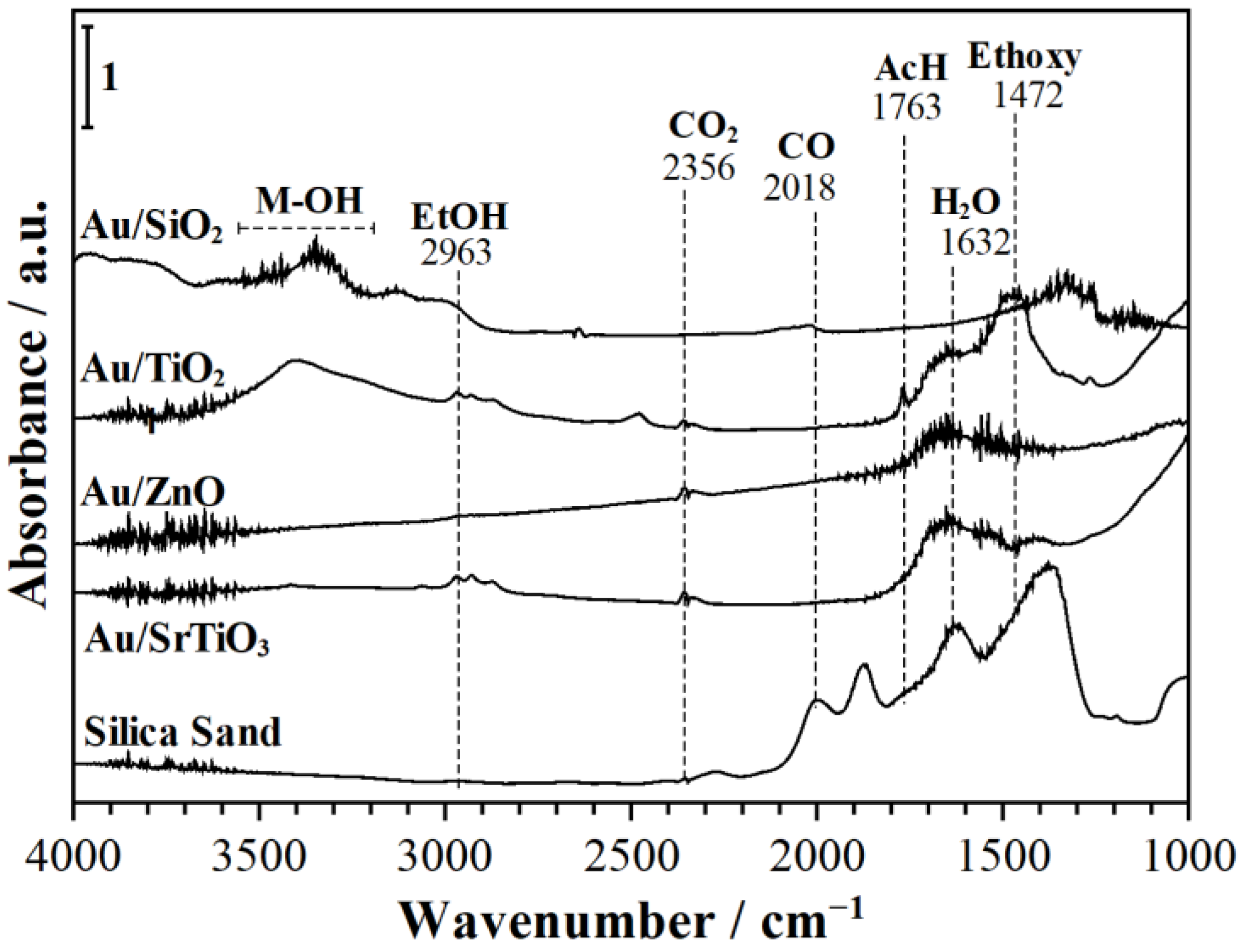
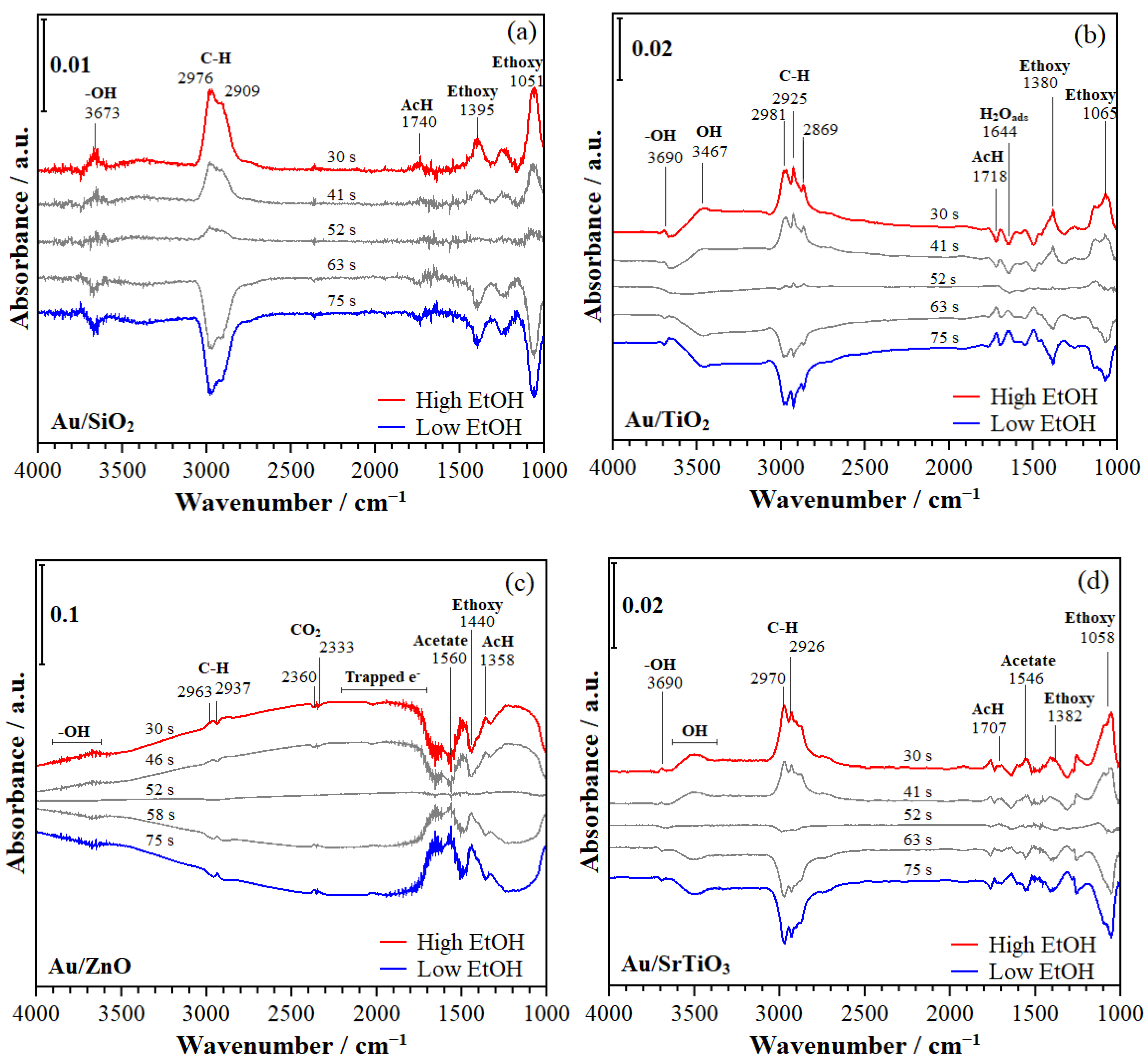
- C-H stretching (C-H). (i) During EtOH modulation (Figure 7), 2976, 2909 cm−1 (on Au/SiO2); 2981, 2925, 2869 cm−1 (on Au/TiO2); 2963, 2937 cm−1 (on Au/ZnO); and 2870, 2926 cm−1 (on Au/SrTiO3). (ii) During O2 modulation (Figure 8), 2966, 2874 cm−1 (on Au/TiO2); 2963, 2896 cm−1 (on Au/ZnO); and 2960, 2873 cm−1 (on Au/SrTiO3).
- Hydroxyls stretching (-OH). (i) During EtOH modulation (Figure 7), 3673 cm−1 (on Au/SiO2); 3690 cm−1 (on Au/TiO2); and 3690 cm−1 (on Au/SrTiO3). (ii) During O2 modulation (Figure 8), ~3700 cm−1 (on Au/SiO2); 3690 cm−1 (on TiO2); ~3700 cm−1 (on Au/ZnO); and ~3700 cm−1 (on Au/SrTiO3). These signals could arise from adsorbed ethanol and/or support metal hydroxyls [52,53,54,55,56,61].
- Among other relevant peaks, broad shoulders in the range of 3500–3000, 2500–2000, and ~1275 cm−1 are indicative of ethanol, water, and hydrogen being bonded to the surface hydroxyl groups (OH), trapped electrons (especially during O2 modulations on Au/TiO2 and Au/ZnO), and carbonates, respectively.
