Abstract
As multifunctional platform molecules, α-keto acids and their esters hold significant value in pharmaceutical synthesis, functional materials, and metabolic processes. Conventional chemical synthesis routes for these compounds are well-established and efficient but often rely on precious metals, high-pressure conditions, and hazardous reagents, leading to considerable environmental costs. In contrast, catalytic biomass conversion pathways—utilizing renewable feedstocks under mild conditions—offer promising alternatives. However, issues such as catalyst deactivation and undesired side reactions remain to be addressed. This review systematically summarizes recent advances in both traditional chemical and catalytic biomass-based synthesis of α-keto acids and their esters, with a particular emphasis on green synthetic routes derived from renewable resources. Finally, current challenges and future perspectives in the field are briefly discussed.
1. Introduction
α-Keto acids and their esters are versatile platform molecules naturally abundant and widely used in organic synthesis. These compounds exhibit excellent stability and are easy to store, transport, and handle [1,2] (Figure 1). Structurally, α-keto acids contain both a keto carbonyl group and a carboxylic acid group, enabling them to participate in diverse reactions such as esterification, nucleophilic addition to carbonyls, and reduction [3,4,5,6,7,8,9,10,11,12,13]. Their chemical versatility allows for the synthesis of a wide range of value-added compounds, including natural product analogs, pharmaceuticals, and agrochemicals [14].
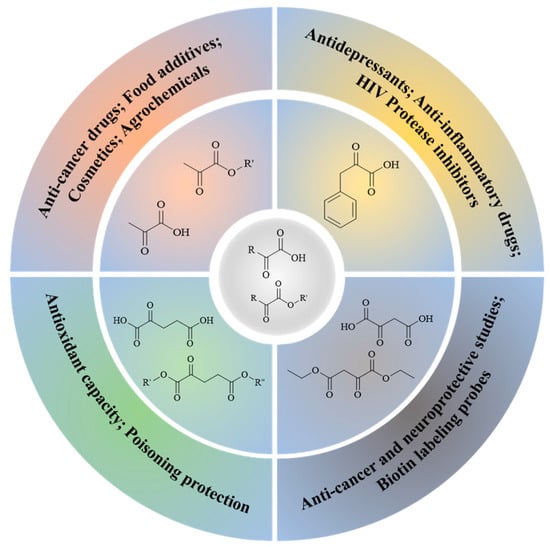
Figure 1.
Applications of typical α-keto acids and their esters.
Pyruvic acid and its esters, as the most representative α-keto acids, are extensively utilized as intermediates in the production of antiviral and anticancer drugs, food additives, antioxidants, and cosmetic ingredients [15,16,17,18]. Pyruvic acid can undergo alcoholic fermentation to yield acetaldehyde and carbon dioxide or serve as a substrate for transamination to generate alanine [19]. Phenylpyruvic acid, another notable α-keto acid, is an established biomarker for phenylketonuria (PKU) and plays a role in the synthesis of antidepressants and HIV protease inhibitors [20,21,22,23,24]. Through enzymatic decarboxylation, phenylpyruvic acid can yield phenylacetaldehyde—a key component of rose fragrance—which may be further reduced to β-phenylethanol for use in food and cosmetics [25,26]. In addition to these applications, other biologically relevant α-keto acids, such as oxaloacetate and α-ketoglutarate (AKG), play central roles in metabolic cycles. Oxaloacetate is a key intermediate in the tricarboxylic acid (TCA) cycle and can be transaminated to aspartic acid [27,28,29], with potential therapeutic applications in treating neurodegenerative diseases and metabolic disorders [30,31]. AKG, another TCA intermediate, is involved in glutamate biosynthesis and exhibits antioxidative and protective effects in models of oxidative stress and metabolic dysfunction [32,33,34,35]. Figure 2 illustrates the different synthetic routes for the aforementioned typical α-keto acids (esters), which will be exemplified below.
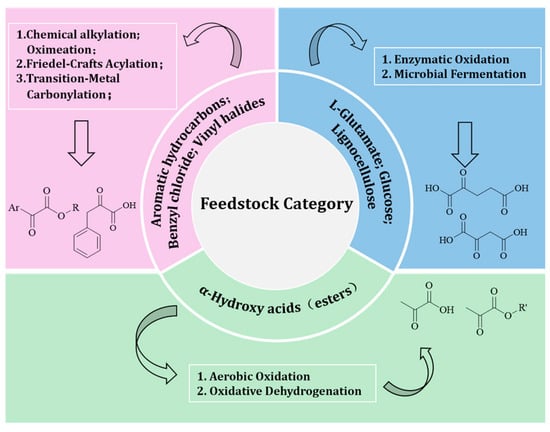
Figure 2.
Primary synthetic pathways for typical α-keto acids (esters).
Chemically, α-keto acids can be synthesized from simple dicarbonyl compounds such as diethyl oxalate and malonate. For instance, malonate derivatives undergo a three-step sequence of alkylation, oximation, and carbonylation to yield α-keto esters [36]. Aryl α-keto esters can be prepared via Friedel–Crafts acylation of aromatic hydrocarbons with ethyl oxalyl chloride [37]. Though these methods involve multiple steps, the readily available reagents and operational robustness make them suitable for industrial applications. Transition-metal-catalyzed carbonylation methods—using substrates such as benzyl chloride or vinyl halides and CO in the presence of Co or Pd catalysts—have also proven effective. Li et al. reported a mild and high-yielding bicarbonylation of benzyl chloride and CO, catalyzed by CoCl2 in the presence of calcium hydroxide and potassium pyridine-2-carboxylate [38].
In recent years, enzymatic synthesis of α-keto acids has attracted growing attention. L-amino acids serve as common starting materials in this approach. For example, Sun et al. developed a nanoreactor integrating single-atom iron nanozymes and L-glutamate oxidase to significantly enhance AKG production [39]. Engineered microorganisms such as Corynebacterium glutamicum and Escherichia coli have also been used to biosynthesize α-keto acids from sugars like glucose via the TCA cycle [40]. Non-food biomass, such as lignocellulose, can serve as a carbon source after pretreatment, aligning with the principles of green chemistry [41].
α-Hydroxy acids, including chiral variants, are important intermediates in asymmetric synthesis and fine chemical production. Among them, lactic acid is considered a top platform chemical derived from lignocellulosic biomass [42,43]. Recent efforts have focused on the aerobic oxidation of ethyl lactate (EL) to produce ethyl pyruvate (EP), a high-value derivative. Although this reaction exhibits high atom economy, product degradation through overoxidation and decarboxylation remains a key limitation [44]. Pt nanoparticles supported on CdS have been shown to promote C–C coupling reactions of α-hydroxy acids, while atomically dispersed Pt catalysts enable highly selective α-keto acid synthesis by selectively activating hydroxyl groups [45]. In addition, Bi-based catalysts (e.g., Bi3+ carboxylates) can catalyze the oxidative dehydrogenation of α-hydroxy acids to α-keto acids under mild aerobic conditions without requiring strong oxidants [46].
To provide a comprehensive and systematic understanding of the green synthesis of α-keto acids and their esters, this review is conceptually organized into three thematic modules based on synthetic strategies and catalytic systems. First, conventional chemical synthesis routes are summarized to highlight their historical evolution and existing limitations. Second, catalytic biomass-based synthesis approaches are categorized and discussed in detail, including gas-phase oxidation, liquid-phase oxidation, and photocatalytic transformation, with a particular emphasis on catalyst development and reaction mechanisms. Third, key challenges are analyzed from methodological, catalytic, and process-engineering perspectives, followed by a forward-looking outlook.
2. Conventional Synthesis Routes of α-Keto Acids
The synthesis of α-keto acid compounds can be achieved through a variety of methodologies, including Friedel-Crafts acylation, the Grignard reagent method, oxidation, metal-catalyzed bicarbonylation, electrochemical synthesis, and others. The product obtained from the reaction is typically α-keto acid esters, which can undergo hydrolysis to yield α-keto acids.
As early as 1881, Erlenmeyer conducted the first pyruvic acid synthesis experiments, where tartaric acid generated pyruvic acid via dehydration. This reaction necessitated the use of excess potassium bisulfate (KHSO4) as a dehydrating agent. However, this approach resulted in atomic inefficiency and environmental pollution. The product, pyruvate, could be synthesized as EP by esterification. Nevertheless, the industrialization of the method is hindered by the precise adjustment of reaction conditions, including pH, temperature, low yield in null time, and high cost of product isolation and purification.
The oxidation strategy typically utilizes selenium dioxide [47,48,49,50,51,52], potassium permanganate [53], and osmium tetroxide [54] as oxidizing agents. α-keto acids can be obtained by oxidizing carbon unsaturated bonds or methyl ketones. These methods are highly efficient and exhibit a high atom economy. However, the toxicity of the oxidizing agents used is high, and the violent oxidizing conditions result in a decreased functional group tolerance, which limits the development of such methods. In 1891, Claus and Neukranz employed KMnO4 to oxidize acetophenone first, where the C(sp3)-H bond of acetophenone was oxidized under alkaline conditions, leading to a 70% yield of α-phenylglyoxalic acid (PGA) [55]. In 1939, Hurd et al. reported the use of KMnO4 as an effective oxidizing agent for the conversion of styrene in PGA, yielding a 55% yield for α-keto acid [56]. In 1944, Oakwood et al. reported a molar-scale synthesis of benzoyl cyanide by hydrolysis with an aqueous solution of hydrochloric acid [57]. While this method is considered highly reliable, it should be noted that the reaction may require up to five days to complete, and products were isolated in yields ranging from 73% to 77%.
A more versatile and widely used route is through organometallic acylation reactions. Friedel–Crafts acylation of arenes with oxalyl chloride derivatives in the presence of Lewis acids offers a straightforward path to α-keto acid esters, though the outcome is highly sensitive to electronic effects of ring substituents. In the 1980s, Grignard and aryllithium reagents were shown to react efficiently with diethyl oxalate or acylimidazoles, offering access to diverse α-keto acid esters in good yields. While highly efficient, these reagents often require careful handling and, in the case of aryllithiums, benefit from flow reactor systems to address their high reactivity and improve safety [58]. In the same year, Weinstock et al. added diethyl oxalate directly to Grignard’s reagent, and α-keto acid esters could be easily obtained by a simple one-step reaction with good substrate tolerance [59].
The development of transition-metal-catalyzed bicarbonylation has significantly expanded the synthetic toolbox. Palladium- or cobalt-catalyzed carbonylation of aryl halides under a CO atmosphere (at high or atmospheric pressure) enables selective formation of α-keto acid esters, often in the presence of alcohols or base. These methods offer improved selectivity and substrate scope but may involve costly catalysts and high-pressure operations. In 1987, Tanaka et al. achieved the bicarbonylation of aryl halides using a palladium-catalyzed strategy under a CO atmosphere at a pressure of 150 atm [60]. The corresponding α-keto acid esters were obtained in the presence of alcohols in the system. In the subsequent year, Itoh et al. accomplished the bicarbonylation of α-bromonaphthalene, bromobenzene, and iodobenzene in a catalytic system derived from the treatment of cobalt dichloride with sodium sulfide or sodium borohydride in an atmospheric pressure CO atmosphere, in the presence of calcium hydroxide and iodomethane. This process yielded α-keto acids with notable selectivity [61].
In recent years, electrochemical synthesis has emerged as a sustainable and atom-economical alternative. For example, anodic oxidation of aryl methyl ketones in methanol can directly yield α-keto acid methyl esters, with potassium iodide improving system stability and reaction cleanliness [62]. These electrochemical approaches are attractive for their operational simplicity, safety, and environmental compatibility and represent a promising direction for green synthesis.
Recent reviews highlight innovations in sustainable synthesis, such as electrocatalytic decarboxylation [63], and alongside emerging bio-electrocatalytic hybrid systems [64], offering solutions to limitations of classical methods. Conventional synthetic routes to α-keto acids span from early dehydration and oxidation protocols to modern organometallic and catalytic systems. While classical methods offer historical insight, they are limited by harsh conditions and low sustainability. In contrast, recent developments in catalytic and electrochemical techniques provide more efficient, selective, and environmentally benign pathways. Future efforts are expected to focus on enhancing green chemistry metrics and broadening substrate compatibility to meet the demands of practical and industrial synthesis.
3. Biomass Synthesis Routes—Exemplified by the Synthesis of Typical α-Keto Acids and Their Esters
Recent advances in biomass-derived synthesis pathways have led to the development of greener and more scalable alternatives for the preparation of α-keto acids and their esters. Compared to traditional methods such as microbial fermentation and high-valent iodine reagent-dependent oxidation, these new strategies offer improved atom economy, milder reaction conditions, and better alignment with the principles of sustainable chemistry. For instance, Hong et al. reported a one-pot strategy for synthesizing AKG and methane triacetic acid (MTA) from biomass-derived pyruvic acid and glyoxylic acid [65]. This method integrates cross-hydroxyaldol condensation with Pd/TiO2-catalyzed hydrodeoxygenation, affording yields of 85.4% and 86.2% for AKG and MTA, respectively. The process avoids the use of strong acids, bases, and toxic solvents and achieves superior carbon atom economy compared to conventional synthetic routes. In another approach, Luo et al. developed a metal-free oxidative esterification reaction employing potassium xanthate as both an alkoxide donor and reaction promoter [66]. By modulating solvent composition, temperature, and additives, this method enables the selective formation of α-keto acid esters and conventional esters. Crucially, the reaction proceeds without the need for transition metals or high-valent iodine reagents, which are common in traditional oxidative esterifications, thereby enhancing environmental compatibility and safety.
