Abstract
M-edge X-ray absorption spectroscopy (XAS), which probes 3p→3d transitions in first-row transition metals, provides detailed insights into oxidation states, spin-states, and local electronic structure with high element and orbital specificity. Operating in the extreme ultraviolet (XUV) region, this technique provides sharp multiplet-resolved features with high sensitivity to ligand field and covalency effects. Compared to K- and L-edge XAS, M-edge spectra exhibit significantly narrower full widths at half maximum (typically 0.3–0.5 eV versus >1 eV at the L-edge and >1.5–2 eV at the K-edge), owing to longer 3p core-hole lifetimes. M-edge measurements are also more surface-sensitive due to the lower photon energy range, making them particularly well-suited for probing thin films, interfaces, and surface-bound species. The advent of tabletop high-harmonic generation (HHG) sources has enabled femtosecond time-resolved M-edge measurements, allowing direct observation of ultrafast photoinduced processes such as charge transfer and spin crossover dynamics. This review presents an overview of the fundamental principles, experimental advances, and current theoretical approaches for interpreting M-edge spectra. We further discuss a range of applications in catalysis, materials science, and coordination chemistry, highlighting the technique’s growing impact and potential for future studies.
1. Introduction
X-ray absorption spectroscopy (XAS) is a widely used and element-specific technique for probing the coordination environment and electronic structure of materials [1,2]. X-ray absorption spectra consist mainly of two regions: the X-ray absorption near-edge structure (XANES), which reflects transitions of core-level electrons to unoccupied bound states, and the extended X-ray absorption fine structure (EXAFS), which is dominated by transitions to continuum states [3]. XANES offers critical insights into the oxidation states, coordination environment, and local symmetry surrounding metal centers in metalloproteins [4], catalytic systems [5,6,7], semiconductors [8], etc. X-ray absorption edges are further categorized based on the principal quantum number of the core electron that is excited. For nd transition metals ( n = 3, 4, 5), the K-edge corresponds to transitions from the 1s core orbital to unoccupied (n + 1)p orbitals—for example, 1s→4p for 3d metals. In many transition metal complexes, a pre-edge feature is also observed just below the main K-edge. This pre-edge arises from the transition of an electron from 1s to valence nd orbital, which are formally electric-dipole forbidden but can gain intensity through nd–(n + 1)p orbital mixing in non-centrosymmetric environments. K-edges fall in the hard X-ray region and are extensively used due to their deep penetration and compatibility with in situ and operando measurements. Transitions from the 2p core levels to valence nd orbitals define the L-edges—specifically, the L2 (2p1/2) and L3 (2p3/2) edges. These are located in the soft X-ray region and are particularly important for transition metals, as they directly probe 2p→ nd transitions with high sensitivity to the electronic structure and spin–orbit effects. Finally, transitions from the 3p core electrons to valence nd orbitals, define the M edges, such as the M2,3-edge (3p1/2 and 3p3/2), which appear in the vacuum ultraviolet to soft X-ray region [9].
M-edge X-ray absorption spectroscopy is uniquely suited for analyzing surface-sensitive phenomena and chemically sensitive 3d orbitals [10]. Operating in the extreme ultraviolet (XUV) range ( 30–80 eV), M-edge XAS captures fine multiplet features associated with spin–orbit and ligand field interactions, offering insights into oxidation states, spin configurations, and coordination number of the metal center [11,12,13]. This makes M-edge XAS particularly valuable for catalytic systems, where tracking changes in the oxidation state, spin, and coordination environment under reaction conditions is essential for understanding active site dynamics. Compared to the more commonly employed L-edge (2p → nd) spectroscopy, the M-edge boasts shorter core-hole lifetimes, which results in sharper spectral features [14]. Additionally, the lower photon energy means that M-edge spectroscopy is particularly sensitive to the top few nanometers of a sample, making it ideal for studying thin films, surfaces, and molecular adsorbates. A key advantage of this technique is that M-edge spectra can be acquired not only at synchrotron facilities but also using more accessible XUV spectrometers that are suitable for standard laboratory setups [13]. Recent advances in high harmonic generation (HHG) now enable time-resolved M-edge spectroscopy, expanding the scope to ultrafast chemical dynamics and magnetism [15,16,17,18,19]. In parallel, computational methods from ligand field theory to ab initio simulations have improved the accuracy of spectral interpretation, cementing M-edge spectroscopy as a crucial tool for transition metal research [20,21,22,23,24]. In addition to this, to access M-edge transitions under conditions where X-ray absorption is limited, complementary spectroscopic techniques such as X-ray Raman Scattering (XRS), Resonant Inelastic X-ray Scattering (RIXS), and Electron Energy Loss Spectroscopy (EELS), etc., provide critical alternatives and extensions. XRS is a powerful technique for accessing soft X-ray absorption edge information comparable to that obtained from soft XAS but with significantly greater bulk sensitivity [25,26]. This enables in situ studies under extreme conditions such as high pressure and high temperature. Consequently, XRS provides a valuable means of probing 3d-state occupancy through M-edge spectroscopy in environments inaccessible to traditional soft X-ray methods. Additionally, RIXS, also known as resonant X-ray Raman Spectroscopy, enables element- and excitation-specific probing of occupied 3d states with greater selectivity than XAS [27]. While XAS reveals the unoccupied density of states, RIXS complements it by providing access to the occupied electronic structure [28]. EELS offers similar core-level sensitivity using high-energy electron beams, making it especially valuable in transmission electron microscopy (TEM) for nanoscale spatial resolution. Like RIXS and XAS, EELS can probe both unoccupied and low-energy excited states, providing complementary electronic structure information with high spatial precision [29].
