
Effect of Hydrogen-Donor of Heavy Crude Oil Catalytic Aquathermolysis in the Presence of a Nickel-Based Catalyst

 ,
,  , ,
, ,  and
and 
Abstract
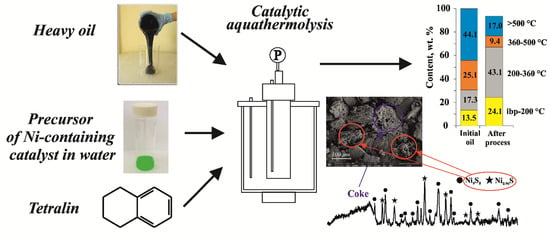
1. Introduction
2. Results and Discussion
3. Research Methods
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- REN21. Renewables 2020 Global Status Report (INIS-FR--20-1110). France. 2020. Available online: https://www.globalwomennet.org/wp-content/uploads/2020/06/GSR2020_Full_Report_with_Endnotes.pdf (accessed on 1 September 2022).
- Zhou, Z.; Slaný, M.; Kuzielová, E.; Zhang, W.; Ma, L.; Dong, S.; Zhang, J.; Chen, G. Influence of Reservoir Minerals and Ethanol on Catalytic Aquathermolysis of Heavy Oil. Fuel 2022, 307, 121871. [Google Scholar] [CrossRef]
- Morelos-Santos, O.; Reyes de la Torre, A.I.; Melo-Banda, J.A.; Schacht-Hernández, P.; Portales-Martínez, B.; Soto-Escalante, I.; José-Yacamán, M. A Novel Direct Method in One-Step for Catalytic Heavy Crude Oil Upgrading Using Iron Oxide Nanoparticles. Catal. Today 2022, 392–393, 60–71. [Google Scholar] [CrossRef]
- Li, Z.; Li, Y.; Xu, H.; Jarvis, J.; Meng, S.; Song, H. Effect of Methane Presence on Catalytic Heavy Oil Partial Upgrading. Fuel 2021, 297, 120733. [Google Scholar] [CrossRef]
- Hosseinpour, M.; Hajialirezaei, A.H.; Soltani, M.; Nathwani, J. Thermodynamic Analysis of In-Situ Hydrogen from Hot Compressed Water for Heavy Oil Upgrading. Int. J. Hydrog. Energy 2019, 44, 27671–27684. [Google Scholar] [CrossRef]
- Rana, M.S.; Sámano, V.; Ancheyta, J.; Diaz, J.A.I. A Review of Recent Advances on Process Technologies for Upgrading of Heavy Oils and Residua. Fuel 2007, 86, 1216–1231. [Google Scholar] [CrossRef]
- Kang, K.H.; Kim, G.T.; Park, S.; Seo, P.W.; Seo, H.; Lee, C.W. A Review on the Mo-Precursors for Catalytic Hydroconversion of Heavy Oil. J. Ind. Eng. Chem. 2019, 76, 1–16. [Google Scholar] [CrossRef]
- León, A.Y.; Guzmán, M.A.; Picón, H.; Laverde, C.D.; Molina, V.D. Reactivity of Vacuum Residues by Thermogravimetric Analysis and Nuclear Magnetic Resonance Spectroscopy. Energy Fuels 2020, 34, 9231–9242. [Google Scholar] [CrossRef]
- Lakhova, A.; Petrov, S.; Ibragimova, D.; Kayukova, G.; Safiulina, A.; Shinkarev, A.; Okekwe, R. Aquathermolysis of Heavy Oil Using Nano Oxides of Metals. J. Pet. Sci. Eng. 2017, 153, 385–390. [Google Scholar] [CrossRef]
- Djimasbe, R.; Varfolomeev, M.A.; Al-Muntaser, A.A.; Yuan, C.; Feoktistov, D.A.; Suwaid, M.A.; Kirgizov, A.J.; Davletshin, R.R.; Zinnatullin, A.L.; Fatou, S.D.; et al. Oil Dispersed Nickel-Based Catalyst for Catalytic Upgrading of Heavy Oil Using Supercritical Water. Fuel 2022, 313, 122702. [Google Scholar] [CrossRef]