The above examples underscore the increasing feasibility of developing efficient and selective biomass-based synthetic routes for α-keto acid derivatives through innovations in reaction pathway design and reagent system optimization. As important biomass-derived platform compounds, α-hydroxy acids can be converted into corresponding α-keto acid esters via catalytic oxidation or dehydrogenation. This transformation route aligns well with green chemistry principles and offers promising scalability for industrial applications. The implementation of heterogeneous catalytic systems for the efficient conversion of α-hydroxy acids into α-keto acid esters represents a practical and sustainable strategy for upgrading biomass into high-value products.
3.1. Gas-Phase Reactions
Gas-phase oxidation has emerged as a promising and sustainable strategy for the synthesis of pyruvic acid and its esters, owing to its alignment with green chemistry principles—such as minimal solvent use, high energy efficiency, and compatibility with continuous-flow operation. In this process, molecular oxygen (O2) serves as the primary oxidant, and its effective utilization depends on the catalyst’s ability to activate O2 through mechanisms including lattice oxygen participation and the generation of oxygen vacancies. These features contribute to the environmental friendliness, low cost, and scalability of the gas-phase approach.
Compared to liquid-phase oxidation systems, which often suffer from catalyst deactivation and the formation of complex by-products, gas-phase reactions offer enhanced control over the reaction pathway. This is achieved through the rational design of gas–solid interfaces and the engineering of multiphase catalytic systems, enabling the selective transformation of lactic acid (or its esters) into pyruvic acid (or esters) via dehydrogenation and related processes. Additionally, the careful tuning of reaction parameters—such as temperature and oxygen partial pressure—can effectively suppress deep oxidation and minimize carbon loss.
The reaction pathway, whether dominated by dehydrogenation or oxidative decarboxylation, is closely governed by the physicochemical properties of the catalyst, including metal composition, surface acidity, and the density of oxygen vacancies. Therefore, establishing a comprehensive understanding of the structure–activity relationships across different catalyst systems is essential for the rational design of selective and durable gas-phase oxidation catalysts. The following sections summarize representative catalyst systems developed for gas-phase oxidation of lactic acid (or its esters), with a focus on their composition, performance, and underlying reaction mechanisms.
3.1.1. Precious Metal Catalysts
Gold-based catalysts have demonstrated significant efficacy in oxidation reactions involving CO, cyclohexane, and alcohols, utilizing both molecular oxygen and ambient air as oxidants [67,68,69]. Lu et al. developed an Au/NiAl-MMO catalyst, comprising gold nanoparticles supported on nickel-aluminum mixed metal oxides (NiAl-MMO), for the aerobic oxidation of EL using real air as the oxidant [70]. At 240 °C, this system achieved a 72.6% conversion of EL with 88.3% selectivity toward EP, along with notable long-term stability in continuous fixed-bed operations. Mechanistic studies revealed a Mars–van Krevelen (MvK) mechanism, highlighting oxygen activation as the key step in the catalytic cycle.
Despite their high activity and selectivity, precious metal catalysts generally suffer from high costs and scarcity, motivating the exploration of more economical alternatives.
3.1.2. Transition Metal Oxide Catalysts
Transition metal oxides (e.g., Mo-, Fe-, Ni-, and V-based systems) function as active matrices for gas-phase catalytic oxidation. Their performance is optimized through crystal phase engineering, exposure of highly active facets, surface modification with polymers, heterojunction construction, and lattice doping. The catalytic activity originates from the redox cycling of metal cations (e.g., Mo6+/Mo5+, Fe3+/Fe2+, Ni2+/Ni3+) and the activation capability of defect sites (e.g., oxygen vacancies) toward reactants. This structure-performance correlation provides a theoretical foundation for designing efficient gas-phase oxidation processes toward α-keto acid synthesis.
Pure-Phase Metal Oxides
Wang et al. highlighted the critical influence of crystalline phase on catalytic performance by comparing hexagonal (h-MoO3) and orthorhombic (α-MoO3) molybdenum trioxide in the oxidative dehydrogenation of lactic acid [71]. The hexagonal phase exhibited superior catalytic activity and selectivity (82.0% conversion and 82.3% selectivity at 230 °C), which was attributed to the exposure of the (110) crystal surface that enhances lattice oxygen mobility and oxidative capacity. The catalyst maintained stability over extended reaction times, confirming the robustness of phase engineering as a strategy to improve catalyst performance.
Yin et al. proposed a phosphorus-doped iron-molybdenum bimetallic oxide catalyst prepared via hydrothermal synthesis and air calcination [72]. This catalyst leveraged strong Fe3+-MoO3 interactions and phosphorus-induced Fe2O3 dispersion to facilitate electron transfer, markedly improving oxidation capacity and stabilizing pyruvate selectivity above 70% during prolonged operation. Complementary research into α-Fe2O3 catalysts demonstrated that crystal facet modulation, particularly exposing the (001) facet, substantially enhances lactic acid conversion and pyruvate selectivity by favoring adsorption of lactic acid molecules and stabilizing active sites. Jia et al. engineered a PVP-modified α-Fe2O3 catalyst that enhances oxidative dehydrogenation of lactic acid (LA) to pyruvic acid (PA) through preferential (110) facet exposure and controlled oxygen vacancy generation [73]. PVP modification optimizes Fe2O3 surface adsorption capacity and active site synergy, achieving 94.6% LA conversion with 91.6% PA selectivity in air. This performance surpasses conventional FePO4 (60% conversion, 62% selectivity) and Pb-Pt catalysts (70.7% conversion, 80.6% selectivity). Mechanistic studies demonstrate that oxygen vacancies facilitate LA adsorption/activation and PA desorption while suppressing byproducts (e.g., acrylic acid, acetaldehyde).
Composite Metal Oxide
Industrial relevance is underscored by the work of Huchede et al., who developed a gas-phase process for EP production through oxidative dehydrogenation of EL [74,75]. Their comprehensive catalyst screening identified phosphomolybdenum heteropolyacid catalysts with iron-resistant cations as optimal, achieving high selectivity and productivity. Catalyst optimization through cesium doping improved surface area and active site density but reduced intrinsic activity, highlighting a trade-off between structural properties and catalytic efficiency. Similarly, vanadium-iron antimonates demonstrated high catalytic performance at elevated temperatures, with surface V5+ species identified as key active sites closely correlated to bulk composition.
Lomate et al. reported that NiNb mixed oxides deliver efficient oxidative dehydrogenation of lactic acid, with catalytic performance tunable via Ni/Nb ratios [76]. Increasing niobium content enhances surface area, porosity, and reducibility, favoring pyruvic acid formation while suppressing by-products such as acetaldehyde, which arises mainly from thermal decarboxylation.
Heterojunction Composite Materials
In 1993, Sugiyama et al. provided a systematic investigation into SnO2-MoO3 binary oxide catalysts for the gas-phase oxidation of EL to EP, revealing key mechanistic insights [77]. XPS analysis demonstrated a reduction in the Mo 3d5/2 binding energy from 232.9 eV (Mo6+) to 231.8 eV (Mo5+) after reaction, indicating active participation of lattice oxygen in the catalytic cycle alongside gaseous oxygen. This catalyst achieved over 75% conversion and 90% selectivity at 250 °C, significantly outperforming single-component MoO3 and other binary oxides such as Fe2O3-MoO3.
Further studies by Hayashi et al. investigated the role of oxidation states in TeO2-MoO3 catalysts during the gas-phase selective oxidation of EL to EP [78]. Their work demonstrated that surface oxidation states, particularly the valence states of Te and Mo, critically govern the reaction. Furthermore, active sites with specific oxidation states likely dominate the highly selective conversion to EP. Oxidation states and surface composition also play decisive roles, as demonstrated by Hayashi et al. [79]. through their work on TeO2-MoO3 catalysts. The valence states of Te and Mo were found to govern active site formation, influencing selectivity in EL oxidation. Subsequent investigations into tellurium molybdates with various metal cations (Co, Mn, Zn) revealed that catalytic activity follows the trend Co > Mn > Zn, emphasizing the importance of metal synergy and spatial arrangement of active sites. Notably, individual components such as ZnMoO4 and TeO2 showed negligible activity, underscoring the necessity of their intimate interaction within the mixed oxide framework.
3.1.3. Phosphate/Pyrophosphate-Based Catalyst
Phosphate-based catalysts feature a rigid structural framework composed of phosphate (PO43−) or pyrophosphate (P2O74−) units. Their advantages include high thermal stability and tunable Brønsted/Lewis acid sites, which facilitate substrate adsorption and direct the selective oxidation of alcohols. The synergy between phosphate tetrahedra and metal centers (e.g., Fe3+-O-P bonds) enhances hole mobility while suppressing over-oxidation side reactions, thus improving α-keto acid selectivity. The structural and oxidation state diversity of iron phosphate catalysts was extensively explored by Ai et al., revealing that mixed-valence catalysts containing both Fe2+ and Fe3+ exhibited enhanced activity and stability [80]. A stable M-phase catalyst emerged under prolonged reaction conditions, delivering optimal performance at moderate oxygen feed rates. The promotional effect of trace molybdenum doping further enhanced catalytic performance, with a Mo/Fe atomic ratio range identified as critical for maximizing activity and selectivity, whereas doping with vanadium or tungsten did not yield similar benefits. The research group also investigated the enhancement effect of trace molybdenum (Mo6+) doping on Mo/Fe (PO4)3 iron phosphate catalysts for the oxidative dehydrogenation of lactic acid to pyruvic acid. Within a molybdenum-to-iron atomic ratio (Mo/Fe) range of 0.01–0.3, Mo6+ doping significantly improved catalytic activity and selectivity, with performance being independent of the molybdenum precursor [81]. A lactic acid conversion of 70.1% and a pyruvic acid yield of 63.2% were achieved. In contrast, neither vanadium (V5+) nor tungsten (W6+) doping exhibited comparable enhancement effects.
In parallel, Yin et al. introduced a novel phosphorus-doped iron-molybdenum bimetallic oxide catalyst (FeMoO/P) for the oxidative dehydrogenation of lactic acid for the preparation of pyruvic acid [82]. The catalyst was synthesized via hydrothermal preparation followed by calcination in air. This process fostered strong interactions between Fe3+ and MoO3. Fe3+ doping induced lattice displacements in MoO3, forming a surface solid solution, while the introduction of phosphorus enhanced the dispersion of Fe3O3 on the MoO3 surface. Electron transfer between Fe and Mo significantly improved the catalyst’s oxidation capability, rendering the activation of α-OH groups a key reaction step (Figure 3a). Notably, in situ tail gas analysis revealed no H2 production during the reaction and indicated the direct participation of lattice oxygen as the active oxygen species in the dehydrogenation process. Pyruvate selectivity remained stable at >70% throughout a continuous 60-h reaction.
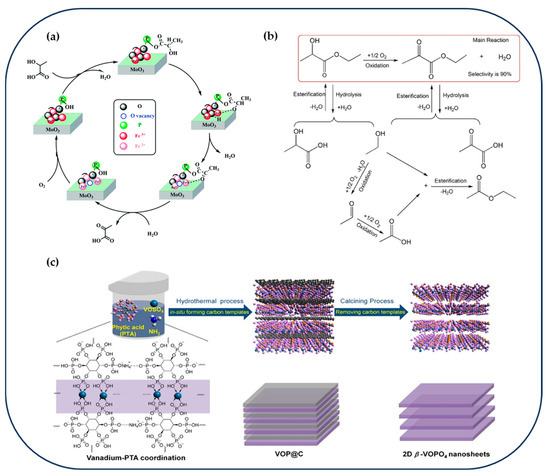
Figure 3.
(a) Reaction mechanism for the oxidative dehydrogenation of LA to PA over the FeMoO/P catalyst [82]. Copyright 2020 Royal Society of Chemistry. (b) Possible reaction pathways in the ODH reaction of EL over MoVNbOx-based catalysts [83]. Copyright 2016 Elsevier. (c) Schematic summary of the synthesis procedure for 2D β-VOPO4 nanosheets [84]. Copyright 2020 ACS Publications.
Building on this, Zhang et al. introduced a novel self-exfoliation synthesis strategy using phytate (PTA) ligands to prepare ultrathin β-VOPO4 nanosheets with an orthorhombic crystal structure [84]. The synthesis involved the assembly of PTA with vanadium oxysulfate (VOSO4) into a layered precursor, followed by hydrothermal carbonization to remove the interlayer carbon template and subsequent calcination. Characterization by AFM and HRTEM revealed nanosheets approximately 6 nm thick (7–8 atomic layers) with exposed (101), (102), and (110) facets. XPS analysis indicated a high concentration of V4+ species (~40%), contributing to abundant V4+/V5+ redox pairs. This catalyst achieved over 90% EL conversion with EP selectivity exceeding 80%, maintaining stable performance for 80 h of continuous operation. Mechanistic investigations suggested that the high catalytic activity is primarily driven by enriched surface oxygen vacancies and the V4+/V5+ redox couple, which promote lattice oxygen involvement consistent with a Mars–van Krevelen mechanism (Figure 3c).