2. Fundamental Principles
For first row 3d transition metals, M-edge absorption arises from the transition of an electron from 3p core orbitals to 3d valence orbitals. This feature is collectively referred to as the M2,3-edge. These transitions are electric dipole allowed and strongly influenced by multiplet effects due to strong 3p–3d Coulomb and exchange interactions. The spin–orbit coupling in the 3p core shell splits the M-edge into M2 (3p1/2) and M3 (3p3/2) components. The resultant XAS spectrum is rich with fine structure due to crystal field splitting, exchange coupling, and metal–ligand covalency. Because the 3p orbitals have less spatial extension than 2p orbitals, the 3p–3d overlap is weaker, yet the final-state multiplet interactions are still significant. This makes M-edge spectroscopy particularly sensitive to changes in electronic configuration, spin state, and symmetry.
3. Experimental Techniques
The advancement of tabletop extreme ultraviolet (XUV) and soft X-ray light sources has enabled researchers to perform core-level spectroscopic measurements with ultrafast temporal resolution. These next-generation light sources, based on HHG and other nonlinear optical techniques such as optical parametric chirped-pulse amplifiers (OPCPA) [30], can produce light pulses with durations ranging from femtosecond (10−15 seconds) to attosecond (10−18 seconds) regime [31,32]. Recent advances in tabletop HHG sources have enabled the generation of XUV pulses with durations as short as 20–50 femtoseconds, dramatically enhancing the temporal resolution of M-edge spectroscopy compared to earlier synchrotron-based methods. This ultrafast capability is crucial for capturing transient electronic and spin dynamics in transition-metal systems with element and orbital specificity. In addition to temporal resolution, the stability of HHG sources plays a crucial role in M-edge spectroscopy. Modern systems typically maintain spectral drift within ±0.1 eV and energy fluctuations below 5% over several hours, which is sufficient to ensure reliable energy referencing and consistent spectral features in both steady-state and time-resolved measurements [33]. In particular, M-edge transitions of 3d transition metals, which involve 3p → 3d excitations, fall within the energy range of 30–80 eV and are well-matched to XUV photon energies generated by HHG [13,15,34,35]. Beyond these XUV energies, significant recent progress has been made in tabletop soft X-ray sources extending into the water window spectral region (200–600 eV). Time-resolved measurements utilizing such water window soft X-rays from laser-driven HHG sources have already been demonstrated highlighting their capability for femtosecond-resolved spectroscopies on a laboratory scale [36]. These higher energies can be attained by high-energy OPCPA instruments employing HHG or metal plasma techniques [37]. HHG using few millijoule Ti:sapphire lasers are routinely used to produce XUV photons in the 50–100 eV range [38]. In addition to HHG-based sources, free-electron lasers (FELs) provide an alternative route to ultrafast M-edge spectroscopy, offering high brightness and tunability over a broad energy range [39]. This variety of light sources has made it possible to track ultrafast changes in local electronic structure, spin state, and oxidation state with element and orbital specificity. Complementary to these ultrafast soft X-ray sources, XRS relies on high-brightness hard X-ray synchrotron beams and crystal analyzer spectrometers, enabling bulk-sensitive measurements under ambient and extreme conditions without vacuum requirements [40]. RIXS employs tunable synchrotron or free-electron laser sources combined with high-resolution photon analyzers to achieve meV-scale energy resolution [41]. EELS uses transmission electron microscopes equipped with electron guns and spectrometers, offering nanoscale spatial resolution in laboratory settings [42].
4. Theoretical Modeling
The rising utilization of soft X-ray and XUV techniques has created a growing need for accurate simulation of M-edge XANES using theoretical methods. Due to strong p-d interactions leading to multiplet effects, single-particle models are insufficient for accurate modeling of M-edge spectra [43]. Standard time-dependent density functional theory (TDDFT) cannot capture spin–orbit coupling and electron–electron Coulombic correlation. A semi-empirical charge transfer multiplet (CTM) theory emerges as a traditional approach to dealing with multiplet effects [8]. It builds upon traditional atomic multiplet theory [44] by incorporating ligand field effects and metal–ligand charge transfer interactions, enabling it to describe both the localized and covalent aspect of transition metal–ligand bonding. As a semi-empirical model, CTM theory relies on parameters fitted to experimental spectra or estimated from complementary methods (e.g., DFT or Hartree–Fock), rather than solving the full many-body Schrödinger equation ab initio. Despite this, it provides a computationally efficient and physically insightful way to reproduce and interpret the complex multiplet structures and covalency effects observed in XAS, Resonant Inelastic X-ray Scattering (RIXS), and Electron Energy Loss Spectroscopy (EELS), especially for first-row transition metals where these effects are most pronounced [8,43]. The theory is implemented in CTM4XAS program, which allows users to explore how changes in symmetry, covalency, and spin state affect spectral shapes, aiding in the interpretation of experimental data [20]. The software has been further extended to simulate the M2,3-edge spectra of the transition metal complexes by Veis et al. The M2,3-edge spectra of the ground-state of neutral ferrocene simulated using CTM4XAS is shown in Figure 1a and is observed to be in good agreement with the experiments [45].

Figure 1.
(a) Comparison of M2,3-edge absorption spectra of ground-state of neutral ferrocene from experiments and CTM simulations. (b) Computed 4c-DR-TDDFT and experimental XAS spectra of MoS4 at the Mo M4,5-edge. (c) Comparison of XAS spectra of MoS4 from 1eX2C, amfX2C, and 4c Hamiltonian. (d) M-edge spectra for three different iron clusters, [FeS12], [Fe6S38], and [Fe19S88], obtained from DFT/ROCIS and compared with experimental data. (e) Experimental and computed M2,3-edge of first row transition metal oxides. The solid and dashed lines represent the experimental and computed data, respectively. The different parts of the figure are reproduced with permissions from References [21,22,23,24,45] and are combined in a single figure.