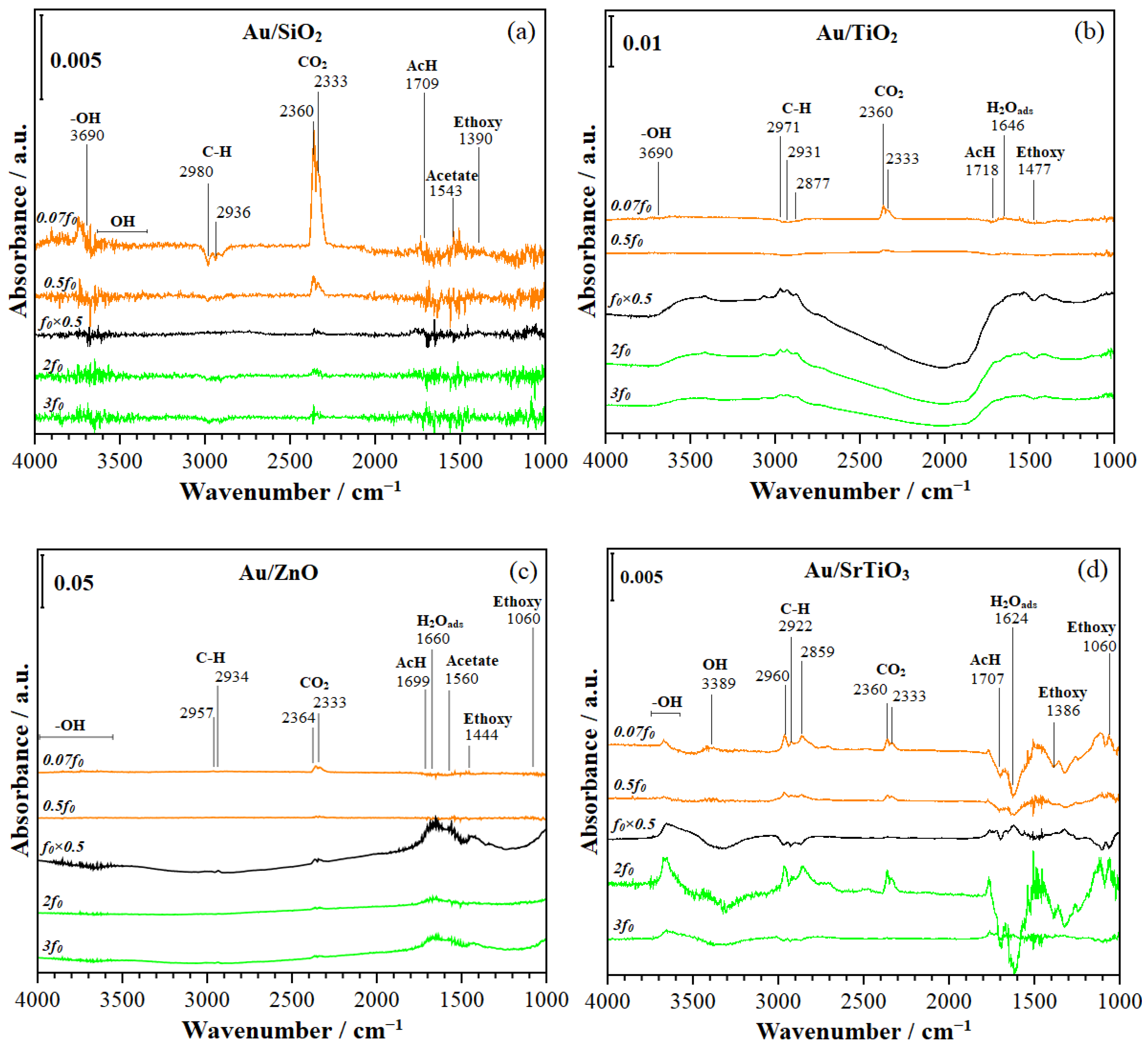
2.2.6. Phase-Domain Trace Plots During ME-PSD-DRIFTS with PSD at 0.07f0, 0.5f0, 2f0, and 3f0
2.3. Modulation Excitation–Phase Sensitive Detection–Diffuse Reflectance UV–Vis Spectroscopy (ME–PSD–DRUV–Vis)
2.4. Possible Reaction Intermediates and Reaction Scheme During Ethanol Oxidation
- Reactant adsorption. Ethanol oxidation reaction on metal oxide supported gold catalysts proceeds through the dissociative adsorption of ethanol on the catalyst surface (Figure 7 and Figure 8), more likely on Au. This was demonstrated by DFT calculations on Au5/TiO2(101) [68]. The preferable adsorption of O2 at the Au–support interface, as mentioned in Section 2.3, was also evidenced by an in situ gold maximum plasmon peak shift (MaPPS) methodology, described in Refs. [68,69] (Scheme 1, steps 1 and 5).
- Ethanol dehydrogenation. Adsorbed ethanol dehydrogenates to form ethoxy species (at 1000–1200 cm−1 described in Section 2.2.4) in a fast elementary step, which is likely equilibrated and forms H species. This proton can transfer charge to Au, as evidenced from the signals due to trapped electrons at 2000–2500 cm−1 in Figures S23 and S24 (Scheme 1, step 2).
- Oxygen reaction at the interface. The H species formed on gold can diffuse rapidly to the Au–support interface, where it can react with O2 (Section 2.3) to form hydroperoxyl species (OOH) in a fast step (Scheme 1, step 5), as proposed recently via a charge-transfer spectrokinetic analysis (CT–SKAn) methodology [68]. This fast reaction is also evidenced in phase-domain plots obtained during O2 modulation in Section 2.2.6. O2 activation at the metal-support interface in the presence of charge transfer from Au can also occur dissociatively to form On− and OH species (Scheme 1, step 6). The CT–SKAn methodology recently showed that Au/TiO2, OOH, On−, and OH are kinetically relevant reaction intermediates for ethanol oxidation [68].
- Acetaldehyde formation. Adsorbed ethoxy species can be oxidized at the Au–support interface with activated O2 species, most likely with atomic oxygen [32,68] to form acetaldehyde and H2O, which are adsorbed at the surface, as evidenced from bands in the 1700–1740 cm−1 and 3300–3500 cm−1, respectively, as detailed in Section 2.2.6 (Scheme 1, steps 3, 9).
- Acetic acid formation. The ME–PSD–DRIFTS results point to the further oxidation of acetaldehyde to acetic acid (Scheme 1, step 7), primarily on Au/SrTiO3, which more strongly adsorbs acetaldehyde (Scheme 1, step 3) on the catalyst surface. MES spectral dynamic changes point to adsorbed acetaldehyde undergoing different surface processes. In one process, weakly adsorbed acetaldehyde desorbs to the bulk phase via a fast elementary step (Scheme 1, step 4). In the second process, strongly bonded acetaldehyde is oxidized to acetic acid in a slow step, as evidenced from MS (Section 2.2.2) and discussed in Section 2.2.6 (Scheme 1, step 7). Au/SrTiO3, which exhibited the highest activity for acetate (acetic acid and ethyl acetate) formation, also showed the rapid desorption of these species (Scheme 1, steps 8 and 13), preventing combustion to CO2 (Section 2.2.6, Scheme 1, steps 10 and 11).