Transition metal oxide catalysts have demonstrated excellent performance in the gas-phase oxidative dehydrogenation of lactic acid (esters) to pyruvic acid (esters). The modulation of crystalline phases, such as the exposure of specific facets in hexagonal MoO3, significantly enhances catalytic activity and selectivity. Doping with elements like Mo, Fe, V, and Nb optimizes the electronic structure and distribution of active sites, thereby improving catalyst stability and efficiency. Niobium oxides, in particular, exhibit increased specific surface area and reducibility through compositional tuning, which favors selective product formation. Overall, these multicomponent oxide catalysts leverage structural control and electron transfer mechanisms to achieve efficient and stable lactic acid oxidation. Future research should integrate mechanistic insights to further refine catalyst design for greener catalytic processes.
3.1.4. Supported Multi-Component Catalyst
Supported catalysts achieve high dispersion and enhanced stability of active sites by confining polymetallic components within high-surface-area carriers (SiO2, TiO2). This architecture is particularly effective for mediating cascade oxidation of complex substrates in gas-phase systems. Such designs markedly expand compositional flexibility while offering scalable pathways for industrial implementation.
Zhao et al., on MoVNbOx catalysts and their TiO2-supported variants, elucidated the positive impact of TiO2 loading on active component dispersion, reaction rates, and activation energy [83]. The reaction mechanism aligns with the Mars–van Krevelen redox model, confirming first-order dependence on oxygen partial pressure (Figure 3b). Meanwhile, Liu et al. demonstrated that quaternary P-Mo-V-Nb/SiO2 catalysts outperform binary and ternary analogs due to a balanced combination of oxidation capacity, redox cycling, and surface acidity modulation [85]. Phosphorus doping plays a crucial role in dispersing active sites and mitigating over-oxidation, while the weakly acidic surface inhibits decarboxylation side reactions, collectively enhancing selectivity and yield.
Lu et al. developed a silica-supported copper catalyst (Cu/SiO2 d EP selectivity of 91.1% at 320 °C [86]. The superior activity was attributed to the abundant Cu0/Cu+ interfacial sites present on the catalyst surface, which synergistically facilitated the activation of both O-H and C-H bonds in EL. Importantly, H/D kinetic isotope effect experiments confirmed that C-H bond activation constitutes a critical step in the reaction mechanism.
Similarly, Zhang et al. explored V2O5/TiO2 catalysts for the oxidative dehydrogenation of EL, highlighting the pivotal role of V–O–Ti bonds in tuning catalytic activity [87]. By adjusting the vanadium surface density, they modulated the morphology of vanadium oxide species from monomeric VOx units to two-dimensional polyvanadate and bulk crystalline phases. Monomeric VOx species, favored at lower vanadium loadings, exhibited enhanced EP yields, whereas higher loadings led to the formation of polymorphic and crystalline structures, resulting in decreased activity. Comparative studies on various supports (TiO2, MgO, Al2O3, ZrO2, CeO2) further confirmed the superior performance of the TiO2-based system. In situ DRIFT spectroscopy combined with mass spectrometry elucidated two distinct reaction pathways: ethyl acetate formation via a 2-hydroxypropionate intermediate under anaerobic conditions and EP formation under aerobic conditions facilitated by chemisorbed oxygen. This work underscores the intricate structure-performance relationship governed by vanadium speciation and support effects.
In summary, these studies highlight the crucial influence of catalyst surface structure, metal oxidation states, and active site interfaces in non-precious metal catalysts for EL oxidation (see Table 1). The findings emphasize that precise control over active site morphology and electronic properties is essential for optimizing catalyst performance, stability, and selectivity. Continued efforts integrating advanced synthesis methods and in situ characterization will be vital for further improving catalyst design in sustainable oxidation processes.

Table 1.
Catalytic gas-phase oxidation of lactic acid (ester) to pyruvic acid (ester) over various catalysts.
3.2. Liquid-Phase Reactions
Compared to gas-phase processes, liquid-phase oxidation offers a more practical and milder approach for the synthesis of α-keto acids (esters) from biomass-derived substrates. Gas-phase reactions typically require harsh conditions—such as elevated temperatures (200–300 °C) or high pressures—to overcome the activation energy barrier associated with molecular oxygen, which often results in catalyst deactivation via carbon deposition or metal sintering. Furthermore, limited mass transfer efficiency at the gas–solid interface and the increased likelihood of side reactions under high-temperature conditions pose additional challenges. These limitations necessitate advanced catalyst design strategies, such as pore structure optimization, surface acidity modulation, or integration of redox cycles to enhance selectivity and stability, as well as the development of continuous flow systems to improve process efficiency. In contrast, liquid-phase systems can utilize milder oxidants and offer better control over reaction parameters. As shown in Figure 4, hydrogen peroxide (H2O2) is frequently employed as a green oxidant in conjunction with acidic molecular sieves (e.g., TS-1) to enable non-radical pathways with high selectivity. However, H2O2 is prone to thermal decomposition and side reactions such as hydrolysis or decarboxylation under elevated temperatures, necessitating stringent control of reaction conditions [44,88,89]. Tert-butyl hydroperoxide (TBHP), owing to its high reactivity and solubility in organic solvents, is another commonly used oxidant. Nevertheless, it requires exogenous addition and is susceptible to radical-induced side reactions, which can be mitigated through stabilizer additives or stepwise feeding protocols [90,91,92,93,94]. Molecular oxygen (O2), as an ideal green oxidant, offers excellent atom economy and environmental compatibility, particularly when sourced directly from air. Its application in continuous flow systems is advantageous, but its activation requires high-temperature, high-pressure conditions and highly active catalytic systems to overcome kinetic limitations [95,96,97,98]. A classification framework based on oxidant properties provides a rational basis for catalyst and process design—guiding decisions on active site engineering, support modification, and the optimization of operational parameters such as temperature, solvent system, and oxidant delivery. Looking forward, the development of efficient catalytic systems that enable the direct utilization of molecular oxygen under mild conditions remains a critical direction for advancing sustainable α-keto acid (ester) synthesis technologies.
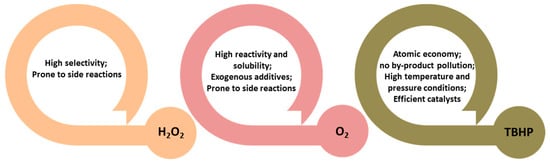
Figure 4.
Characterization of different types of oxidants for the synthesis of α-keto acids (esters) in liquid-phase oxidation reactions.
Based on these considerations, the following section will systematically introduce and categorize the different types of oxidants used in liquid-phase oxidation processes, analyzing their mechanisms, advantages, and limitations to guide the rational selection and design of efficient oxidation systems.
3.2.1. Hydrogen Peroxide
H2O2 has been widely employed as a green oxidant in the liquid-phase oxidation of EL, owing to its high reactivity and the potential for non-radical oxidation pathways. Lu et al. developed a TS-1/H2O2 catalytic system, which achieved 100% conversion of EL and 97.8% EP yield under mild conditions of solvent-free and 50 °C [44]. Mechanistic studies identified Ti–OOH species as the active centers responsible for the selective oxidation, operating via a non-radical pathway (Figure 5a). However, temperature was found to significantly influence the reaction outcome: although elevated temperatures (e.g., 70 °C) enhanced the conversion rate, they also promoted side reactions such as EP hydrolysis and oxidative decarboxylation, leading to the formation of acetic acid and CO2. Notably, TS-1 exhibited excellent recyclability, maintaining its activity over 10 consecutive cycles without significant deactivation. To further explore structure–performance relationships, Wang et al. synthesized two vanadium-aluminophosphate molecular sieves with different topologies—AFI-type VAPO-5 and AEL-type VAPO-11—via hydrothermal methods and systematically compared their catalytic behavior [99]. VAPO-5 demonstrated superior catalytic performance, attributed to its higher acid site density and effective isomorphous substitution of V5+ into P sites. A detailed reaction mechanism was proposed, involving the generation of reactive oxygen species from H2O2 on the molecular sieve surface and the subsequent stepwise oxidation of EL to EP. Despite these promising results, challenges remain regarding the stability of the molecular sieves and the undesired decomposition of H2O2, which require further investigation to enable long-term catalytic operation.
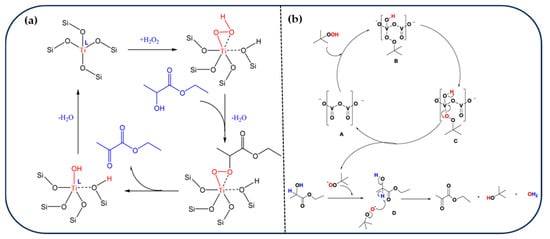
Figure 5.
(a) Reaction Pathway for ELA Oxidation over TS-1 with H2O2 as Oxidant [44]. Copyright 2018 ACS Publications. (b) Plausible reaction mechanism for oxidative dehydrogenation of EL to EP using V2O5 catalyst [100]. Copyright 2019 Elsevier.
3.2.2. TBHP
TBHP, due to its high solubility and reactivity in organic solvents, has been widely employed as an effective oxidant in liquid-phase oxidation systems. Doke et al. conducted a systematic comparison between homogeneous (VO(acac)2) and heterogeneous (V2O5) vanadium-based catalytic systems to evaluate their performance in the oxidative transformation of EL [100]. The homogeneous VO(acac)2 system demonstrated high catalytic activity under ambient conditions in acetonitrile, achieving 83% conversion with 100% selectivity toward EP (EP). However, it suffered from non-recyclability and limited operational stability. In contrast, the heterogeneous V2O5 catalyst required stepwise TBHP addition at elevated temperatures (80 °C) to suppress oxidant decomposition. Despite a longer reaction time (10 h), it achieved a higher conversion (98%) with sustained 100% selectivity over at least five catalytic cycles. Radical trapping experiments confirmed a free-radical mechanism for both systems, though the higher thermal energy input required for V2O5 increased the overall energy consumption (Figure 5b). To further assess the universality of vanadium-based systems, Doke et al. compared the performance of various metal catalysts (Mo, Cu, Fe) in TBHP-mediated oxidation. Molybdenum-based catalysts exhibited 92% conversion in organic media but were deactivated in aqueous environments. VO(acac)2 retained high activity in aqueous TBHP (82% conversion), and interestingly, reducing the solvent volume enhanced conversion (up to 98%) but led to side reactions such as EP polymerization, lowering selectivity to 80%. In a complementary study, Wang et al. reported for the first time the use of oxidized multiwalled carbon nanotubes (oCNTs) as a metal-free catalyst for the oxidative dehydrogenation of EL under mild conditions (90 °C, atmospheric pressure), achieving 93.6% selectivity [101]. Through detailed mechanistic analysis, the authors identified ketonic carbonyl groups (C=O) as the catalytically active sites and demonstrated dynamic surface transformations between defects and oxygen-containing functionalities during the reaction. The low selectivity observed with graphite oxides (<15%) was attributed to excessive carboxylic acid groups, whereas defect engineering via high-temperature treatment of oCNTs improved the distribution of active sites and selectivity. These findings provide valuable insights into the rational design of carbon-based catalysts for selective oxidation in fine chemical synthesis.
3.2.3. Molecular Oxygen
Molecular oxygen (O2) has emerged as an attractive green oxidant for liquid-phase catalytic systems, offering significant advantages in sustainability and cost-efficiency. Unlike exogenous oxidants such as H2O2 or TBHP, O2 can be directly sourced from air, thereby reducing raw material costs and eliminating concerns over secondary pollutant formation. Moreover, molecular oxygen participates directly in the dehydrogenation pathway, which inherently avoids the side reactions—such as hydrolysis and decarboxylation—that are commonly induced by the thermal instability of H2O2 or TBHP at elevated temperatures. This direct involvement not only enhances the atom economy but also significantly improves the selectivity toward target products by suppressing the formation of by-products like acetic acid and CO2. The following section systematically reviews recent advances in the catalytic oxidation of lactate esters to pyruvate esters using molecular oxygen, organized by catalyst classification, with an emphasis on the performance and underlying mechanisms of various catalyst systems.
Precious Metal Catalysts
Ai et al. first introduced a palladium-doped iron phosphate catalyst for the oxidative dehydrogenation of lactic acid to pyruvic acid. The incorporation of a small amount of palladium (0.8 wt%) significantly enhanced catalytic activity, achieving a ten-fold increase compared to pure iron phosphate, although initial selectivity was low with carbon oxides as the predominant by-products [102]. Over prolonged reaction times (~8 h), selectivity gradually improved to approximately 80%, comparable to that of the undoped catalyst, indicating that palladium primarily accelerates the reaction via enhanced surface redox capacity. Feng et al. further optimized pyruvic acid selectivity by employing Sn(IV)-doped MCM-41 supported Pd nanoparticles for the oxidation of 1,2-propanediol [103]. Their study underscored the crucial role of carrier modification in improving metal dispersion and promoting acid-base synergistic effects. Notably, Sn incorporation enhanced Pd dispersion and increased carrier basicity, with an optimal Si/Sn ratio of 12 yielding the highest pyruvic acid production. However, excessive Sn loading (Si/Sn = 6) led to SnO2 crystallization and consequent activity loss. The Sn(IV) species facilitated C–H bond activation via the formation of Sn-diol complexes, presenting a novel strategy for designing non-precious metal auxiliaries. Chu et al. systematically compared noble metals (Pt, Pd, Ru, etc.) supported on carbon for the catalytic oxidation of lactic acid methyl ester under alkali-free conditions [104]. Their findings revealed a correlation between the electronic states of noble metals and reaction pathways. Ru/activated carbon exhibited the highest selectivity (90%) and initial turnover frequency (TOF), an order of magnitude greater than other metals. Kinetic analysis suggested that the rate-determining step involved a Langmuir-Hinshelwood mechanism between surface-adsorbed substrates and oxygen species. The metallic state of Ru (84.3% Ru⁰) synergistically inhibited side reactions mediated by acidic and basic sites; however, its high cost restricts widespread practical application. Zhang et al. evaluated Pb-Pt bimetallic catalysts supported on various carbon materials (activated carbon, carbon black, etc.) and found that carbon black-supported 3Pb-1Pt/CB catalysts achieved a 60% pyruvic acid yield within 20 min, with a TOF of 769 h−1 [105]. The relatively low graphitization degree and small particle size of carbon black facilitated metal dispersion and enhanced reactant diffusion.