However, being a semi-empirical method, CTM relies heavily on user-defined parameters limiting its predictive power, especially for systems with unknown electronic structure. The four-component damped response time-dependent DFT (4c-DR-TDDFT) approach developed by Konečný et al. offers a variational and nonperturbative treatment of both scalar and spin–orbit (SO) relativistic effects within the Dirac–Coulomb framework [21]. Unlike conventional linear-response TDDFT, which often relies on perturbative or approximate SO treatments and suffers from limitations in targeting high-energy core excitations, the 4c-DR-TDDFT method calculates the spectrum directly in the frequency domain with controllable resolution and spectral broadening. The molybdenum M4,5-edges of MoS42− calculated from 4c-DR-TDDFT shows good agreement with the experimental spectra as shown in Figure 1b. Although four-component Dirac–Coulomb damped response TDDFT (4c-DR-TDDFT) is considered the gold standard, it remains computationally demanding, especially for large systems containing heavy elements. The relativistic two-component approaches based on an (extended) atomic mean-field exact two-component Hamiltonian framework (amfX2C and eamfX2C) are proposed by Konečný et al. [22]. It offers a significant advance by reproducing all key spectral features—such as shape, position, and spin–orbit splitting—of the 4c reference at a fraction of the computational cost as shown in Figure 1c. It shows that although 1eX2C Hamiltonian overestimates spin–orbit splitting, amfX2C reproduces the results well in comparison to 4c. The restricted open-shell configuration interaction singles (ROCIS/DFT) and pair-natural orbital (PNO)-ROCIS/DFT methods within an embedded cluster framework to model M- and L-edge X-ray absorption spectra of solid-state transition metal oxides has been developed by Kubas et al. [23]. The approach allows for efficient and accurate calculation of core-level spectra in large clusters such as [FeS12]10−, [Fe6S38]26− and [Fe19S88]48− (Figure 1d), capturing key features such as oxidation state, spin state, and local coordination environment. Klein et al. introduced an ab initio Bethe–Salpeter equation (BSE) approach for modeling XUV absorption spectra in solid-state transition metal oxides including TiO2, Cr2O3, MnO2, Fe2O3, CO3O4, NiO, CuO, and ZnO (Figure 1e), enabling accurate prediction of both ground and excited-state M2,3-edge features. The method captures key many-body effects, such as core–hole screening and angular momentum coupling, providing physical insight into spectral changes following photoexcitation in solids [24]. However, DFT methods often suffer from functional dependence, and both DFT and BSE approaches are inherently single-reference in nature. As a result, they struggle to accurately capture multiplet structures or states arising from near-degeneracies that are common in strongly correlated systems.
5. Applications in Transition Metal Chemistry
Recent advances in M-edge X-ray absorption and transient absorption spectroscopy have enabled direct probing of ultrafast electronic dynamics, oxidation states, and spin-state transitions in transition-metal systems with unprecedented temporal and element specificity. In addition, X-ray Raman scattering (XRS) has emerged as a complementary technique capable of accessing M-edge transitions under conditions not accessible to conventional soft X-ray absorption, such as in high-pressure cells or bulk samples. Broadly, M-edge spectroscopy applications can be categorized into three classes: (1) steady-state studies, focused on static oxidation states; (2) ultrafast time-resolved investigations, that track intermediates with femtosecond resolution; and (3) complementary techniques like, X-ray Raman scattering (XRS), Resonance Inelastic X-ray Scattering (RIXS), and Electron Energy-Loss Spectroscopy (EELS).
5.1. Steady-State Applications of M-Edge Spectroscopy
In steady-state M-edge X-ray absorption spectroscopy, measurements are performed under static conditions to investigate the ground-state electronic structure of transition metal centers. These studies provide detailed information on oxidation states, spin states, and ligand field environments, and are particularly valuable for understanding the geometric and electronic properties of materials and complexes at equilibrium. Aken et al. demonstrated the use of Fe M2,3-edge spectroscopy to quantitatively determine the Fe3+/∑Fe ratio in oxide and silicate minerals with high spatial resolution [12]. They established a robust correlation between the pre-edge/main peak intensity ratio and the oxidation state of iron, enabling valence-specific imaging down to the nanometer scale. The electronic structures of molecular transition metal complexes of Fe and Co are effectively probed by Zhang and co-workers revealing distinct spectral signatures sensitive to oxidation state, spin state, and ligand field [11]. Wang et al. studied M2,3-edge XAS of various complexes of Co, Ni, and Cu and revealed different chemical shifts reflecting different electronic structure and charge compensation. Moreover, spectral multiplet features across Ni halide series become less resolved with increasing covalency which is attributed to delocalization of p orbitals and broader Ni 3d states due to increased p-d mixing [46].
5.2. Ultrafast Time-Resolved M-Edge Spectroscopy
Ultrafast M-Edge spectroscopy captures real-time evolution of excited-state electronic and spin configurations in transition metal systems. They enable the identification of short-lived intermediates and dynamic pathways that are inaccessible through steady-state techniques. Vura-Weis et al. used femtosecond M2,3-edge transient absorption spectroscopy to probe photoinduced charge-transfer dynamics in -Fe2O3 thin films [13].
As shown in Figure 2, panel A presents the simulated XUV absorption spectra of the Fe3+ ground state, d–d excited state, and Fe2+ ligand-to-metal charge transfer (LMCT) state, while panel B displays their corresponding difference spectra. The simulated LMCT difference spectrum closely matches the experimental spectrum in shape and spacing, while the d–d spectrum differs significantly, particularly showing a 60–70 eV feature absent in the experiment. This reveals LMCT from O 2p to Fe 3d orbitals, leading to the formation of a transient Fe2+ state distinguishable from d–d excitations via oxidation-state-specific spectral signatures. This Fe2+ state decayed to a long-lived trap state within 240 fs, demonstrating the technique’s sensitivity to ultrafast charge-transfer dynamics. The study of charge-transfer processes offers a promising avenue for investigating photocatalytic materials, where tabletop M-edge spectroscopy enables real-time observation of electron flow and intermediate states at 3d metal centers. The femtosecond M2,3-edge XUV transient absorption spectroscopy to directly resolve the photoinduced charge transfer and hot carrier relaxation dynamics in spinel Co3O4 was done by Jiang and co-workers [34]. A ligand-to-metal charge transfer (LMCT) from O2−(2p) to Co3+(3d eg), forming a transient state that decays rapidly via Auger relaxation and hot carrier relaxation to the band edge was observed. The study was further extended to the study of charge-carrier relaxation dynamics of Co3O4-methanol interface by Baker et al. [35]. By probing the Co M2,3-edge (∼60–70 eV), the study achieved oxidation-state-specific tracking of charge carriers, enabling direct observation of hole transfer from Co3O4 to methanol and the associated surface-mediated recombination pathway. The ligand-to-metal charge transfer was observed for the initial states in both the vacuum and methanol as shown in Figure 3.