- ME–PSD–DRIFTS also indicated strong CO2 adsorption on all catalysts, along with a strong affinity for water (Scheme 1, step 9), further evidenced by the slow water desorption (Scheme 1, step 12) detected by MS (Section 2.2.2).
3. Materials and Methods
3.1. Materials
3.2. Catalyst Preparation
3.3. Catalyst Characterization
3.3.1. ME–PSD Experimental Setup
3.3.2. ME–PSD–DRIFTS Experimental Conditions During Ethanol Modulation
3.3.3. ME–PSD–DRIFTS Experimental Conditions During Oxygen Modulation
3.3.4. Modulation Excitation–Phase Sensitive Detection Methodology
- The reactant concentration is varied in a periodic fashion while simultaneously collecting infrared or UV–Vis spectra.
- IR and/or UV–Vis spectra are swiftly and consistently acquired at regular intervals (~1–2 spectra per second) to capture surface alterations coinciding with periodic changes in gas concentrations. The spectra are plotted in what is called the time domain.
- These time-domain spectra are processed via the Fourier transform (FT) equation, transforming time-domain data, f(t), into the frequency domain, F(ω).However, the above equation is solved in a form to deal with equally spaced spectral datasets, which is called the discrete Fourier transform (DFT). The solution to the DFT is performed with the fast Fourier transform (FFT) algorithm via the “fft” function in Python (https://www.python.org/). The resultant data will present the spectral response as a function of frequency, which is commonly referred to as the frequency domain.
- At this stage, one sorts the frequencies in ascending order (i.e., with the fft shift function in Python) and selects a frequency or range of frequencies of interest to filter the different species contributions to the spectra. Usually, the fundamental frequency (e.g., 1/90 Hz) is chosen to filter out reactive species that respond to that modulation frequency.
- Then, the filtered data in the frequency domain, F(ω), is processed via the inverse Fourier transform (IFT) to recreate the original data into an averaged f(t), where this t corresponds to the time of a single modulation period.Technically, this spectral response can be plotted against time (of a single period, in this case from 0 to 90 s), radians (0 to 2π), or degrees (0 to 360°). This is the reason why the data is said to be in the phase domain, and it contains spectral contributions from species that respond to modulation at the chosen frequency or frequencies.The above IFT equation is solved for discrete datasets via its inverse discrete Fourier transform (IDFT). We perform this transformation within Python using the “ifft” function, which effectively reverts the “filtered” data in the frequency domain back into the time domain of a single period (i.e., phase domain).
- The resulting “phase domain” dataset represents an average across all periods and in this work, for simplicity, it is plotted in time units. The specificity of the spectral response will depend on the frequency or frequency range chosen for filtering. When the filtered frequency is the fundamental one, the data obtained in the “phase domain” correspond to the sinusoidal contribution to the waveform, irrespective of the waveform shape of the system species.
3.4. Catalytic Activity Testing
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AcOH | Acetic acid |
| DFT | Discrete Fourier Transform |
| EtOH | Ethanol |
| EtOAc | Ethyl acetate |
| FBR | Fixed Bed Reactor |
| FFT | Fast Fourier Transform |
| FT | Fourier Transform |
| FTIR | Fourier Transform Infrared |
| IDFT | Inverse Discrete Fourier Transform |
| IR | Infrared |
| MaPPS | Maximum Plasmon Peak Shifts |
| ME | Modulation Excitation |
| ME–PSD–DRIFTS | Modulation Excitation–Phase Sensitive Detection–Diffuse Reflectance Infrared Fourier Transform Spectroscopy |
| ME–PSD–DRUV–Vis | Modulation Excitation–Phase Sensitive Detection–Diffuse Reflectance Ultraviolet Visible Spectroscopy |
| MES | Modulation Excitation Spectroscopy |
| MS | Mass spectrometry |