Non-Noble Metal Oxide Catalysts
Ramos-Fernandez et al. found that TiO2-catalyzed oxidation of EL followed a free radical mechanism, with activated carbon acting as a solid radical scavenger to inhibit the liquid-phase side reaction, and the selectivity was enhanced to 75% [106]. Molecular oxygen acted as the terminal oxidant, and the reaction displayed efficient performance even in inexpensive solvents. Zhang et al. synthesized vanadium–titanium mesoporous materials (Meso-VTN) via a one-pot synthesis and identified the V4+–O–Ti bond as the key active site [107]. In situ DRIFTS and DFT analyses confirmed EL adsorption and β-H deprotonation on the catalyst surface. Molecular oxygen was shown to regenerate surface oxygen vacancies, thus sustaining the catalytic cycle (Figure 6a). NH4VO3 was identified as the optimal vanadium precursor to form highly dispersed V4+ species, outperforming conventional impregnation methods for VOx/TiO2 catalysts. Liu et al. proposed that the MoO3–Mo–O acid–base pair on TiO2 catalyzes the dehydrogenation of lactic acid. Mo species were highly dispersed on TiO2, forming tetrahedral Mo sites, and the acid–base pair consisting of Mo=O and TiO2 basic sites facilitated C–H bond activation [108] (Figure 6b). While Mo aggregation led to undesired side reactions, a selectivity of 80% was achieved at 200 °C using 2 wt% MoO3/TiO2. Under oxidizing conditions (presence of O2), the hydrolysis–reoxidation cycle dominated, whereas anaerobic conditions favored bimolecular hydrogen transfer leading to by-products such as propionic acid.
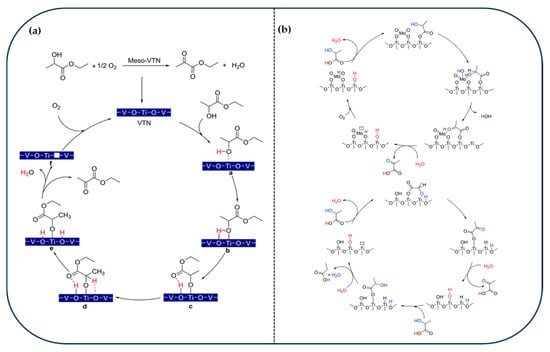
Figure 6.
(a) Proposed Catalytic Cycle for the Oxidative Dehydrogenation of EL to EP in the Presence of Meso-VTN [107]. Copyright 2018 ACS Publications. (b) Proposed catalytic reaction mechanisms for the aerobic and anaerobic conversion of LA over MoO3/TiO2 catalysts [108]. Copyright 2017 Royal Society of Chemistry.
Nitrogen-Doped/Nitrogen-Containing Carrier-Loaded Vanadium Catalysts
Liu et al. developed carbon nitride (CN)-doped VOx/SBA-15 catalysts containing pyridinic nitrogen species, which significantly enhanced the oxidation activity of EL, achieving 93% conversion and 98.7% selectivity at 130 °C over 3 h with 2.6 wt% V loading [109]. The incorporation of CN improved the carrier’s oxygen affinity and vanadium dispersion, as confirmed by increased surface oxygen mobility observed in O2-TPD measurements. This study elucidated the influence of different nitrogen species on selectivity modulation and provided guidance for designing nitrogen-doped catalysts. In another approach, Liu et al. employed biomass-derived chitin as a nitrogen-carbon precursor to synthesize nitrogen-doped carbon-supported VxOy/NC_P catalysts via a one-pot pyrolysis method, resulting in 97.3% conversion and 97.8% selectivity at 120 °C for 2 h [110]. Compared to chemically synthesized CN, chitin-derived nitrogen carbon (NC) featured higher pyridinic nitrogen content and a rich mesoporous structure. The combination of high surface area, abundant pyridinic nitrogen, and stabilized low-valent vanadium species (V3+/V4+) synergistically promoted O2 activation. The rigid chitin polymer backbone was proposed to facilitate the formation of a defective carbon matrix, enhancing the stabilization of low-valent vanadium. Zhang et al. further advanced this concept by developing homogeneous multiphase systems utilizing polypyridine carbon carriers loaded with vanadium, demonstrating that pyridinic nitrogen substantially improved EL oxidation activity in CN-doped VOx/P4VP catalysts [111]. Similarly, Wu et al. reported g-C3N4-supported V2O5 catalysts, where strong carrier–active component interactions enhanced the mobility of active oxygen species, significantly boosting catalytic activity. The best-performing catalyst, 13V2O5/g-C3N4, achieved 96.2% conversion and 85.6% selectivity at 130 °C, with only a 10% activity loss over four cycles [112].
Multiple Non-Precious Metal Synergistic Catalysts
Hariharan et al. synthesized Zn/Ni co-loaded mesoporous SBA-15 catalysts for continuous flow oxidation of EL, achieving 97.7% conversion and 99.5% selectivity [113]. High Ni loading enhanced catalytic activity, while Zn modulated the electronic structure, effectively suppressing side reactions. NH3-TPD analyses confirmed strong interactions between metal species and the carrier, which improved catalyst reducibility. Additionally, the mesoporous structure facilitated efficient mass transfer, highlighting the potential of this catalyst system for biomass valorization into high-value chemicals such as pharmaceutical intermediates. Ju et al. reported an efficient Zn(NO3)2/VOC2O4 catalytic system for the selective oxidation of α-hydroxy acid esters to α-keto acid esters under molecular oxygen and mild conditions [114]. The system exhibited up to 99% conversion and selectivity for substrates including methyl DL-mandelate and lactate esters, effectively minimizing side reactions like C–C bond cleavage. The unique catalytic behavior of Zn(NO3)2 was attributed to HNO3 gas generated via its interaction with VOC2O4, differing mechanistically from Fe(NO3)3-based systems that produce N2O3/N2O4 mixed gases.
Microreactor and Continuous Flow Process Optimization
In process intensification, Zhang et al. developed a Cu/keto-ABNO catalytic system integrated into a micro-packed-bed reactor, which reduced reaction time to 1/15 of conventional batch processes and increased space–time yield six-fold under continuous flow conditions [115]. Kinetic modeling revealed that enhanced mass transfer was the primary factor improving reaction efficiency, providing critical insights for industrial-scale continuous production optimization. Addressing limitations of traditional pyruvic acid synthesis, Yasukawa et al. introduced a microfluidic gas–liquid slug flow reactor featuring a double mixing zone combined with vanadium catalysis [116]. VOCl3 emerged as the optimal catalyst, achieving 31% yield via oxygen oxidation. The catalytic cycle involves VOCl3 reacting with lactate to form the active intermediate VO2Cl, which undergoes β-hydrogen elimination to yield pyruvate; subsequently, low-activity V(OH)2Cl is re-oxidized by oxygen. The slug flow enhances oxygen mass transfer through internal circulation and interface renewal, resulting in a 56% yield of EP in a 1.0 mm diameter reactor and reducing reaction time to minutes. Madhanagopal et al. designed a novel manganese-zinc bimetallic catalyst supported on mesoporous activated carbon (AC/Mn/Zn) for selective EL oxidation to EP [117]. Characterization confirmed the presence of both Brønsted and Lewis acid sites, critical for catalytic activity. Under optimized conditions (90 °C, WHSV 1.0 h−1, atmospheric pressure, air as oxidant), the catalyst achieved 91% conversion and 90% selectivity, outperforming comparable catalysts. Prolonged reaction times, however, favored by-product formation.
Molecular oxygen, as a green and cost-effective oxidant, demonstrates significant advantages in liquid-phase catalytic oxidation systems. Compared with conventional oxidants such as hydrogen peroxide and tert-butyl hydroperoxide, molecular oxygen not only reduces raw material costs and minimizes secondary pollution risks but also directly participates in the dehydrogenation pathway, effectively suppressing side reactions and enhancing both the selectivity and atom economy of target products. Current research covers precious metal catalysts, non-precious metal oxides, nitrogen-doped carrier-supported vanadium catalysts, and various non-precious metal synergistic catalyst systems, systematically revealing the effects of catalyst structures and active sites on reaction activity and selectivity (See Table 2). Notably, carrier modification, nitrogen doping, and multi-metal synergistic regulation significantly improve the generation and migration efficiency of active species. Furthermore, the introduction of microreactors and continuous flow processes greatly enhances mass transfer efficiency and process safety, offering new approaches and technological pathways for industrial application. Future studies should focus on designing low-cost, highly stable catalysts and integrating green processes to promote the efficient and sustainable production of biomass-derived chemicals using molecular oxygen.
Table 2.
Synthesis of pyruvic acid (esters) from lactic acid (esters) using molecular oxygen as the oxidizing agent in a liquid-phase catalytic system.
3.3. Photocatalytic Reaction
Photocatalytic oxidation has emerged as a promising and environmentally benign approach for the green synthesis of pyruvic acid (esters) in recent years. Compared to conventional gas-phase oxidation—often characterized by high temperatures, elevated pressures, and significant energy consumption—and liquid-phase oxidation methods that typically require strong oxidants and generate complex by-products, photocatalysis offers efficient substrate conversion under ambient conditions. This process leverages light-driven redox reactions to activate substrates while circumventing the need for toxic reagents and excessive energy input [118,119,120]. The core of photocatalytic oxidation lies in the precise regulation of electron transfer pathways and reactive oxygen species (ROS), such as hydroxyl radicals (·OH) and superoxide anions (O2·−), under photoexcitation. These species selectively cleave and oxidize the C–OH bond in precursors like lactic acid (esters), enabling the production of high-purity α-keto acids.
Notably, Han et al. first demonstrated visible-light-induced cleavage of the C=C bond in allylamine ketones, efficiently synthesizing enamine ketones at room temperature in ambient air [121]. This approach utilized air as the oxygen source, exhibited excellent selectivity for monoesterification of polyhydroxy substrates (e.g., diols, triols), and was compatible with natural product modifications. Recent advances have expanded from sole reliance on light energy to combined photothermal synergistic catalysis, optimized reaction interface microenvironments, and real-time in situ characterization techniques. These developments have substantially improved catalytic efficiency and product selectivity.
Selecting highly efficient catalysts is central to photocatalytic oxidation reactions. Given the critical influence of catalyst type, a systematic classification of existing photocatalytic oxidation catalysts is essential. This facilitates understanding their mechanisms and provides the basis for designing/screening optimal catalysts for specific keto acid substrates.
3.3.1. Sulfur-Based Catalysts
Sulfur-containing photocatalysts play multifaceted roles by modulating light absorption, enhancing charge separation, increasing surface reactivity, and improving catalyst stability.
CdS-Based Catalysts
CdS, with its narrow band gap, efficiently harvests visible light and facilitates product selectivity through direct hole oxidation and an indirect free radical pathway. Its band structure aligns well with the oxidation mechanism of lactic acid (esters). Miao et al. further developed a direct photoreforming strategy using Pd-CdS catalysts to convert real PLA plastics into pyruvic acid with concurrent hydrogen production under visible light, achieving 95.9% pyruvate selectivity and stable operation over 100 h without alkaline pretreatment [122]. Pd sites serve a dual function by enhancing hydrogen evolution and suppressing undesirable side reactions such as lactic acid coupling (Figure 7a)
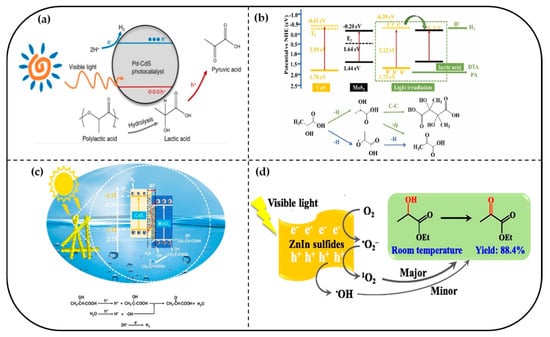
Figure 7.
The reaction pathways of lactic acid (esters) on different photocatalysts. (a) Schematic diagram of the direct photoreforming process of PLA over Pd-CdS [122]. Copyright 2024 ACS Publications. (b) Mechanistic diagram of CdM-20 photocatalytic hydrolysis for hydrogen production coupled to selective oxidation of lactic acid [123]. Copyright 2024 Elsevier. (c) Schematic representation of possible reaction pathways for CdS/MoS2 photoreforming of lactic acid [124]. Copyright 2024 Elsevier. (d) Proposed reaction mechanism for photocatalytic oxidation of EL to EP over ZIS-4 [125]. Copyright 2022 Elsevier.