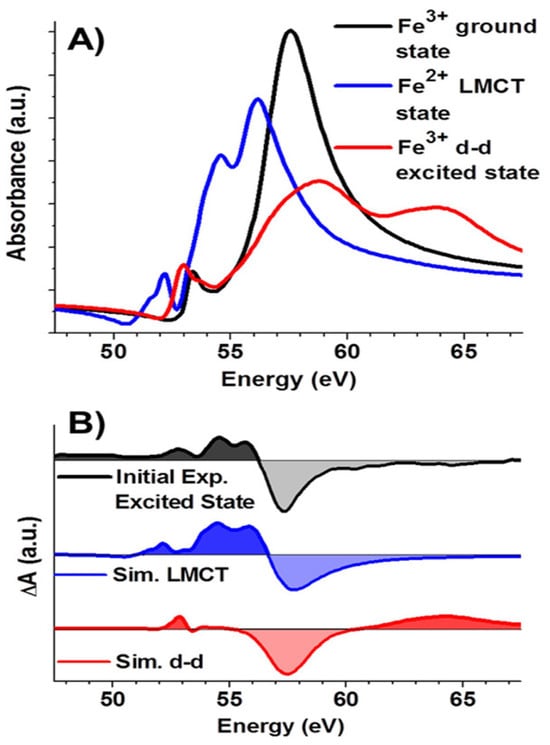
Figure 2.
(A) Simulated XUV absorption spectra of Fe3+ in ground state, d-d excited state, and Fe2+ LMCT state (B) Comparison of simulated excited-state minus ground-state difference spectra with the experimental initial excited-state difference spectrum. The figure is reproduced with permission from Reference [13].

Figure 3.
Experimental transient spectra for Co3O4 initial states in vacuum and methanol and comparison to the simulated difference spectra showing LMCT transition from O2−→Co3+. The figure is reproduced with permission from Reference [35].
By probing carrier relaxation at catalytic interfaces, this technique provides mechanistic insight into how surface states modulate the kinetics of charge injection and recombination, offering valuable guidance for the rational design of more effective photocatalysts. The femtosecond M-edge spectroscopy to directly track ultrafast photoinduced valence tautomerism in a cobalt–dioxolene complex is studied by Ash et al. [16]. Vura-Weis and co-workers tracked the ultrafast dynamics of [Fe(phen)3]2+ complex employing tabletop femtosecond M-edge XANES, enabling the direct observation of a short-lived metal-centered triplet intermediate during spin-crossover [17].
Ryland et al. carried out femtosecond M-edge XANES studies of FeTPPCl and show that 400 nm photoexcitation generates an Fe(II) ligand-to-metal charge transfer (LMCT) state within 70 fs, which decays to a vibrationally hot ground state in 1.13 ps, bypassing any detectable metal-centered (d,d) states as shown in Figure 4 [47]. This rapid deactivation pathway limits the time window for engaging the metal center in photochemical transformations, suggesting that efficient catalytic activity—such as axial ligand binding, electron transfer, or substrate activation—must occur within the first picosecond. These insights underscore the importance of ultrafast metal-centered probes in evaluating the photochemical viability of metalloporphyrins in catalytic applications.
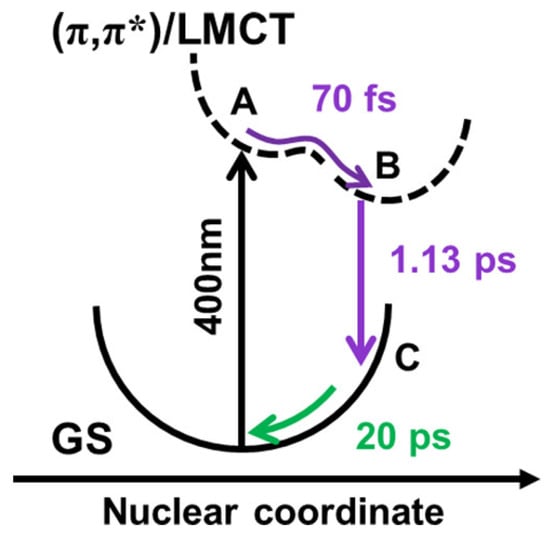
Figure 4.
Schematic of potential energy surface of FeTPPCl. Species A and B represent local and global minima on a single excited-state potential energy surface. Back-electron transfer leads to the formation of species C, a vibrationally excited ground-state structure that is dark to XUV excitation. The figure is reproduced with permission from Reference [47].
Similarly, ultrafast M-edge XANES spectroscopy reveals that photoexcitation of nickel(II) octaethylporphyrin (NiOEP) leads to sub-100 fs intersystem crossing, directly forming a vibrationally hot metal-centered triplet (d,d) state without detectable charge-transfer intermediates, As shown in Figure 5, CTM simulations confirm that the transient spectral features match triplet states, underscoring the ultrafast spin-state switching in first-row transition-metal complexes [18]. Time-resolved M2,3-edge XANES spectroscopy enabled direct observation of ultrafast spin-state dynamics in Co4O4 cubane, a model for water oxidation catalysts. The study demonstrated femtosecond intersystem crossing to a metal-centered quintet and its subsequent bifurcation into ballistic back-ISC or relaxation to a long-lived triplet [48]. The photodynamics of the -oxo iron porphyrins [(TPPFe)2O], photocatalyst for oxidative organic transformations, is investigated using M-edge XANES, revealing the formation of a ligand-centered Fe(III) ion pair as the origin of low quantum yield. This highlights the utility of M-edge XANES in elucidating reaction pathways and identifying loss mechanisms in photocatalytic systems [49].