| NP | Nanoparticle |
| NTP | Normal temperature and pressure |
| PD | Phase Domain |
| PSD | Phase Sensitive Detection |
| RAcetaldehyde | Production rate of acetaldehyde (mol/gcat/s) |
| RAcetates | Production rate of acetates, acetic acid + ethyl acetate (mol/gcat/s) |
| SAcH | C2-carbon selectivity to acetaldehyde |
| SAcetates | C2-carbon selectivity to acetates, acetic acid + ethyl acetate |
| TRIC | Transition Relative Intensity Changes |
| UV–Vis | Ultraviolet-Visible |
References
- Cherepakhin, V.; Williams, T.J. Direct oxidation of primary alcohols to carboxylic acids. Synthesis 2021, 53, 1023–1034. [Google Scholar]
- Morris, H.E. Reactions of ethyl alcohol. Chem. Rev. 1932, 10, 465–506. [Google Scholar]
- Chen, J.; Fang, W.; Zhang, Q.; Deng, W.; Wang, Y. A comparative study of size effects in the Au-catalyzed oxidative and non-oxidative dehydrogenation of benzyl alcohol. Chem.—Asian J. 2014, 9, 2187–2196. [Google Scholar]
- Evans, W.L.; Day, J.E. The oxidation of ethyl alcohol by means of potassium permanganate. J. Am. Chem. Soc. 1919, 41, 1267–1285. [Google Scholar]
- Mallat, T.; Baiker, A. Oxidation of alcohols with molecular oxygen on solid catalysts. Chem. Rev. 2004, 104, 3037–3058. [Google Scholar]
- Najafishirtari, S.; Friedel Ortega, K.; Douthwaite, M.; Pattisson, S.; Hutchings, G.J.; Bondue, C.J.; Tschulik, K.; Waffel, D.; Peng, B.; Deitermann, M. A perspective on heterogeneous catalysts for the selective oxidation of alcohols. Chem.-A Eur. J. 2021, 27, 16809–16833. [Google Scholar]
- Yamaguchi, K.; Kim, J.W.; He, J.; Mizuno, N. Aerobic alcohol oxidation catalyzed by supported ruthenium hydroxides. J. Catal. 2009, 268, 343–349. [Google Scholar]
- Markó, I.E.; Giles, P.R.; Tsukazaki, M.; Chellé-Regnaut, I.; Urch, C.J.; Brown, S.M. Efficient, aerobic, ruthenium-catalyzed oxidation of alcohols into aldehydes and ketones. J. Am. Chem. Soc. 1997, 119, 12661–12662. [Google Scholar]
- Ray, R.; Chandra, S.; Maiti, D.; Lahiri, G.K. Simple and efficient ruthenium-catalyzed oxidation of primary alcohols with molecular oxygen. Chem.- A Eur. J. 2016, 22, 8814–8822. [Google Scholar]
- Kolobova, E.; Pestryakov, A.; Bogdanchikova, N.; Corberán, V.C. Silver catalysts for liquid-phase oxidation of alcohols in green chemistry: Challenges and outlook. Catal. Today 2019, 333, 81–88. [Google Scholar]
- Solomon, J.; Madix, R.J. Kinetics and mechanism of the oxidation of allyl alcohol on silver (110). J. Phy. Chem. 1987, 91, 6241–6244. [Google Scholar]
- Pestryakov, A.N.; Bogdanchikova, N.; Knop-Gericke, A. Alcohol selective oxidation over modified foam-silver catalysts. Catal. Today 2004, 91, 49–52. [Google Scholar]
- Ryland, B.L.; McCann, S.D.; Brunold, T.C.; Stahl, S.S. Mechanism of alcohol oxidation mediated by copper (II) and nitroxyl radicals. J. Am. Chem. Soc. 2014, 136, 12166–12173. [Google Scholar]
- Silva, T.F.; Martins, L.M. Recent advances in copper catalyzed alcohol oxidation in homogeneous medium. Molecules 2020, 25, 748. [Google Scholar] [CrossRef]
- Boronat, M.; Corma, A.; Illas, F.; Radilla, J.; Ródenas, T.; Sabater, M.J. Mechanism of selective alcohol oxidation to aldehydes on gold catalysts: Influence of surface roughness on reactivity. J. Catal. 2011, 278, 50–58. [Google Scholar]
- Kilpiö, T.; Behravesh, E.; Russo, V.; Eränen, K.; Salmi, T. Physical modeling of the laboratory-scale packed bed reactor for partial gas-phase oxidation of alcohol using gold nanoparticles as the heterogeneous catalyst. Chem. Eng. Res. Des. 2017, 117, 448–459. [Google Scholar]
- Hui, Y.; Zhang, S.; Wang, W. Recent progress in catalytic oxidative transformations of alcohols by supported gold nanoparticles. ASC 2019, 361, 2215–2235. [Google Scholar]
- Muzart, J. Palladium-catalysed oxidation of primary and secondary alcohols. Tetrahedron 2003, 59, 5789–5816. [Google Scholar]
- Tamaru, Y.; Yamada, Y.; Inoue, K.; Yamamoto, Y.; Yoshida, Z. Oxidation of primary and secondary alcohols by the catalysis of palladium. J. Org. Chem. 1983, 48, 1286–1292. [Google Scholar]