Cai et al. designed bifunctional CdS–MoO2 catalysts to couple lactic acid oxidation with H2 generation. The heterojunction exploits CdS’s photoactivity and MoO2’s oxidation capability, achieving >90% lactic acid conversion and two-fold-enhanced H2 yield (Figure 7b). Mechanistically, h+ on CdS activates lactic acid to form radicals, while MoO2 shifts selectivity toward propionic acid (PA) by suppressing coupling or favoring α-O-H cleavage [123]. Liang et al. constructed a layered 1T/2H-MoS2/CdS heterojunction composite, wherein the heterojunction interface regulates photogenerated carrier distribution and directs selective photoreforming of lactic acid. In this catalytic system, electrons accumulating at MoO2 reduction centers drive efficient hydrogen evolution, while holes on CdS surfaces selectively activate α-C–H and α-O–H bonds to generate carbon- and oxygen-centered radicals. These radicals subsequently mediate the selective formation of tartaric acid derivatives and pyruvic acid (with yields up to 75% and 80%, respectively, Figure 7c); concurrently, the dispersion of MoO2 facilitates parallel H2 evolution and lactic acid oxidation via radical-intermediate pathways [124].
ZnIn2S4-Based Catalysts
ZnIn2S4 photocatalysts precisely regulate oxidation pathways via synergistic effects of photogenerated holes and reactive oxygen species. Their layered structure exposes abundant active sites, while synergistic interactions between In, Zn, and sulfur terminals enhance substrate adsorption and electron transfer [126,127,128]. Lu et al. reported the first efficient, selective oxidation of EL to EP at room temperature under visible light, achieving 100% substrate conversion and 88.4% product yield by tuning Zn/In ratios to optimize specific surface area and electron-hole separation [125]. Radical trapping and mechanistic studies identified singlet oxygen (1O2) generation and C(OH)–H bond cleavage as key steps, with the latter likely rate-determining (Figure 7d). Roostaei et al. developed a hollow-structured NiP–ZnIn2S4 heterojunction photocatalyst, enabling co-production of hydrogen (381 μmol/h) and pyruvic acid (97.8% selectivity). NiP co-catalysts improve charge separation efficiency, yielding an apparent quantum efficiency of 11.1% under 365 nm light [129]. Subsequently, the same group fabricated a 0D–2D CoP–ZnIn2S4 heterojunction catalyst with enhanced spatial charge separation and bifunctionality, achieving a hydrogen evolution rate of 233.78 μmol/h and lactic acid conversion to pyruvic acid with up to 93.9% selectivity and 80.9% total conversion. Economic analyses underscore the commercial viability of this cogeneration approach, with a pyruvate recovery rate of 99% [130].
3.3.2. Molybdenum-Based Catalysts
Molybdenum’s unique electronic structure, multiple valence states, and diverse compound forms significantly contribute to enhanced light absorption, charge separation, and surface reactivity in photocatalysis [131,132,133]. Although less explored for lactic acid (ester) oxidation, molybdenum-based catalysts hold substantial potential in green catalytic processes. Chen et al. demonstrated that subsurface molybdenum vacancies in Bi2MoO6 enhance photocatalytic oxidation of EL to EP. These vacancies induce hole trapping by neighboring oxygen atoms and create built-in electric fields that improve photogenerated carrier separation and promote singlet oxygen (1O2) formation. The modified Bi2MoO6 achieved over 99% conversion and 90.2% product yield within 3 h of irradiation, outperforming pristine Bi2MoO6 (28.6% conversion), with good catalyst stability and applicability to other hydroxy compounds [134].
Photocatalytic oxidation represents a green, mild, and efficient alternative to traditional high-temperature and high-pressure oxidation processes, enabling selective transformation of biomass-derived substrates into valuable α-keto acids under ambient conditions. Advances in sulfur-based and molybdenum-based photocatalysts, including CdS, ZnIn2S4, and Bi2MoO6 systems, have demonstrated enhanced light absorption, charge separation, and active species generation, significantly improving conversion efficiencies and product selectivities (See Table 3). Nevertheless, challenges remain, such as limited visible-light quantum efficiencies, catalyst stability in continuous operation, and economic feasibility for scale-up. Future research should emphasize the development of full-spectrum-responsive materials, defect engineering, integrated reaction-separation technologies, and life-cycle-optimized systems to facilitate the transition of photocatalytic oxidation from laboratory studies to industrial applications.
Table 3.
Research on the photocatalytic oxidation of lactic acid (ester) to α-keto acid (ester) catalyzed by multiple catalysts.
4. Difficulties and Challenges
Although significant progress has been made in the synthesis of α-keto acids and their esters, multidimensional challenges remain across methodological innovation, catalyst design, oxidant optimization, and green process development, requiring integrated and systematic breakthroughs.
Regarding synthetic methodologies, traditional chemical routes such as Friedel-Crafts acylation (e.g., AlCl3-catalyzed direct acylation of oxalyl chloride monoethyl ester with aromatics [37]) are operationally straightforward but suffer from limitations due to electronic effects of aryl substituents (strong electron-donating groups cause a sharp drop in yield) and environmental issues associated with Lewis acid post-treatment. Bi-carbonylation reactions (e.g., CoCl2-catalyzed benzyl chloride carbonylation to phenylpyruvic acid [38]) boast high atom economy but face high-pressure CO conditions (>50 atm) that impose equipment costs and safety challenges. In contrast, biomass catalytic routes (such as oxidative dehydrogenation of lactic acid to pyruvate) align with green chemistry principles, yet liquid-phase oxidation employing H2O2 and TBHP induces side reactions (e.g., over-oxidation of EL yielding acetate or decarboxylation products), and oxidant costs represent 30–50% of total expenses (with TBHP nearly three times more costly than H2O2 at a 50 mmol scale). Vapor phase oxidation (VPO), while efficient and able to use airborne oxygen directly, demands high temperatures, resulting in elevated energy consumption, and carbon deposition on catalyst surfaces causes active site blockage requiring frequent regeneration, undermining continuous operation stability. Photocatalysis achieves high selectivity under mild conditions but suffers from insufficient quantum efficiency and unresolved mass transfer limitations in photoreactor design.
Catalyst-wise, the electronic structures and coordination environments of active centers in multiphase catalysts undergo dynamic evolution during reactions (e.g., metal valence fluctuations, interfacial restructuring between carrier and active components), yet current in situ characterization techniques fall short in revealing these processes precisely, hindering mechanistic understanding—such as the unresolved dynamic reconfiguration mechanism of active sites in V2O5/TiO2 systems [87]. Catalyst deactivation remains a major bottleneck for industrial application; for example, MoO3/TiO2 loses over 50% activity after 500 h of continuous operation due to Mo species migration and agglomeration plus surface carbon accumulation [108]. Homogeneous catalysts like VO(acac)2 achieve up to 98% conversion of EL in TBHP systems but pose challenges in separation and recovery, and vanadium toxicity restricts industrial adoption. Photocatalysts exhibit high carrier recombination rates and unclear oxygen migration mechanisms within metal oxide lattices, limiting rational design. Sulfur-containing metal chalcogenide photocatalysts such as ZnIn2S4 suffer from oxidation of interlayer sulfur terminals to SO32− during reaction, increasing carrier recombination centers. Although CdS can efficiently harvest visible light, dynamic annihilation of sulfur vacancies also intensifies carrier recombination [125,129,130]. Noble metal catalysts (e.g., Au) deliver outstanding activity, but their high cost and toxicity hinder scale-up [70], while non-precious metal systems (e.g., Fe- and Mo-based) suffer from active site sintering and metal leaching, compromising stability. Furthermore, current research tends to emphasize material synthesis and catalytic activity data while overlooking the upstream and downstream integration of biomass valorization. For example, limited attention is paid to the compatibility of catalysts with complex biomass hydrolysates containing impurities such as lignin fragments, minerals, or sugars, which may poison active sites or alter redox dynamics. Similarly, little work has been done to couple α-keto acid synthesis with downstream product separation or functionalization, making it difficult to evaluate the overall efficiency and feasibility of proposed systems in real biorefinery scenarios.
Regarding oxidant selection, H2O2 performs excellently in liquid-phase oxidation due to in situ generation of hydroxyl radicals but requires stringent temperature control to prevent thermal decomposition, and residual H2O2 quenching steps increase wastewater chemical oxygen demand (COD). Direct activation of molecular oxygen is greener but limited by low O2 mass transfer coefficients in the liquid phase, necessitating microbubble generators or supercritical CO2 to improve solubility, which escalates equipment complexity and operating costs.
From an industrial perspective, traditional methods (e.g., Friedel-Crafts acylation) offer operational simplicity but incur hidden costs from waste treatment (e.g., 25% production cost for acid neutralization). In contrast, catalytic biomass routes align with green chemistry principles but face scalability barriers due to catalyst stability issues (e.g., Mo species agglomeration in MoO3/TiO2) [108]. Photocatalysis enables ambient-condition synthesis but suffers from low quantum efficiency, while gas-phase oxidation minimizes solvent use but requires energy-intensive temperatures. A techno-economic balance must be struck between atom economy (traditional bicarbonylation: >80% yield) and sustainability (O2-based liquid-phase systems: near-zero toxic byproducts). To facilitate readers’ more intuitive understanding, in Table 4, we analyzed and summarized the differences between the traditional methods of α-keto acids (esters) and the new green synthetic routes from different perspectives.
Table 4.
Comparative analysis of α-keto acid synthesis routes.
From safety and economic perspectives, traditional sulfuric acid dehydration methods (e.g., pyruvic acid from tartaric acid) require concentrated sulfuric acid in a 1:3 molar ratio, causing severe equipment corrosion and costly acid waste treatment (accounting for 25% of production costs). Biomass pretreatment (e.g., acid or enzymatic hydrolysis) accounts for over 60% of total energy consumption, and product separation/purification (e.g., pyruvate/ethyl acetate azeotropic systems) depends on molecular sieve membranes or extractive distillation, further increasing complexity and cost. The bioaccumulation risks of nano-catalysts (e.g., CdS, carbon nanotubes) remain poorly understood, with industrial spent catalyst recovery rates under 60%. Acidic sludge and heavy metal pollutants from conventional processes require complex reprocessing, raising environmental remediation costs, while strong acids/bases and toxic intermediates present safety hazards.
5. Summary and Prospect
The synthesis of α-keto acids and their esters has evolved from classical multi-step chemical transformations to greener catalytic processes leveraging biomass-derived substrates and renewable oxidants. Traditional chemical methods offer high yields and scalability but face growing scrutiny over sustainability and safety. In contrast, catalytic oxidation of α-hydroxy acids under mild conditions—particularly in gas-phase, liquid-phase, and photocatalytic systems—has emerged as a viable strategy for green synthesis, though each pathway presents its own limitations in catalyst stability, oxidant efficiency, and reaction selectivity.
To address these challenges and transition toward industrial implementation, future research should focus on the following strategic directions: 1. Rational catalyst design through multi-scale understanding. Developing robust catalysts with high selectivity and long-term operational stability requires precise control over surface structure, oxidation state, and active site environment. Advances in single-atom catalysts, heterostructure engineering, and oxygen-vacancy regulation will be critical. Moreover, in situ/operando characterization combined with DFT and machine-learning-guided modeling can provide mechanistic insight to guide design, enabling the prediction of structure–activity relationships under real reaction conditions. 2. Integrated process and reactor engineering. Efforts should move beyond batch-mode proof-of-concept studies toward continuous flow systems and modular reactor platforms. Gas–liquid-solid microreactors, membrane-assisted separations, and photothermal co-driven systems hold potential to bridge the gap between catalytic performance and industrial throughput. Particular attention should be given to improving oxidant utilization (e.g., O2) via interfacial engineering and intensifying mass/heat transfer under ambient conditions. 3. Green oxidant systems and redox cycling. Transitioning to environmentally benign oxidants necessitates developing catalysts that can directly activate molecular oxygen or in situ-generated reactive oxygen species (ROS) without external oxidants. Designing catalysts capable of selective redox cycling may improve O2 activation efficiency while minimizing overoxidation or decarboxylation side reactions. 4. Expanding renewable feedstock scope. Incorporating lignin derivatives, food/agricultural waste, and even plastic waste as carbon sources for α-keto acid synthesis offers a sustainable path forward. Valorization of such unconventional substrates requires multifunctional catalysts and tandem reaction strategies capable of handling complex feedstock compositions and reaction intermediates. 5. Lifecycle and sustainability assessment. Beyond synthetic efficiency, future technologies must be evaluated through comprehensive techno-economic analysis (TEA), life-cycle assessment (LCA), and environmental risk analysis. This includes understanding the fate of nanocatalysts, waste treatment feasibility, and energy input–output balance. Establishing such metrics will be essential to guide scalable, low-carbon synthesis technologies. In conclusion, the future of α-keto acid synthesis lies in the convergence of catalytic innovation, process intensification, and systems-level sustainability. Interdisciplinary efforts integrating chemistry, materials science, and chemical engineering will be key to advancing from laboratory-scale reactions to practical, low-emission, and cost-effective manufacturing platforms.
From an industrial perspective, bridging the gap between academic catalysis research and real-world application demands process integration across feedstock pretreatment, catalytic conversion, and downstream purification. The development of continuous-flow systems, modular reactors, and scalable green oxidant supply chains will be essential to enable practical deployment. Moreover, techno-economic analysis (TEA) and lifecycle assessment (LCA) should be embedded into early-stage catalyst and process development to ensure that green synthesis is not only environmentally benign but also commercially viable.