Figure 5.
(A) Ligand field multiplet simulations of singlet and triplet (d,d) excited-state spectra. (B) Simulated difference spectra (excited minus ground) for each excited state. (C) Electronic configurations of the potential excited states. The figure is reproduced with permission from Reference [18].
5.3. Complementary Techniques
XRS, RIXS, and EELS are complementary techniques that provide bulk-sensitive and high-resolution insights into the electronic structure and low-energy excitations of 3d transition metal systems under diverse conditions. Nyrow et al. demonstrated the application of Fe M2,3-edge X-ray Raman scattering to investigate the pressure-induced spin transition from a high-spin to a low-spin state in FeS over a range of pressures [50]. Comparison with atomic multiplet simulations using CTM4XAS enabled the quantification of crystal field splitting parameters, demonstrating the method’s ability to probe spin-state transitions under high-pressure conditions. M2,3-edge RIXS measurements have been conducted on NiO, NiF2, and two additional covalent complexes and revealed distinct d-d transition patterns providing element-specific insights into their occupied 3d electronic structures and ligand field environments [46].
Together, these studies underscore the unique power of M-edge spectroscopy, particularly when combined with tabletop XUV sources, for investigating metal-centered dynamics, oxidation-state changes, and interfacial charge transfer processes across a broad range of materials and molecular systems. Its ability to directly probe 3d valence orbitals with high spectral resolution and ultrafast time sensitivity makes M-edge spectroscopy irreplaceable by conventional techniques such as K-edge XAS or optical methods, especially for capturing transient and surface-specific electronic phenomena.
6. Conclusions and Outlook
M-edge X-ray absorption spectroscopy has emerged as a powerful technique for probing the electronic structure, spin state, and oxidation state of first-row transition metal complexes with high sensitivity and element specificity. The advent of tabletop femtosecond XUV sources, particularly those based on high-harmonic generation (HHG), has enabled time-resolved M-edge measurements. These capabilities are now being extended to in situ and operando conditions, broadening the technique’s relevance to catalysis, energy materials, and environmental systems. However, significant technical challenges remain, including the stability of HHG sources under extreme temperature and pressure, signal attenuation due to limited photon penetration depth of low-energy XUV photons in complex reaction environments. Overcoming these obstacles through advances in source robustness, experimental design, and complementary techniques is essential to fully harness M-edge spectroscopy for real-world catalytic and materials applications. Despite this, M-edge XAS offers a powerful means to investigate redox non-innocence in ligand frameworks, which plays a central role in modulating the activity and selectivity of transition-metal-based catalysts. By directly probing the metal 3d orbitals with oxidation- and spin-state sensitivity, M-edge spectroscopy can disentangle metal- versus ligand-centered redox processes. Moreover, spin-crossover often critically influences catalytic pathways by altering the reactivity and selectivity of metal centers. The ability of M-edge X-ray absorption spectroscopy to sensitively probe spin crossover and spin-state transitions holds great promise for advancing our understanding of catalytic mechanisms. Direct observation of reactive intermediates in photoredox catalysis remains a key challenge for unraveling mechanistic pathways. M-edge X-ray absorption spectroscopy, with its element- and oxidation-state specificity and ultrafast time resolution, offers a powerful approach to capture transient species and electronic changes during photocatalytic cycles. By enabling real-time tracking of metal center oxidation and spin states, M-edge measurements can illuminate how light-induced electron transfer drives catalytic transformations. Another exciting future direction involves applying M-edge spectroscopy to multi-component catalyst systems, where its element-specific sensitivity may enable direct differentiation between coexisting metal centers such as Fe and Co.
On the computational side, in most of the previous reports, M-edge spectra have been interpreted using semi-empirical charge transfer multiplet (CTM) models or, more recently, relativistic TDDFT approaches. The two-component multireference restricted active space configuration interaction (X2C-MRRASCI) method developed by Jenkins et al. has proven effective in capturing spin–orbit coupling and multiconfigurational effects for L-edge XAS of iron complexes [51]. Its extension to the M2,3-edge region holds strong potential for accurately simulating 3d transition-metal complexes, particularly in cases involving spin-crossover, redox-active ligands, or time-resolved spectroscopy. Similarly, relativistic multireference wavefunction calculations with the restricted active space framework incorporating spin–orbit coupling (RAS-SO) have been employed by Sergentu et al. to analyze metal–ligand covalency in actinide complexes using M-edge XANES [52]. This methodology provides a robust theoretical foundation for interpreting complex spectral features and could be effectively extended to 3d transition metal systems to enhance the simulation and understanding of their M-edge spectra. The recent development of exact-two-component multiconfiguration pair-density functional theory (X2C-MC-PDFT) provides an efficient and accurate approach for treating systems with strong relativistic effects and both static and dynamic electron correlation [53]. By variationally including scalar relativity and spin–orbit coupling within the active space, X2C-MC-PDFT significantly improves predictions of ground- and excited-state fine-structure splittings compared to traditional methods, while remaining computationally practical. This method’s ability to capture state-specific correlation and relativistic effects makes it a promising tool for interpreting X-ray spectroscopies and electronic structure in transition metal and actinide complexes, and its extension to noncollinear functionals could further enhance its accuracy for systems with strong spin–orbit coupling. Likewise, machine learning tools [54] trained on theoretical and experimental spectra could assist in automated interpretation and inverse design of transition metal complexes but have not been widely applied to M-edge data. Exploring these directions could significantly enhance the accuracy and scalability of M-edge spectral analysis, especially for complex excited states and dynamic systems.