- Privalov, T.; Linde, C.; Zetterberg, K.; Moberg, C. Theoretical studies of the mechanism of aerobic alcohol oxidation with palladium catalyst systems. Organometallics 2005, 24, 885–893. [Google Scholar]
- Avgouropoulos, G.; Oikonomopoulos, E.; Kanistras, D.; Ioannides, T. Complete oxidation of ethanol over alkali-promoted Pt/Al2O3 catalysts. Appl. Catal. B Environ. 2006, 65, 62–69. [Google Scholar]
- Kaichev, V.V.; Chesalov, Y.A.; Saraev, A.A.; Klyushin, A.Y.; Knop-Gericke, A.; Andrushkevich, T.V.; Bukhtiyarov, V.I. Redox mechanism for selective oxidation of ethanol over monolayer V2O5/TiO2 catalysts. J. Catal. 2016, 338, 82–93. [Google Scholar]
- Li, X.; Iglesia, E. Selective catalytic oxidation of ethanol to acetic acid on dispersed Mo-V-Nb mixed oxides. Chem.-A Eur. J. 2007, 13, 9324–9330. [Google Scholar]
- Yan, S.; Liang, X.; Song, H.; Ma, S.; Lu, Y. Synthesis of porous CeO2-SnO2 nanosheets gas sensors with enhanced sensitivity. Ceram. Int. 2018, 44, 358–363. [Google Scholar]
- Sadovskaya, E.; Goncharov, V.; Popova, G.; Ishchenko, E.; Frolov, D.; Fedorova, A.; Andrushkevich, T. Mo-V-Te-Nb oxide catalysts: Reactivity of different oxygen species in partial and deep oxidation. J. Mol. Catal. A Chem. 2014, 392, 61–66. [Google Scholar]
- Fujitani, T.; Nakamura, I.; Haruta, M. Role of water in CO oxidation on gold catalysts. Catal. Lett. 2014, 144, 1475–1486. [Google Scholar]
- Widmann, D.; Behm, R.J. Activation of molecular oxygen and the nature of the active oxygen species for CO oxidation on oxide supported Au catalysts. Accounts Chem. Res. 2014, 47, 740–749. [Google Scholar]
- Kung, M.C.; Davis, R.J.; Kung, H.H. Understanding Au-catalyzed low-temperature CO oxidation. J. Phys. Chem. C 2007, 111, 11767–11775. [Google Scholar]
- Min, B.K.; Friend, C.M. Heterogeneous gold-based catalysis for green chemistry: Low-temperature CO oxidation and propene oxidation. Chem. Rev. 2007, 107, 2709–2724. [Google Scholar]
- Deng, X.; Min, B.K.; Liu, X.; Friend, C.M. Partial oxidation of propene on oxygen-covered Au (111). J. Phys. Chem. B 2006, 110, 15982–15987. [Google Scholar]
- Liu, X.; Xu, B.; Haubrich, J.; Madix, R.J.; Friend, C.M. Surface-mediated self-coupling of ethanol on gold. J. Am. Chem. Soc. 2009, 131, 5757–5759. [Google Scholar] [PubMed]
- Behravesh, E.; Melander, M.M.; Wärnå, J.; Salmi, T.; Honkala, K.; Murzin, D.Y. Oxidative dehydrogenation of ethanol on gold: Combination of kinetic experiments and computation approach to unravel the reaction mechanism. J. Catal. 2021, 394, 193–205. [Google Scholar]
- Takei, T.; Iguchi, N.; Haruta, M. Support effect in the gas phase oxidation of ethanol over nanoparticulate gold catalysts. New J. Chem. 2011, 35, 2227–2233. [Google Scholar]
- Cárdenas-Lizana, F.; Keane, M.A. The development of gold catalysts for use in hydrogenation reactions. J. Mater. Sci. 2013, 48, 543–564. [Google Scholar]
- Pan, M.; Gong, J.; Dong, G.; Mullins, C.B. Model studies with gold: A versatile oxidation and hydrogenation catalyst. Accounts Chem. Res. 2014, 47, 750–760. [Google Scholar]
- Guan, Y.; Hensen, E.J. Ethanol dehydrogenation by gold catalysts: The effect of the gold particle size and the presence of oxygen. App. Catal. A General 2009, 361, 49–56. [Google Scholar]
- Sun, K.-Q.; Luo, S.-W.; Xu, N.; Xu, B.-Q. Gold nano-size effect in Au/SiO2 for selective ethanol oxidation in aqueous solution. Catal. Lett. 2008, 124, 238–242. [Google Scholar]
- Behravesh, E.; Kumar, N.; Balme, Q.; Roine, J.; Salonen, J.; Schukarev, A.; Mikkola, J.-P.; Peurla, M.; Aho, A.; Eränen, K. Synthesis and characterization of Au nano particles supported catalysts for partial oxidation of ethanol: Influence of solution pH, Au nanoparticle size, support structure and acidity. J. Catal. 2017, 353, 223–238. [Google Scholar]
- Li, J.; Yang, Y.; Wang, Q.; Cheng, X.; Luo, Y.; An, B.; Bai, J.; Wang, Y.; Xie, E. Design of size-controlled Au nanoparticles loaded on the surface of ZnO for ethanol detection. CrystEngComm 2021, 23, 783–792. [Google Scholar]