Author Contributions
C.S.: Conceptualization, investigation, and writing—review and editing; K.H.: Investigation, visualization, and resources; X.L.: Conceptualization, project administration, supervision, and writing—review and editing; Y.X.: Conceptualization, supervision, and writing—review and editing; F.Y. and T.L.: Conceptualization, project administration, supervision, and writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (22379131), and the China Postdoctoral Science Foundation (2024M762993).
Data Availability Statement
The original contributions presented in the study are included in the article; further inquiries can be directed to the corresponding researchers.
Acknowledgments
The researchers thank the financial support from the funding sources mentioned above.
Conflicts of Interest
Author Yongming Xu was employed by the company of China Tobacco Henan Industrial Co., Ltd. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Wang, Z.; La, B.; Fortunak, J.M.; Meng, X.-J.; Kabalka, G.W. Enantioselective synthesis of α-hydroxy carboxylic acids: Direct conversion of α-oxocarboxylic acids to enantiomerically enriched α-hydroxy carboxylic acids via neighboring group control. Tetrahedron Lett. 1998, 39, 5501–5504. [Google Scholar] [CrossRef]
- Song, T.; Ma, Z.; Wang, X.; Yang, Y. Synthesis of α-keto acids via oxidation of alkenes catalyzed by a bifunctional iron nanocomposite. Org. Lett. 2021, 23, 5917–5921. [Google Scholar] [CrossRef]
- Penteado, F.; Lopes, E.F.; Alves, D.; Perin, G.; Jacob, R.G.; Lenardão, E.J. α-keto acids: Acylating agents in organic synthesis. Chem. Rev. 2019, 119, 7113–7278. [Google Scholar] [CrossRef] [PubMed]
- Earle, R.H.; Hurst, D.T.; Viney, M. Synthesis and hydrolysis of some fused-ring β-lactams. J. Chem. Soc. C 1969, 16, 2093–2098. [Google Scholar] [CrossRef]
- Draber, W.; Timmler, H.; Dickoré, K.; Donner, W. Synthese und reaktionen von 3-Alkyl-4-amino-l,2,4-triazin-5-onen. Justus Liebigs Ann. Chemie 1976, 1976, 2206–2221. [Google Scholar] [CrossRef]
- Domagala, J.M. A mild, rapid, and convenient esterification of α-keto acids. Tetrahedron Lett. 1980, 21, 4997–5000. [Google Scholar] [CrossRef]
- Ibrahim, Y.A.; Eid, M.M.; Badawy, M.A.; Abdel-Hady, S.A.L. Reaction of 4-aryl-1,2,4-triazines with hydrazine. J. Heterocycl. Chem. 1981, 18, 953–956. [Google Scholar] [CrossRef]
- Styles, V.L.; Morrison, R.W., Jr. Pyrimido[4,5-c]pyridazines. 4. cyclizations with alpha-oxo acids. J. Org. Chem. 1982, 47, 585–587. [Google Scholar] [CrossRef]
- Clerici, A.; Porta, O. A novel reaction type promoted by aqueous titanium trichloride. Synthesis of unsymmetrical 1,2-diols. J. Org. Chem. 1982, 47, 2852–2856. [Google Scholar] [CrossRef]
- Cohen, M.J.; McNelis, E. Oxidative decarboxylation of propiolic acids. J. Org. Chem. 1984, 49, 515–518. [Google Scholar] [CrossRef]
- Bris, M.-T.L. Réaction de l’amino-2 Nitro-5 Phénol et Du Diamino-2,5 Phénol avec quelques acides et esters α-cétoniques. Synthèse d’amino-7 benzoxazines-1,4 ones-2. J. Heterocycl. Chem. 1984, 21, 551–555. [Google Scholar] [CrossRef]
- Arndt, F.; Franke, W.; Klose, W.; Lorenz, J.; Schwarz, K. Synthesen von thiazolo-und [1,3]thiazino[1,2,4]triazinonen. Justus Liebigs Ann. Chem. 1984, 1984, 1302–1307. [Google Scholar] [CrossRef]
- Bris, M.-T.L. Synthesis and properties of some 7-dimethylamino-1,4-benzoxazin-2-ones. J. Heterocycl. Chem. 1985, 22, 1275–1280. [Google Scholar] [CrossRef]
- Shirley, H.J.; Koyioni, M.; Muncan, F.; Donohoe, T.J. Synthesis of lamellarin alkaloids using orthoester-masked α-keto acids. Chem. Sci. 2019, 10, 4334–4338. [Google Scholar] [CrossRef]
- Mandzhieva, I.; Adelabu, I.; Chekmenev, E.Y.; Theis, T. Proton-only sensing of hyperpolarized [1,2-13C2]pyruvate. ACS Sens. 2022, 7, 3773–3781. [Google Scholar] [CrossRef]
- Hara, T.; Iio, M.; Izuchi, R.; Tsukiyama, T.; Yokoi, F. Synthesis of pyruvate-1-11C as a radiopharmaceutical for tumor imaging. Eur. J. Nucl. Med. 1985, 11, 275–278. [Google Scholar] [CrossRef]
- Liu, S.-Q. Practical implications of lactate and pyruvate metabolism by lactic acid bacteria in food and beverage fermentations. Int. J. Food Microbiol. 2003, 83, 115–131. [Google Scholar] [CrossRef]
- Pundir, C.S.; Malik, M.; Chaudhary, R. Quantification of pyruvate with special emphasis on biosensors: A review. Microchem. J. 2019, 146, 1102–1112. [Google Scholar] [CrossRef]
- Zangari, J.; Petrelli, F.; Maillot, B.; Martinou, J.-C. The multifaceted pyruvate metabolism: Role of the mitochondrial pyruvate Carrier. Biomolecules 2020, 10, 1068. [Google Scholar] [CrossRef]
- van Spronsen, F.J.; Blau, N.; Harding, C.; Burlina, A.; Longo, N.; Bosch, A.M. Phenylketonuria. Nat. Rev. Dis. Primers 2021, 7, 36. [Google Scholar] [CrossRef]
- Hu, Y.; Wang, Y.; Wang, R.; Zhang, W.; Hua, R. Designing stimuli-responsive up conversion nanoparticles based on an inner filter effect mimetic immunoassay for phenylketonuria accuracy diagnosis. Colloids Surf. B 2022, 217, 112642. [Google Scholar] [CrossRef] [PubMed]
- Pantaleone, D.P.; Geller, A.M.; Taylor, P.P. Purification and characterization of an L-amino acid deaminase used to prepare unnatural amino acids. J. Mol. Catal. B Enzym. 2001, 11, 795–803. [Google Scholar] [CrossRef]
- Li, X.; Jiang, B.; Pan, B. Biotransformation of phenylpyruvic acid to phenyllactic acid by growing and resting cells of a lactobacillus Sp. Biotechnol. Lett. 2007, 29, 593–597. [Google Scholar] [CrossRef][Green Version]
- Hou, Y.; Hossain, G.S.; Li, J.; Shin, H.; Liu, L.; Du, G. Production of phenylpyruvic acid from L-phenylalanine using an L-amino acid deaminase from proteus mirabilis: Comparison of enzymatic and whole-cell biotransformation approaches. Appl. Microbiol. Biotechnol. 2015, 99, 8391–8402. [Google Scholar] [CrossRef]
- Coban, H.B.; Demirci, A.; Patterson, P.H.; Elias, R.J. Screening of phenylpyruvic acid producers and optimization of culture conditions in bench scale bioreactors. Bioprocess Biosyst. Eng. 2014, 37, 2343–2352. [Google Scholar] [CrossRef]
- Rodríguez-Pazo, N.; Vázquez-Araújo, L.; Pérez-Rodríguez, N.; Cortés-Diéguez, S.; Domínguez, J.M. Cell-free supernatants obtained from fermentation of cheese whey hydrolyzates and phenylpyruvic acid by lactobacillus plantarum as a source of antimicrobial compounds, bacteriocins, and natural aromas. Appl. Biochem. Biotechnol. 2013, 171, 1042–1060. [Google Scholar] [CrossRef]
- Brekke, E.; Walls, A.B.; Nørfeldt, L.; Schousboe, A.; Waagepetersen, H.S.; Sonnewald, U. Direct measurement of backflux between oxaloacetate and fumarate following pyruvate carboxylation. Glia 2012, 60, 147–158. [Google Scholar] [CrossRef]
- Li, J.; Tang, G.; Dao, F.; Deng, K.; Li, P.; Huang, J.; Xie, J.; Jiang, J. Recyclable malate dehydrogenase nanocatalyst based on chitosan for oxaloacetic acid green synthesis. Microchem. J. 2024, 197, 109787. [Google Scholar] [CrossRef]
- Hannya, A.; Nishimura, T.; Matsushita, I.; Tsubota, J.; Kawata, Y. Efficient production and secretion of oxaloacetate from halomonas sp. KM-1 under aerobic conditions. AMB Express 2017, 7, 209. [Google Scholar] [CrossRef]
- Gupte, R.; Keselman, P.L.; Christian, S.K.; Hu, J.; Swerdlow, R.H.; Harris, J.L. Does oxaloacetate preserve mitochondrial function in the aged brain? Alzheimers Dement. 2020, 16, e037386. [Google Scholar] [CrossRef]
- Campos, F.; Sobrino, T.; Ramos-Cabrer, P.; Castillo, J. Oxaloacetate: A novel neuroprotective for acute ischemic stroke. Int. J. Biochem. Cell Biol. 2012, 44, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Nikulin, M.; Drobot, V.; Švedas, V.; Krasnikov, B.F. Preparative biocatalytic synthesis of α-ketoglutaramate. Int. J. Mol. Sci. 2021, 22, 12748. [Google Scholar] [CrossRef] [PubMed]
- Niu, S.; Liu, F.; Wang, Y.; Rao, B.; Wang, Y. A study on the efficient preparation of α-ketoglutarate with L-glutamate oxidase. Molecules 2024, 29, 1861. [Google Scholar] [CrossRef] [PubMed]
- Doucette, C.D.; Schwab, D.J.; Wingreen, N.S.; Rabinowitz, J.D. α-ketoglutarate coordinates carbon and nitrogen utilization via enzyme I inhibition. Nat. Chem. Biol. 2011, 7, 894–901. [Google Scholar] [CrossRef]
- Shen, D.; Kruger, L.; Deatherage, T.; Denton, T.T. Synthesis of α-ketoglutaramic acid. Anal. Biochem. 2020, 607, 113862. [Google Scholar] [CrossRef]
- Nemoto, T.; Ohshima, T.; Shibasaki, M. Catalytic asymmetric synthesis of α,β-epoxy esters, aldehydes, amides, and γ,δ-epoxy β-keto esters: Unique reactivity of α,β-unsaturated carboxylic acid imidazolides. J. Am. Chem. Soc. 2001, 123, 9474–9475. [Google Scholar] [CrossRef]
- Xiang, J.M.; Li, B.L. Solvent-free synthesis of some ethyl arylglyoxylates. Chin. Chem. Lett. 2009, 20, 55–57. [Google Scholar] [CrossRef]
- Li, G.-X.; Li, L.; Huang, H.-M.; Cai, H.-Q. One pot formation of catalyst and double carbonylation of benzyl chloride. J. Mol. Catal. A Chem. 2003, 193, 97–102. [Google Scholar]
- Sun, Y.; Jing, X.; Xu, B.; Liu, H.; Chen, M.; Wu, Q.; Huang, Z.; Zheng, L.; Bi, X.; Nie, Y.; et al. A single-atom iron nanozyme reactor for α-Ketoglutarate Synthesis. Chem. Eng. J. 2023, 466, 143269. [Google Scholar] [CrossRef]
- Li, M.; Wu, Y.; Zhao, B.-H.; Cheng, C.; Zhao, J.; Liu, C.; Zhang, B. Electrosynthesis of amino acids from NO and α-keto acids using two decoupled flow reactors. Nat. Catal. 2023, 6, 906–915. [Google Scholar] [CrossRef]
- Xiao, Y.; Lim, C.W.; Chang, J.; Yuan, Q.; Wang, L.; Yan, N. Electrocatalytic amino acid synthesis from biomass-derivable keto acids over ball milled carbon nanotubes. Green Chem. 2023, 25, 3117–3126. [Google Scholar] [CrossRef]
- Bozell, J.J.; Petersen, G.R. Technology development for the production of biobased products from biorefinery carbohydrates—The US department of energy’s “Top 10” revisited. Green Chem. 2010, 12, 539–554. [Google Scholar] [CrossRef]
- Ma, J.; Jin, D.; Li, Y.; Xiao, D.; Jiao, G.; Liu, Q.; Guo, Y.; Xiao, L.; Chen, X.; Li, X.; et al. Photocatalytic conversion of biomass-based monosaccharides to lactic acid by ultrathin porous oxygen doped carbon nitride. Appl. Catal. B Environ. 2021, 283, 119520. [Google Scholar] [CrossRef]
- Lu, T.L.; Zou, J.P.; Zhan, Y.Z.; Yang, X.M.; Wen, Y.Q.; Wang, X.Y.; Zhou, L.P.; Xu, J. Highly efficient oxidation of EL to EP catalyzed by TS-1 under mild conditions. ACS Catal. 2018, 8, 1287–1296. [Google Scholar] [CrossRef]