Author Contributions
R.K.—conceptualization, writing original draft, review, and editing. C.L.—conceptualization, funding acquisition, supervision, review, and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This paper is supported by United States Department of Energy (DOE), Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Biosciences, under contract No. DE-AC02-06CH11357. R. K. acknowledges the support from the Computational Chemical Sciences Program, under Award DE-SC0023382, funded by the U.S. Department of Energy, Office of Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| XUV | extreme ultraviolet |
| HHG | high-harmonic generation |
| XAS | X-ray absorption spectroscopy |
| XANES | X-ray absorption near-edge structure |
| EXAFS | extended X-ray absorption fine structure |
| OPCPA | optical parametric chirped-pulse amplifiers |
| CTM | charge transfer multiplet |
| TDDFT | time-dependent density functional theory |
| RIXS | Resonant Inelastic X-ray Scattering |
| EELS | Electron Energy Loss Spectroscopy |
| 4c-DR-TDDFT | four-component damped response time-dependent DFT |
| SO | spin–orbit |
| amfX2C | atomic mean-field exact two-component Hamiltonian framework |
| eamfX2C | extended atomic mean-field exact two-component Hamiltonian framework |
| ROCIS | restricted open-shell configuration interaction singles |
| BSE | Bethe–Salpeter equation |
| LMCT | ligand-to-metal charge transfer |
| XRS | X-ray Raman scattering |
| TEM | Transmission electron microscopy |
| FEL | Free electron lasers |
References
- Blumberg, W. X-ray absorption spectrometry as a tool for the study of molecular structure. Inorg. Chim. Acta 1983, 79, 31. [Google Scholar] [CrossRef]
- van Bokhoven, J.A. Recent developments in X-ray absorption spectroscopy. Phys. Chem. Chem. Phys. 2010, 12, 5502. [Google Scholar] [CrossRef]
- Newton, M.A.; Zimmermann, P.; van Bokhoven, J.A. X-Ray Absorption Spectroscopy (XAS): XANES and EXAFS. In Springer Handbook of Advanced Catalyst Characterization; Springer: Cham, Switzerland, 2023; pp. 565–600. [Google Scholar]
- Solomon, E.I.; Szilagyi, R.K.; DeBeer George, S.; Basumallick, L. Electronic structures of metal sites in proteins and models: Contributions to function in blue copper proteins. Chem. Rev. 2004, 104, 419–458. [Google Scholar] [CrossRef]
- Bordiga, S.; Groppo, E.; Agostini, G.; van Bokhoven, J.A.; Lamberti, C. Reactivity of surface species in heterogeneous catalysts probed by in situ X-ray absorption techniques. Chem. Rev. 2013, 113, 1736–1850. [Google Scholar] [CrossRef] [PubMed]
- Khurana, R.; Liu, C. Unveiling the Redox Noninnocence of Metallocorroles: Exploring K-Edge X-ray Absorption Near-Edge Spectroscopy with a Multiconfigurational Wave Function Approach. J. Phys. Chem. Lett. 2024, 15, 10985–10995. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Kim, Y.L.; Khurana, R.; Havenridge, S.; Patel, P.; Liu, C. Recent advances in X-ray absorption near edge structure (XANES) simulations for catalysis: Theories and applications. Ann. Rep. Comput. Chem. 2024, 20, 157–187. [Google Scholar]
- De Groot, F.; Kotani, A. Core Level Spectroscopy of Solids; CRC Press: Boca Raton, FL, USA, 2008. [Google Scholar]
- Penner-Hahn, J.E. X-ray absorption spectroscopy. eLS. 2001. [CrossRef]
- Vura-Weis, J. Femtosecond Extreme Ultraviolet Absorption Spectroscopy of Transition Metal Complexes. Annu. Rev. Phys. Chem. 2025, 76, 455–470. [Google Scholar] [CrossRef]
- Zhang, K.; Lin, M.F.; Ryland, E.S.; Verkamp, M.A.; Benke, K.; De Groot, F.M.; Girolami, G.S.; Vura-Weis, J. Shrinking the synchrotron: Tabletop extreme ultraviolet absorption of transition-metal complexes. J. Phys. Chem. Lett. 2016, 7, 3383–3387. [Google Scholar] [CrossRef]
- Van Aken, P.; Styrsa, V.; Liebscher, B.; Woodland, A.; Redhammer, G. Microanalysis of Fe3+/ΣFe in oxide and silicate minerals by investigation of electron energy-loss near-edge structures (ELNES) at the Fe M2,3 edge. Phys. Chem. Miner. 1999, 26, 584–590. [Google Scholar] [CrossRef]
- Vura-Weis, J.; Jiang, C.M.; Liu, C.; Gao, H.; Lucas, J.M.; De Groot, F.M.; Yang, P.; Alivisatos, A.P.; Leone, S.R. Femtosecond M2,3-edge spectroscopy of transition-metal oxides: Photoinduced oxidation state change in α-Fe2O3. J. Phys. Chem. Lett. 2013, 4, 3667–3671. [Google Scholar] [CrossRef]
- Fano, U. Effects of configuration interaction on intensities and phase shifts. Phys. Rev. 1961, 124, 1866. [Google Scholar] [CrossRef]
- La-O-Vorakiat, C.; Siemens, M.; Murnane, M.M.; Kapteyn, H.C.; Mathias, S.; Aeschlimann, M.; Grychtol, P.; Adam, R.; Schneider, C.M.; Shaw, J.M.; et al. Ultrafast Demagnetization Dynamics at the M Edges of Magnetic Elements Observed Using a Tabletop High-Harmonic Soft X-Ray Source. Phys. Rev. Lett. 2009, 103, 257402. [Google Scholar] [CrossRef]