- Tabakova, T.; Boccuzzi, F.; Manzoli, M.; Sobczak, J.; Idakiev, V.; Andreeva, D. Effect of synthesis procedure on the low-temperature WGS activity of Au/ceria catalysts. Appl. Catal. B Environ. 2004, 49, 73–81. [Google Scholar]
- Tamaru, K. Dynamic Heterogeneous Catalysis; Academic Press: New York, NY, USA, 1978; pp. 1–140. [Google Scholar]
- Schlögl, R. Heterogeneous catalysis. Angew. Chem. Int. Ed. 2015, 54, 3465–3520. [Google Scholar]
- Satterfield, C.N. Heterogeneous Catalysis in Practice; McGraw-Hill: New York, NY, USA, 1980; pp. 1–416. [Google Scholar]
- Armaroli, T.; Bécue, T.; Gautier, S. Diffuse reflection infrared spectroscopy (DRIFTS): Application to the in situ analysis of catalysts. Oil Gas Sci. Technol.-Rev. 2004, 59, 215–237. [Google Scholar]
- Haw, J.F. In-Situ Spectroscopy in Heterogeneous Catalysis; Wiley-VCH: Weinheim, Germany, 2002; pp. 1–276. [Google Scholar]
- Baurecht, D.; Fringeli, U.P. Quantitative modulated excitation Fourier transform infrared spectroscopy. Rev. Sci. Instruments 2001, 72, 3782–3792. [Google Scholar]
- Baurecht, D.; Porth, I.; Fringeli, U. A new method of phase sensitive detection in modulation spectroscopy applied to temperature induced folding and unfolding of RNase A. Vib. Spectrosc. 2002, 30, 85–92. [Google Scholar]
- Srinivasan, P.D.; Patil, B.S.; Zhu, H.; Bravo-Suárez, J.J. Application of modulation excitation-phase sensitive detection-DRIFTS for in situ/operando characterization of heterogeneous catalysts. React. Chem. Eng. 2019, 4, 862–883. [Google Scholar]
- Pandey, R.K.; Lakshminarayanan, V. Ethanol electrocatalysis on gold and conducting polymer nanocomposites: A study of the kinetic parameters. Appl. Catal. B Environ. 2012, 125, 271–281. [Google Scholar]
- Li, J.; Kazakov, A.; Chaos, M.; Dryer, F.L. Chemical kinetics of ethanol oxidation. In Proceedings of the 5th US Combustion Meeting, San Diego, CA, USA, 25–28 March 2007. [Google Scholar]
- Gonzalez-Yañez, E.O.; Fuentes, G.A.; Hernández-Terán, M.E.; Fierro-Gonzalez, J.C. Influence of supported gold particles on the surface reactions of ethanol on TiO2. Appl. Catal. A Gen. 2013, 464, 374–383. [Google Scholar]
- Tan, T.H.; Scott, J.; Ng, Y.H.; Taylor, R.A.; Aguey-Zinsou, K.-F.; Amal, R. C–C cleavage by Au/TiO2 during ethanol oxidation: Understanding bandgap photoexcitation and plasmonically mediated charge transfer via quantitative in situ DRIFTS. ACS Catal. 2016, 6, 8021–8029. [Google Scholar]
- Cornejo-Romero, J.; Solis-Garcia, A.; Vega-Diaz, S.M.; Fierro-Gonzalez, J.C. Reverse hydrogen spillover during ethanol dehydrogenation on TiO2-supported gold catalysts. Mol. Catal. 2017, 433, 391–402. [Google Scholar]
- Green, I.X.; Tang, W.; Neurock, M.; Yates, J.T., Jr. Localized partial oxidation of acetic acid at the dual perimeter sites of the Au/TiO2 catalyst formation of gold ketenylidene. J. Am. Chem. Soc. 2012, 134, 13569–13572. [Google Scholar]
- Chang, C.-A.; Ray, B.; Paul, D.K.; Demydov, D.; Klabunde, K.J. Photocatalytic reaction of acetaldehyde over SrTiO3 nanoparticles. J. Mol. Catal. A Chem. 2008, 281, 99–106. [Google Scholar] [CrossRef]
- Gao, J.; Teplyakov, A.V. Surface species formed during thermal transformation of ethanol on ZnO powder. J. Catal. 2013, 300, 163–173. [Google Scholar] [CrossRef]
- Zheng, Y.; Qi, Y.; Tang, Z.; Tan, J.; Koel, B.E.; Podkolzin, S.G. Spectroscopic observation and structure-insensitivity of hydroxyls on gold. ChemComm. 2022, 58, 4036–4039. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Ma, Y.; Fan, Y.; Shen, J. Fourier transform infrared spectroscopy and oxygen luminescence probing combined study of modified sol–gel derived film. Thin Solid Films 2001, 397, 95–101. [Google Scholar]
- Bhatia, S.; Verma, N.; Bedi, R. Ethanol gas sensor based upon ZnO nanoparticles prepared by different techniques. Results Phys. 2017, 7, 801–806. [Google Scholar] [CrossRef]