- You, Y.; Han, P.; Song, S.; Luo, W.; Zhao, S.; Han, K.; Tian, Y.; Yan, N.; Li, X. Distinct selectivity control in solar-driven bio-based α-hydroxyl acid conversion: A comparison of Pt nanoparticles and atomically dispersed Pt on CdS. Angew. Chem. Int. Ed. 2023, 62, e202306452. [Google Scholar] [CrossRef]
- Coin, C.; Le Boisselier, V.; Favier, I.; Postel, M.; Duñach, E. Bi0/O2 as a new catalytic system for the oxidation of α-ketols, α-hydroxy acids and epoxides. Eur. J. Org. Chem. 2001, 2001, 735–740. [Google Scholar] [CrossRef]
- Zhuang, J.; Wang, C.; Xie, F.; Zhang, W. One-pot efficient synthesis of aryl α-keto esters from aryl-ketones. Tetrahedron 2009, 65, 9797–9800. [Google Scholar] [CrossRef]
- Wadhwa, K.; Yang, C.; West, P.R.; Deming, K.C.; Chemburkar, S.R.; Reddy, R.E. Synthesis of arylglyoxylic acids and their collision-induced dissociation. Synth. Commun. 2008, 38, 4434–4444. [Google Scholar] [CrossRef]
- Hallmann, G.; Hägele, K. Benzofuranderivate. Tryptamin-, serotonin- und melatonin-analoga. Liebigs Ann. Chem. 1963, 662, 147–159. [Google Scholar] [CrossRef]
- Pirrung, M.C.; Tepper, R.J. Photochemistry of substituted benzoylformate esters. A convenient method for the photochemical oxidation of alcohols. J. Org. Chem. 1995, 60, 2461–2465. [Google Scholar] [CrossRef]
- Beebe, X.; Nilius, A.M.; Merta, P.J.; Soni, N.B.; Bui, M.H.; Wagner, R.; Beutel, B.A. Synthesis and SAR evaluation of oxadiazolopyrazines as selective haemophilus influenzae antibacterial agents. Bioorg. Med. Chem. Lett. 2003, 13, 3133–3136. [Google Scholar] [CrossRef] [PubMed]
- Stocks, M.J.; Harrison, R.P.; Teague, S.J. Macrocyclic ring closures employing the intramolecular heck reaction. Tetrahedron Lett. 1995, 36, 6555–6558. [Google Scholar] [CrossRef]
- Tatlock, J.H. Oxidation of alkynyl ethers with potassium permanganate. A new acyl anion equivalent for the preparation of alpha-keto esters. J. Org. Chem. 1995, 60, 6221–6223. [Google Scholar] [CrossRef]
- Bulman Page, P.C.; Rosenthal, S. Simple preparation of α-bromo acyl silanes α-ketoacyl silanes and α-ketoesters from silyl acetylenes. Tetrahedron 1990, 46, 2573–2586. [Google Scholar] [CrossRef]
- Claus, A.; Neukranz, W. Ueber die oxydation der gemischten fettaromatischen ketone durch kaliumpermanganat-kali. J. Prakt. Chem. 1891, 44, 77–85. [Google Scholar] [CrossRef]
- Hurd, C.D.; McNamee, R.W.; Green, F.O. Benzoylformic acid from styrene. J. Am. Chem. Soc. 1939, 61, 2979–2980. [Google Scholar] [CrossRef]
- Oakwood, T.S.; Weisgerber, C.A. Benzoylformic acid. Org. Synth. 1944, 24, 16. [Google Scholar]
- Nimitz, J.S.; Mosher, H.S. A new synthesis of alpha-keto esters and acids. J. Org. Chem. 1981, 46, 211–213. [Google Scholar] [CrossRef]
- Weinstock, L.M.; Currie, R.B.; Lovell, A.V. A General, One-step synthesis of α-keto esters. Synth. Commun. 1981, 11, 943–946. [Google Scholar] [CrossRef]
- Sakakura, T.; Yamashita, H.; Kobayashi, T.; Hayashi, T.; Tanaka, M. Synthesis of alpha-keto esters via palladium-catalyzed double carbonylation. J. Org. Chem. 1987, 52, 5733–5740. [Google Scholar] [CrossRef]
- Itoh, K.; Miura, M.; Nomura, M. Normal pressure double carbonylation of aryl halides using cobalt(II) chloride in the presence of either sodium sulfide or sodium borohydride. Bull. Chem. Soc. Jpn 1988, 61, 4151–4152. [Google Scholar] [CrossRef]
- Zhang, Z.; Su, J.; Zha, Z.; Wang, Z. Electrochemical Synthesis of the aryl α-ketoesters from acetophenones mediated by KI. Chem. Eur. J. 2013, 19, 17711–17714. [Google Scholar] [CrossRef]
- Li, S.; Zhang, K. Xu, Recent advances towards electrochemical transformations of α-keto acids. Chin. Chem. Lett. 2021, 32, 2729–2735. [Google Scholar] [CrossRef]
- Luo, Z.; Yu, S.; Zeng, W.; Zhou, J. Comparative analysis of the chemical and biochemical synthesis of keto acids. Biotechnol. Adv. 2021, 47, 107706. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.-B.; Hua, W.; Liu, L.; Liu, H. Sustainable synthesis of α-ketoglutaric and methanetriacetic acids from biomass feedstocks. Nat. Commun. 2025, 16, 1245. [Google Scholar] [CrossRef]
- Luo, X.; He, R.; Liu, Q.; Gao, Y.; Li, J.; Chen, X.; Zhu, Z.; Huang, Y.; Li, Y. Metal-free oxidative esterification of ketones and potassium xanthates: Selective synthesis of α-ketoesters and esters. J. Org. Chem. 2020, 85, 5220–5230. [Google Scholar] [CrossRef]
- Hughes, M.D.; Xu, Y.-J.; Jenkins, P.; McMorn, P.; Landon, P.; Enache, D.I.; Carley, A.F.; Attard, G.A.; Hutchings, G.J.; King, F.; et al. Tunable gold catalysts for selective hydrocarbon oxidation under mild conditions. Nature 2005, 437, 1132–1135. [Google Scholar] [CrossRef] [PubMed]
- Corma, A.; Concepción, P.; Boronat, M.; Sabater, M.J.; Navas, J.; Yacaman, M.J.; Larios, E.; Posadas, A.; López-Quintela, M.A.; Buceta, D.; et al. Exceptional oxidation activity with size-controlled supported gold clusters of low atomicity. Nat. Chem. 2013, 5, 775–781. [Google Scholar] [CrossRef]
- Agarwal, N.; Freakley, S.J.; McVicker, R.U.; Althahban, S.M.; Dimitratos, N.; He, Q.; Morgan, D.J.; Jenkins, R.L.; Willock, D.J.; Taylor, S.H.; et al. Aqueous Au-Pd colloids catalyze selective CH4 oxidation to CH3OH with O2 under mild conditions. Science 2017, 358, 223–227. [Google Scholar] [CrossRef]
- Lu, S.; Zhang, J.; Wu, Z.; Su, Z.; Huang, J.; Liang, Y.; Xiao, F.-S. Catalytic oxidation of EL to EP over Au-based catalyst using authentic air as oxidant. Catal. Surv. Asia 2022, 26, 211–220. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, C.; Ma, K.; Li, X. Crystal phase-mediated oxidative dehydrogenation of lactic acid to pyruvic acid on MoO3. Surf. Interf. 2023, 42, 103524. [Google Scholar] [CrossRef]
- Yin, C.; Li, X.; Dai, Y.; Chen, Z.; Yang, D.; Liu, R.; Zou, W.; Tang, C.; Dong, L. The facet-regulated oxidative dehydrogenation of lactic acid to pyruvic acid on α-Fe2O3. Green Chem. 2021, 23, 328–332. [Google Scholar] [CrossRef]
- Jia, Z.; Tang, C.; Ma, K.; Li, X. Boosted oxidative dehydrogenation of lactic acid into pyruvic acid on polyvinylpyrrolidone modified Fe2O3. Chem. Commun. 2023, 59, 9235–9238. [Google Scholar] [CrossRef] [PubMed]
- Huchede, M.; Morvan, D.; Bellière-Baca, V.; Millet, J.M.M. Screening of catalysts for the oxidative dehydrogenation of EL to EP, and optimization of the best catalysts. Appl. Catal. A-Gen. 2020, 601, 117619. [Google Scholar] [CrossRef]
- Huchede, M.; Morvan, D.; Vera, R.; Bellière-Baca, V.; Millet, J.M.M. Oxidative dehydrogenation of EL to EP over vanadium and iron antimonates catalysts. Appl. Catal. A-Gen. 2021, 617, 118016. [Google Scholar] [CrossRef]
- Lomate, S.; Bonnotte, T.; Paul, S.; Dumeignil, F.; Katryniok, B. Synthesis of pyruvic acid by vapour phase catalytic oxidative dehydrogenation of lactic acid. J. Mol. Catal. A Chem. 2013, 377, 123–128. [Google Scholar] [CrossRef]
- Sugiyama, S.; Shigemoto, N.; Masaoka, N.; Suetoh, S.; Kawami, H.; Miyaura, K.; Hayashi, H. Vapor-phase oxidation of EL to pyruvate over various oxide catalysts. Bull. Chem. Soc. Jpn. 1993, 66, 1542–1547. [Google Scholar] [CrossRef]
- Hayashi, H.; Shigemoto, N.; Sugiyama, S.; Masaoka, N.; Saitoh, K. X-ray photoelectron spectra for the oxidation state of TeO2-MoO3 catalyst in the vapor-phase selective oxidation of EL to pyruvate. Catal. Lett. 1993, 19, 273–277. [Google Scholar] [CrossRef]
- Hayashi, H. Additive telluromolybdates. Structure and catalysis in oxidation. Catal. Surv. Asia 1999, 3, 43–52. [Google Scholar] [CrossRef]
- Ai, M.; Ohdan, K. Effects of differences in the structures of iron phosphates on the catalytic action in the oxidative dehydrogenation of lactic acid to pyruvic acid. Appl. Catal. A-Gen. 1997, 165, 461–465. [Google Scholar] [CrossRef]
- Ai, M. Catalytic activity of iron phosphate doped with a small amount of molybdenum in the oxidative dehydrogenation of lactic acid to pyruvic acid. Appl. Catal. A-Gen. 2002, 234, 235–243. [Google Scholar] [CrossRef]
- Yin, C.; Li, X.; Chen, Z.; Zou, W.; Peng, Y.; Wei, S.; Tang, C.; Dong, L. Sustainable production of pyruvic acid: Oxidative dehydrogenation of lactic acid over the FeMoO/P catalyst. New J. Chem. 2020, 44, 5884–5894. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, C.; Xu, C.; Li, H.; Huang, H.; Song, L.; Li, X. Kinetics study for the oxidative dehydrogenation of EL to EP over MoVNbOx based catalysts. Chem. Eng. J. 2016, 296, 217–224. [Google Scholar] [CrossRef]
- Zhang, W.; Oulego, P.; Sharma, S.K.; Yang, X.-L.; Li, L.-J.; Rothenberg, G.; Shiju, N.R. Self-exfoliated synthesis of transition metal phosphate nanolayers for selective aerobic oxidation of EL to EP. ACS Catal. 2020, 10, 3958–3967. [Google Scholar] [CrossRef]
- Liu, X.; Wang, Q.; Liu, C.; Wang, X.; Peng, C.; Liu, R.; Yang, C. High-performance vapor-phase selective oxidation of EL to EP over SiO2 supported PMoVNb oxides. Catalysts 2020, 10, 197. [Google Scholar] [CrossRef]
- Lu, S.; Zhang, J.; Meng, H.; Qin, X.; Huang, J.; Liang, Y.; Xiao, F.S. Catalytic direct dehydrogenation of EL to produce EP over a synergetic Cu0/Cu+ interface. Appl. Catal. B Environ. 2023, 325, 122329. [Google Scholar] [CrossRef]
- Zhang, W.; Innocenti, G.; Ferbinteanu, M.; Ramos-Fernandez, E.V.; Sepulveda-Escribano, A.; Wu, H.; Cavani, F.; Rothenberg, G.; Shiju, N.R. Understanding the oxidative dehydrogenation of EL to EP over vanadia/titania. Catal. Sci. Technol. 2018, 8, 3737–3747. [Google Scholar] [CrossRef]
- Wang, X.; Shao, Y.; Pan, J.; Jiang, D.; Cong, Y.; Lv, S.-W. Exploring the green technique based on photocatalysis to produce hydrogen peroxide: Progress and challenge. Chem. Eng. J. 2024, 490, 151923. [Google Scholar] [CrossRef]
- Liu, Y.; Wei, B.; Yang, L.; Han, X.; Wang, Y.; Zhao, X. Recent research progress in hydrogen peroxide synthesized by electrocatalytic process with two-electron transfer: A brief review. J. Environ. Chem. Eng. 2024, 12, 112972. [Google Scholar] [CrossRef]
- Sofi, F.A.; Sharma, R.; Masoodi, M.H.; Tabassum, N. Recent developments in oxidative functionalization reactions promoted by TBAI/TBHP. Synth. Commun. 2021, 51, 3657–3693. [Google Scholar] [CrossRef]
- Varala, R.; Seema, V.; Amanullah, M.; Alam, M.M. Recent advances in TBHP-promoted heterocyclic ring construction via annulation/cyclization. J. Heterocycl. Chem. 2024, 61, 1269–1298. [Google Scholar] [CrossRef]