- Ash, R.; Zhang, K.; Vura-Weis, J. Photoinduced valence tautomerism of a cobalt-dioxolene complex revealed with femtosecond M-edge XANES. J. Chem. Phys. 2019, 151, 104201. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Ash, R.; Girolami, G.S.; Vura-Weis, J. Tracking the metal-centered triplet in photoinduced spin crossover of Fe(phen)32+ with tabletop femtosecond M-edge X-ray absorption near-edge structure spectroscopy. J. Am. Chem. Soc. 2019, 141, 17180–17188. [Google Scholar] [CrossRef] [PubMed]
- Ryland, E.S.; Zhang, K.; Vura-Weis, J. Sub-100 fs intersystem crossing to a metal-centered triplet in Ni(II)OEP observed with M-edge XANES. J. Phys. Chem. A 2019, 123, 5214–5222. [Google Scholar] [CrossRef] [PubMed]
- Richter, J.; Jana, S.; Behrends, R.; Davies, C.S.; Engel, D.W.; Hennecke, M.; Schick, D.; Schmising, C.v.K.; Eisebitt, S. Spectroscopic probe of ultrafast magnetization dynamics in the extreme ultraviolet spectral range. Phys. Rev. B 2025, 111, 214423. [Google Scholar] [CrossRef]
- Stavitski, E.; De Groot, F.M. The CTM4XAS program for EELS and XAS spectral shape analysis of transition metal L edges. Micron 2010, 41, 687–694. [Google Scholar] [CrossRef]
- Konecny, L.; Vicha, J.; Komorovsky, S.; Ruud, K.; Repisky, M. Accurate x-ray absorption spectra near L-and M-edges from relativistic four-component damped response time-dependent density functional theory. Inorg. Chem. 2021, 61, 830–846. [Google Scholar] [CrossRef]
- Konecny, L.; Komorovsky, S.; Vicha, J.; Ruud, K.; Repisky, M. Exact two-component TDDFT with simple two-electron picture-change corrections: X-ray absorption spectra near L-and M-edges of four-component quality at two-component cost. J. Phys. Chem. A 2023, 127, 1360–1376. [Google Scholar] [CrossRef]
- Kubas, A.; Verkamp, M.; Vura-Weis, J.; Neese, F.; Maganas, D. Restricted open-shell configuration interaction singles study on M-and L-edge X-ray absorption spectroscopy of solid chemical systems. J. Chem. Theory Comput. 2018, 14, 4320–4334. [Google Scholar] [CrossRef]
- Klein, I.M.; Krotz, A.; Lee, W.; Michelsen, J.M.; Cushing, S.K. Ab Initio calculations of XUV ground and excited states for first-row transition metal oxides. J. Phys. Chem. C 2023, 127, 1077–1086. [Google Scholar] [CrossRef]
- Schülke, W. Electron Dynamics by Inelastic X-Ray Scattering; OUP Oxford: Oxford, UK, 2007; Volume 7. [Google Scholar]
- Lee, S.K.; Eng, P.J.; Mao, H.k. Probing of pressure-induced bonding transitions in crystalline and amorphous earth materials: Insights from X-ray Raman scattering at high pressure. Rev. Mineral. Geochem. 2014, 78, 139–174. [Google Scholar] [CrossRef]
- Chiuzbăian, S.; Ghiringhelli, G.; Dallera, C.; Grioni, M.; Amann, P.; Wang, X.; Braicovich, L.; Patthey, L. Localized Electronic Excitations in NiO Studied with Resonant Inelastic X-Ray Scattering at the Ni M Threshold: Evidence of Spin Flip. Phys. Rev. Lett. 2005, 95, 197402. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.H.; Luo, Y.; Augustsson, A.; Kashtanov, S.; Rubensson, J.E.; Shuh, D.K.; Ågren, H.; Nordgren, J. Molecular structure of alcohol-water mixtures. Phys. Rev. Lett. 2003, 91, 157401. [Google Scholar] [CrossRef]
- Steiner, P.; Zimmermann, R.; Reinert, F.; Engel, T.; Hüfner, S. 3s-and 3p-core level excitations in 3d-transition metal oxides from electron-energy-loss spectroscopy. Z. Phys. B Condens. Matter 1995, 99, 479–490. [Google Scholar] [CrossRef]
- Hrisafov, S.; Pupeikis, J.; Chevreuil, P.A.; Brunner, F.; Phillips, C.R.; Gallmann, L.; Keller, U. High-power few-cycle near-infrared OPCPA for soft X-ray generation at 100 kHz. Opt. Express 2020, 28, 40145–40154. [Google Scholar] [CrossRef]
- Turgut, E.; La-o Vorakiat, C.; Shaw, J.M.; Grychtol, P.; Nembach, H.T.; Rudolf, D.; Adam, R.; Aeschlimann, M.; Schneider, C.M.; Silva, T.J.; et al. Controlling the Competition between Optically Induced Ultrafast Spin-Flip Scattering and Spin Transport in Magnetic Multilayers. Phys. Rev. Lett. 2013, 110, 197201. [Google Scholar] [CrossRef]
- Goulielmakis, E.; Loh, Z.H.; Wirth, A.; Santra, R.; Rohringer, N.; Yakovlev, V.S.; Zherebtsov, S.; Pfeifer, T.; Azzeer, A.M.; Kling, M.F.; et al. Real-time observation of valence electron motion. Nature 2010, 466, 739–743. [Google Scholar] [CrossRef]
- Ross, A.D. Ultrafast Dynamics Studied by Few-Femtosecond Soft X-Ray Transient Absorption Spectroscopy. Ph.D. Thesis, University of California, Berkeley, CA, USA, 2022. [Google Scholar]
- Jiang, C.M.; Baker, L.R.; Lucas, J.M.; Vura-Weis, J.; Alivisatos, A.P.; Leone, S.R. Characterization of photo-induced charge transfer and hot carrier relaxation pathways in spinel cobalt oxide (Co3O4). J. Phys. Chem. C 2014, 118, 22774–22784. [Google Scholar] [CrossRef]