- Pan, X.; Wang, L.; Ling, F.; Li, Y.; Han, D.; Pang, Q.; Jia, L. A novel biomass assisted synthesis of Au–SrTiO3 as a catalyst for direct hydrogen generation from formaldehyde aqueous solution at low temperature. Int. J. Hydrogen Energy 2015, 40, 1752–1759. [Google Scholar] [CrossRef]
- Horton, R.C.; Herne, T.M.; Myles, D.C. Aldehyde-terminated self-assembled monolayers on gold: Immobilization of amines onto gold surfaces. J. Am. Chem. Soc. 1997, 119, 12980–12981. [Google Scholar] [CrossRef]
- Hartadi, Y.; Widmann, D.; Behm, R.J. CO2 hydrogenation to methanol on supported Au catalysts under moderate reaction conditions: Support and particle size effects. ChemSusChem 2015, 8, 456–465. [Google Scholar] [CrossRef]
- Foo, G.S.; Hood, Z.D.; Wu, Z. Shape effect undermined by surface reconstruction: Ethanol dehydrogenation over shape-controlled SrTiO3 nanocrystals. ACS Catal. 2018, 8, 555–565. [Google Scholar]
- Wiberg, K.B.; Thiel, Y.; Goodman, L.; Leszczynski, J. Acetaldehyde: Harmonic frequencies, force field, and infrared intensities. J. Phys. Chem. 1995, 99, 13850–13864. [Google Scholar] [CrossRef]
- Su, X.; Yang, X.F.; Huang, Y.; Liu, B.; Zhang, T. Single-atom catalysis toward efficient CO(2) conversion to CO and formate products. Acc. Chem. Res. 2019, 52, 656–664. [Google Scholar]
- Tamaki, Y.; Hara, K.; Katoh, R.; Tachiya, M.; Furube, A. Femtosecond visible-to-IR spectroscopy of TiO2 nanocrystalline films: Elucidation of the electron mobility before deep trapping. J. Phys. Chem. C 2009, 113, 11741–11746. [Google Scholar] [CrossRef]
- Panayotov, D.A.; Burrows, S.P.; Morris, J.R. Infrared spectroscopic studies of conduction band and trapped electrons in UV-photoexcited, H-atom n-doped, and thermally reduced TiO2. J. Phys. Chem. C 2012, 116, 4535–4544. [Google Scholar]
- Torres-Velasco, A.; Patil, B.S.; Zhu, H.; Qi, Y.; Podkolzin, S.G.; Bravo-Suarez, J.J. A general method for studying reactive surface species, CT-SKAn: Charge-transfer spectrokinetic analysis. J. Catal. 2024, 434, 115508. [Google Scholar]
- Srinivasan, P.D.; Zhu, H.; Bravo-Suárez, J.J. In situ UV–vis plasmon resonance spectroscopic assessment of oxygen and hydrogen adsorption location on supported gold catalysts. Mol. Catal. 2021, 507, 111572. [Google Scholar] [CrossRef]
- Ahmadi Daryakenari, A.; Ahmadi Daryakenari, M.; Bahari, Y.; Omivar, H. Preparation and Ethanol Sensing Properties of ZnO Nanoparticles via a Novel Sol-Gel Method. Int. Sch. Res. Not. 2012, 2012, 879480. [Google Scholar] [CrossRef]
- Waheed, A.; Wang, X.; Maeda, N.; Meier, D.M.; Ishida, T.; Murayama, T.; Haruta, M.; Baiker, A. Influence of the support in aqueous phase oxidation of ethanol on gold/metal oxide catalysts studied by ATR-IR spectroscopy under working conditions. Catal. Comm. 2021, 148, 106183. [Google Scholar]




 = hydroxyls, and △ = water.
= hydroxyls, and △ = water.
= hydroxyls, and △ = water.
= hydroxyls, and △ = water.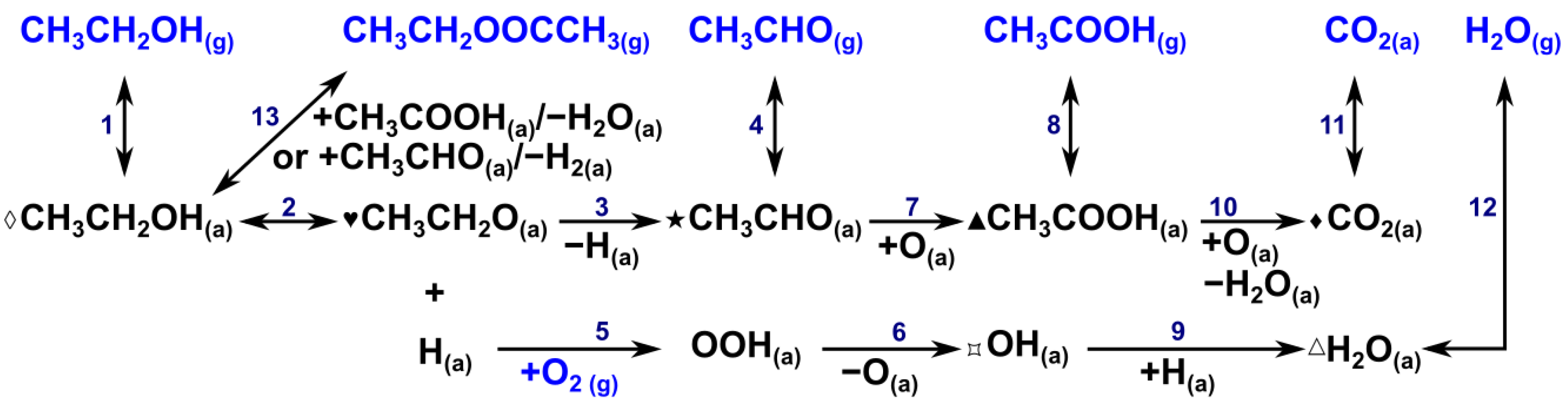
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patil, B.S.; Torres-Velasco, A.; Bravo-Suárez, J.J. Frequency-Resolved Modulation Excitation Spectroscopy Methodology for Identifying Surface Reaction Species in Ethanol Oxidation on Gold Catalysts. Catalysts 2025, 15, 346. https://doi.org/10.3390/catal15040346
Patil BS, Torres-Velasco A, Bravo-Suárez JJ. Frequency-Resolved Modulation Excitation Spectroscopy Methodology for Identifying Surface Reaction Species in Ethanol Oxidation on Gold Catalysts. Catalysts. 2025; 15(4):346. https://doi.org/10.3390/catal15040346
Chicago/Turabian StylePatil, Bhagyesha S., Alejandra Torres-Velasco, and Juan J. Bravo-Suárez. 2025. "Frequency-Resolved Modulation Excitation Spectroscopy Methodology for Identifying Surface Reaction Species in Ethanol Oxidation on Gold Catalysts" Catalysts 15, no. 4: 346. https://doi.org/10.3390/catal15040346
APA StylePatil, B. S., Torres-Velasco, A., & Bravo-Suárez, J. J. (2025). Frequency-Resolved Modulation Excitation Spectroscopy Methodology for Identifying Surface Reaction Species in Ethanol Oxidation on Gold Catalysts. Catalysts, 15(4), 346. https://doi.org/10.3390/catal15040346