- Jyoti; Devi, S.; Manisha; Wadhwa, D.; Sindhu, J.; Kumar, V. TBHP: A sustainable alternative for carbon-oxygen bond formation. Eur. J. Org. Chem. 2024, 27, e202301030. [Google Scholar] [CrossRef]
- Varala, R.; Seema, V.; Hussein, M.; Alam, M.M.; Beda, D.P. Tert-butyl hydroperoxide (TBHP): Recent progress in C−H functionalization and heteroatom-heteroatom bond formations. ChemistrySelect 2024, 9, e202401937. [Google Scholar] [CrossRef]
- Chen, R.; Chen, J.; Zhang, J.; Wan, X. Combination of tetrabutylammonium iodide (TBAI) with tert-butyl hydroperoxide (TBHP): An efficient transition-metal-free system to construct various chemical bonds. Chem. Rec. 2018, 18, 1292–1305. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.-F.; Jiao, N. Oxygenation via C–H/C–C bond activation with molecular oxygen. Acc. Chem. Res. 2017, 50, 1640–1653. [Google Scholar] [CrossRef]
- Hone, C.A.; Kappe, C.O. The use of molecular oxygen for liquid phase aerobic oxidations in continuous flow. Top. Curr. Chem. 2019, 377, 2. [Google Scholar] [CrossRef]
- Javanainen, M.; Vattulainen, I.; Monticelli, L. On atomistic models for molecular oxygen. J. Phys. Chem. B 2017, 121, 518–528. [Google Scholar] [CrossRef]
- Zhou, L.; Liu, Y.; Shi, H.; Qing, Y.; Chen, C.; Shen, L.; Zhou, M.; Li, B.; Lin, H. Molecular oxygen activation: Innovative techniques for environmental remediation. Water Res. 2024, 250, 121075. [Google Scholar] [CrossRef]
- Wang, J.; Feng, X.; Sun, X.; Wang, J.; Shao, H. Impact of VAPO molecular sieve configuration on catalytic oxidation of EL to EP and its reaction mechanism. Polyhedron 2024, 259, 117046. [Google Scholar] [CrossRef]
- Doke, D.S.; Khomane, S.B.; Pandhare, S.L.; Dongare, M.K.; Dumeignil, F.; Umbarkar, S.B. Vanadium-based highly active and selective catalysts for oxidative dehydrogenation of EL to EP. Appl. Catal. A-Gen. 2019, 587, 117246. [Google Scholar] [CrossRef]
- Wang, D.; Liu, W.; Xie, Z.; Tian, S.; Su, D.; Qi, W. Oxidative dehydrogenation of EL over nanocarbon catalysts: Effect of oxygen functionalities and defects. Catal. Today 2020, 347, 96–101. [Google Scholar] [CrossRef]
- Ai, M. Catalytic activity of palladium-doped iron phosphate in the oxidative dehydrogenation of lactic acid to pyruvic acid. Appl. Catal. A-Gen. 2002, 232, 1–6. [Google Scholar] [CrossRef]
- Feng, Y.; Li, W.; Meng, M.; Yin, H.; Mi, J. Mesoporous Sn(IV) doping MCM-41 supported Pd nanoparticles for enhanced selective catalytic oxidation of 1,2-propanediol to pyruvic acid. Appl. Catal. B Environ. 2019, 253, 111–120. [Google Scholar] [CrossRef]
- Chu, Y.; Yan, B.; Yang, X.; Wang, S.; Cao, P.; Li, T.; Yu, H.; Yin, H. Selective oxidation of mEL over carbon-supported noble metal catalysts under base-free conditions. Ind. Eng. Chem. Res. 2021, 60, 14397–14409. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, T.; Ding, Y. Oxidative dehydrogenation of lactic acid to pyruvic acid over Pb-Pt bimetallic supported on carbon materials. Appl. Catal. A-Gen. 2017, 533, 59–65. [Google Scholar] [CrossRef]
- Ramos-Fernandez, E.V.; Geels, N.J.; Shiju, N.R.; Rothenberg, G. Titania-catalysed oxidative dehydrogenation of EL: Effective yet selective free-radical oxidation. Green Chem. 2014, 16, 3358–3363. [Google Scholar] [CrossRef]
- Zhang, W.; Innocenti, G.; Oulego, P.; Gitis, V.; Wu, H.; Ensing, B.; Cavani, F.; Rothenberg, G.; Shiju, N.R. Highly selective oxidation of EL to EP catalyzed by mesoporous vanadia–titania. ACS Catal. 2018, 8, 2365–2374. [Google Scholar] [CrossRef]
- Liu, K.; Huang, X.; Pidko, E.A.; Hensen, E.J.M. MoO3–TiO2 Synergy in oxidative dehydrogenation of lactic acid to pyruvic acid. Green Chem. 2017, 19, 3014–3022. [Google Scholar] [CrossRef]
- Liu, Z.; Xu, J.; Cai, C.; Cai, J.; Dai, Q.; Du, M.; Wen, Z.; Yan, B.; Xue, B. Carbon nitride doped VxOy/SBA-15 as highly efficient catalysts for the oxidation of EL to EP. Appl. Catal. A-Gen. 2024, 671, 119577. [Google Scholar] [CrossRef]
- Liu, Z.; Xu, J.; Wen, Z.; Yan, B.; Xue, B. Chitin-derived NC support vanadia as highly efficient catalyst for the oxidation of EL to EP. Biomass Bioenergy 2024, 190, 107396. [Google Scholar] [CrossRef]
- Zhang, W.; Ensing, B.; Rothenberg, G.; Shiju, N.R. Designing effective solid catalysts for biomass conversion: Aerobic oxidation of EL to EP. Green Chem. 2018, 20, 1866–1873. [Google Scholar] [CrossRef]
- Wu, J.; Hua, W.; Yue, Y.; Gao, Z. Efficient aerobic oxidation of EL to EP over V2O5/g-C3N4 catalysts. ACS Omega 2020, 5, 16200–16207. [Google Scholar] [CrossRef]
- Hariharan, A.; Tamizhdurai, P.; Kavitha, C.; Yadav, K.K.; Kumar, R.S.; Sugumaran, S.; Subramani, A.; Sasikumar, P.; Vimalan, M.; Albakri, G.S.; et al. Zn/Ni catalyst facilitates the oxidation of EL to EP by selective progressions on mesoporous SBA-15 support by a continuous processing approach. Mol. Catal. 2024, 564, 114275. [Google Scholar] [CrossRef]
- Ju, Y.; Du, Z.; Xiao, C.; Li, X.; Li, S. Novel effect of zinc nitrate/vanadyl oxalate for selective catalytic oxidation of α-hydroxy esters to α-keto esters with molecular oxygen: An in situ ATR-IR study. Molecules 2019, 24, 1281. [Google Scholar] [CrossRef]
- Zhang, C.; Luo, J.; Xie, B.; Liu, W.; Zhang, J. Continuous Cu/Keto-ABNO catalyzed aerobic oxidation of EL to EP in a micro-packed bed reactor: Process optimization and reaction kinetics. Chem. Eng. Sci. 2024, 292, 119985. [Google Scholar] [CrossRef]
- Yasukawa, T.; Ninomiya, W.; Ooyachi, K.; Aoki, N.; Mae, K. Efficient oxidative dehydrogenation of lactate to pyruvate using a gas-liquid micro flow system. Ind. Eng. Chem. Res. 2011, 50, 3858–3863. [Google Scholar] [CrossRef]
- Madhanagopal, G.; Premalatha, K.; Poovizhi, P.N.; Sumithra, V.; Mahalingam, S.; Guganathan, L.; Sivakumar, S.; Subramani, A.; Tamizhdurai, P. Effect of a bimetal Mn/Zn catalyst supported on activated carbon for selective oxidation of EL to EP. Carbon Trends 2025, 19, 100472. [Google Scholar] [CrossRef]
- Wang, L.; Bie, C.; Yu, J. Challenges of z-scheme photocatalytic mechanisms. Trends Chem. 2022, 4, 973–983. [Google Scholar] [CrossRef]
- Ramakrishnan, S.B.; Mohammadparast, F.; Dadgar, A.P.; Mou, T.; Le, T.; Wang, B.; Jain, P.K.; Andiappan, M. Photoinduced electron and energy transfer pathways and photocatalytic mechanisms in hybrid plasmonic photocatalysis. Adv. Opt. Mater. 2021, 9, 2101128. [Google Scholar] [CrossRef]
- Swords, W.B.; Chapman, S.J.; Hofstetter, H.; Dunn, A.L.; Yoon, T.P. Variable temperature LED–NMR: Rapid insights into a photocatalytic mechanism from reaction progress kinetic analysis. J. Org. Chem. 2022, 87, 11776–11782. [Google Scholar] [CrossRef]
- Yu, Q.; Zhang, Y.; Wan, J.-P. Ambient and aerobic carbon–carbon bond cleavage toward α-ketoester synthesis by transition-metal-free photocatalysis. Green Chem. 2019, 21, 3436–3441. [Google Scholar] [CrossRef]
- Miao, Y.; Zhao, Y.; Gao, J.; Wang, J.; Zhang, T. Direct photoreforming of real-world polylactic acid plastics into highly selective value-added pyruvic acid under visible light. J. Am. Chem. Soc. 2024, 146, 4842–4850. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Liu, J.; Luo, Y.; Liao, Z.; Li, B.; Xiang, X.; Fang, Y. Bifunctional CdS-MoO2 catalysts for selective oxidation of lactic acid coupled with photocatalytic H2 production. J. Colloid Interface Sci. 2024, 675, 836–847. [Google Scholar] [CrossRef]
- Liang, E.; Liu, X.; Yang, J.F.; Qi, Y.B.; Teng, R.; Cui, W.B.; Xu, M.C.; Luo, S.; Ma, C.H.; Liu, S.X.; et al. 1T/2H-MoS2/CdS heterojunction for the Co-production of lactic acid photoreforming to hydrogen and value-added chemicals. J. Environ. Chem. Eng. 2024, 12, 114287. [Google Scholar] [CrossRef]
- Lu, T.; Yang, Z.; Li, H.; Chen, H.; Xu, J.; Xu, C.C.; Wang, J.; Li, Z.; Zhang, Y. Selective oxidation of EL to EP by a photocatalytic strategy under room temperature. J. Catal. 2022, 410, 103–111. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, Z.; Yang, D.; Wang, L.; Yang, X.; Tang, K.; Yang, H.; Zhan, X.; Wang, Z.; Yang, W. Crystal-interface-mediated self-assembly of ZnIn2S4/CdS S-scheme heterojunctions toward efficient photocatalytic hydrogen production. Carbon Energy 2025, e707. [Google Scholar] [CrossRef]
- Zeng, Z.; Mao, L.; Zhang, R.; Liu, Y.; Ling, Y.; Cai, X.; Zhang, J. A Triple-benefit strategy of microstructure, electronic property and active site modulation on ZnIn2S4 photocatalyst by Sn atom doping. Chem. Eng. J. 2024, 496, 153769. [Google Scholar] [CrossRef]
- Li, W.-Y.; Liu, J.; Li, Z.-R.; Shen, T.-T.; Chen, J.; Hu, Z.-Y.; Yu, W.-B.; Li, Y.; Su, B.-L. Probing the deactivation and regeneration of ZnIn2S4 in photocatalytic degradation of tetracycline. J. Colloid Interface Sci. 2025, 694, 137700. [Google Scholar] [CrossRef]
- Roostaei, T.; Zhao, H.; Eisapour, M.; Zhao, J.; Omidkar, A.; Chen, Z.; Hu, J. Hollow NiP–ZnIn2S4 heterojunction for simultaneous hydrogen and pyruvic acid production from lactic acid photoreforming. Energy Fuels 2023, 37, 16915–16923. [Google Scholar] [CrossRef]
- Roostaei, T.; Rahimpour, M.R.; Eisapour, M.; Zhao, J.; Zhao, H.; Chen, Z.; Hu, J. Coproduction of hydrogen and pyruvic acid on 0D-2D CoP–ZnIn2S4 from lactic acid photoreforming. Mater. Today Chem. 2024, 35, 101882. [Google Scholar] [CrossRef]
- Ma, B.; Li, D.; Wang, X.; Lin, K. Molybdenum-based Co-catalysts in photocatalytic hydrogen production: Categories, structures, and roles. ChemSusChem 2018, 11, 3871–3881. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Li, L.; Liu, N.; Wang, D.; Xue, Y.; Tang, L. Molybdenum disulfide (MoS2) as a Co-catalyst for photocatalytic degradation of organic contaminants: A review. Process Saf. Environ. 2018, 118, 40–58. [Google Scholar] [CrossRef]
- Wu, C.; Zhang, J.; Tong, X.; Yu, P.; Xu, J.-Y.; Wu, J.; Wang, Z.M.; Lou, J.; Chueh, Y.-L. A critical review on enhancement of photocatalytic hydrogen production by molybdenum disulfide: From growth to interfacial activities. Small 2019, 15, 1900578. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Xu, R.; Chen, D.; Lu, T.; Li, H.; Wang, M. Subsurface Mo vacancy in bismuth molybdate promotes photocatalytic oxidation of lactate to pyruvate. ACS Catal. 2024, 14, 1977–1986. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).