- Baker, L.R.; Jiang, C.M.; Kelly, S.T.; Lucas, J.M.; Vura-Weis, J.; Gilles, M.K.; Alivisatos, A.P.; Leone, S.R. Charge carrier dynamics of photoexcited Co3O4 in methanol: Extending high harmonic transient absorption spectroscopy to liquid environments. Nano Lett. 2014, 14, 5883–5890. [Google Scholar] [CrossRef]
- Jarecki, J.; Hennecke, M.; Sidiropoulos, T.; Schnuerer, M.; Eisebitt, S.; Schick, D. Ultrafast energy-dispersive soft-x-ray diffraction in the water window with a laser-driven source. Struct. Dyn. 2024, 11, 054303. [Google Scholar] [CrossRef]
- Weisshaupt, J.; Juvé, V.; Holtz, M.; Ku, S.; Woerner, M.; Elsaesser, T.; Ališauskas, S.; Pugžlys, A.; Baltuška, A. High-brightness table-top hard X-ray source driven by sub-100-femtosecond mid-infrared pulses. Nat. Photonics 2014, 8, 927–930. [Google Scholar] [CrossRef]
- Popmintchev, T.; Chen, M.C.; Popmintchev, D.; Arpin, P.; Brown, S.; Ališauskas, S.; Andriukaitis, G.; Balčiunas, T.; Mücke, O.D.; Pugzlys, A.; et al. Bright coherent ultrahigh harmonics in the keV X-ray regime from mid-infrared femtosecond lasers. Science 2012, 336, 1287–1291. [Google Scholar] [CrossRef] [PubMed]
- McNeil, B.W.; Thompson, N.R. X-ray free-electron lasers. Nat. Photonics 2010, 4, 814–821. [Google Scholar] [CrossRef]
- Sahle, C.J.; Mirone, A.; Niskanen, J.; Inkinen, J.; Krisch, M.; Huotari, S. Planning, performing and analyzing X-ray Raman scattering experiments. Synchrotron Radiat. 2015, 22, 400–409. [Google Scholar] [CrossRef]
- de Groot, F.M.; Haverkort, M.W.; Elnaggar, H.; Juhin, A.; Zhou, K.J.; Glatzel, P. Resonant inelastic X-ray scattering. Nat. Rev. Methods Prim. 2024, 4, 45. [Google Scholar] [CrossRef]
- Egerton, R.F. Electron Energy-Loss Spectroscopy in the Electron Microscope; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2011. [Google Scholar]
- De Groot, F. Multiplet effects in X-ray spectroscopy. Coord. Chem. Rev. 2005, 249, 31–63. [Google Scholar] [CrossRef]
- Cowan, R.D. The Theory of Atomic Structure and Spectra; Univ of California Press: Oakland, CA, USA, 2023; Volume 3. [Google Scholar]
- Zhang, K.; Girolami, G.S.; Vura-Weis, J. Improved charge transfer multiplet method to simulate M-and L-edge X-ray absorption spectra of metal-centered excited states. Synchrotron Radiat. 2018, 25, 1600–1608. [Google Scholar] [CrossRef]
- Wang, H.; Young, A.T.; Guo, J.; Cramer, S.P.; Friedrich, S.; Braun, A.; Gu, W. Soft X-ray absorption spectroscopy and resonant inelastic X-ray scattering spectroscopy below 100 eV: Probing first-row transition-metal M-edges in chemical complexes. Synchrotron Radiat. 2013, 20, 614–619. [Google Scholar] [CrossRef]
- Ryland, E.S.; Lin, M.F.; Verkamp, M.A.; Zhang, K.; Benke, K.; Carlson, M.; Vura-Weis, J. Tabletop femtosecond M-edge X-ray absorption near-edge structure of FeTPPCl: Metalloporphyrin photophysics from the perspective of the metal. J. Am. Chem. Soc. 2018, 140, 4691–4696. [Google Scholar] [CrossRef]
- Shari’Ati, Y.; Vura-Weis, J. Ballistic Δ S= 2 intersystem crossing in a cobalt cubane following ligand-field excitation probed by extreme ultraviolet spectroscopy. Phys. Chem. Chem. Phys. 2021, 23, 26990–26996. [Google Scholar] [CrossRef] [PubMed]
- Sye, K.M.; Leahy, C.A.; Vura-Weis, J. Low quantum efficiency of μ-oxo iron bisporphyrin photocatalysts explained with femtosecond M-edge XANES. Catal. Sci. Technol. 2022, 12, 6092–6097. [Google Scholar] [CrossRef]
- Nyrow, A.; Tse, J.S.; Hiraoka, N.; Desgreniers, S.; Büning, T.; Mende, K.; Tolan, M.; Wilke, M.; Sternemann, C. Pressure induced spin transition revealed by iron M2,3-edge spectroscopy. Appl. Phys. Lett. 2014, 104, 262408. [Google Scholar] [CrossRef]
- Jenkins, A.J.; Hu, H.; Lu, L.; Frisch, M.J.; Li, X. Two-component multireference restricted active space configuration interaction for the computation of l-edge x-ray absorption spectra. J. Chem. Theory Comput. 2021, 18, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Sergentu, D.C.; Duignan, T.J.; Autschbach, J. Ab initio study of covalency in the ground versus core-excited states and X-ray absorption spectra of actinide complexes. J. Phys. Chem. Lett. 2018, 9, 5583–5591. [Google Scholar] [CrossRef]
- Sharma, P.; Jenkins, A.J.; Scalmani, G.; Frisch, M.J.; Truhlar, D.G.; Gagliardi, L.; Li, X. Exact-two-component multiconfiguration pair-density functional theory. J. Chem. Theory Comput. 2022, 18, 2947–2954. [Google Scholar] [CrossRef]
- Guda, A.A.; Guda, S.A.; Martini, A.; Kravtsova, A.; Algasov, A.; Bugaev, A.; Kubrin, S.P.; Guda, L.; Šot, P.; van Bokhoven, J.A.; et al. Understanding X-ray absorption spectra by means of descriptors and machine learning algorithms. NPJ Comput. Mater. 2021, 7, 203. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).