- Grabchenko, M.; Pantaleo, G.; Puleo, F.; Kharlamova, T.S.; Zaikovskii, V.I.; Vodyankina, O.; Liotta, L.F. Design of Ni-Based Catalysts Supported over Binary La-Ce Oxides: Influence of La/Ce Ratio on the Catalytic Performances in DRM. Catal. Today 2021, 382, 71–81. [Google Scholar] [CrossRef]
- Zhao, F.; Liu, Y.; Wu, Y.; Zhao, X.; Tan, L. Study of Catalytic Aquathermolysis of Heavy Oil in the Presence of a Hydrogen Donor. Chem. Technol. Fuels Oils 2012, 48, 273–282. [Google Scholar] [CrossRef]
- Chen, G.; Yuan, W.; Yan, J.; Meng, M.; Guo, Z.; Gu, X.; Zhang, J.; Qu, C.; Song, H.; Jeje, A. Zn(II) Complex Catalyzed Coupling Aquathermolysis of Water-Heavy Oil-Methanol at Low Temperature. Pet. Chem. 2018, 58, 197–202. [Google Scholar] [CrossRef]
- Chen, G.; Yuan, W.; Bai, Y.; Zhao, W.; Gu, X.; Zhang, J.; Jeje, A. Ethanol Enhanced Aquathermolysis of Heavy Oil Catalyzed by a Simple Co(II) Complex at Low Temperature. Pet. Chem. 2017, 57, 389–394. [Google Scholar] [CrossRef]
- Philippov, A.A.; Chibiryaev, A.M.; Martyanov, O.N. Catalyzed Transfer Hydrogenation by 2-Propanol for Highly Selective PAHs Reduction. Catal. Today 2021, 379, 15–22. [Google Scholar] [CrossRef]
- Chesnokov, V.V.; Dik, P.P.; Nikityonok, A.V.; Chichkan, A.S.; Parmon, V.N. Comparative Analysis of the Effects of Hydrogen and Formic Acid on the Vacuum Residue Hydrocracking. Chem. Eng. J. 2022, 449, 137839. [Google Scholar] [CrossRef]
- Hart, A.; Leeke, G.; Greaves, M.; Wood, J. Down-hole heavy crude oil upgrading by CAPRI: Effect of hydrogen and methane gases upon upgrading and coke formation. Fuel 2014, 119, 226–235. [Google Scholar] [CrossRef]
- Kayukova, G.P.; Mikhailova, A.M.; Feoktistov, D.A.; Morozov, V.P.; Vakhin, A.V. Conversion of the Organic Matter of Domanic Shale and Permian Bituminous Rocks in Hydrothermal Catalytic Processes. Energy Fuels 2017, 31, 7789–7799. [Google Scholar] [CrossRef]
- Hosseinpour, M.; Fatemi, S.; Ahmadi, S.J.; Oshima, Y.; Morimoto, M.; Akizuki, M. Isotope Tracing Study on Hydrogen Donating Capability of Supercritical Water Assisted by Formic Acid to Upgrade Heavy Oil: Computer Simulation vs. Experiment. Fuel 2018, 225, 161–173. [Google Scholar] [CrossRef]
- Safaei Mahmoudabadi, Z.; Rashidi, A.; Panahi, M. New Approach to Unsupported ReS2 Nanorod Catalyst for Upgrading of Heavy Crude Oil Using Methane as Hydrogen Source. Int. J. Hydrogen Energy 2021, 46, 5270–5285. [Google Scholar] [CrossRef]
- Zhao, F.; Xu, T.; Zhu, G.; Wang, K.; Xu, X.; Liu, L. A Review on the Role of Hydrogen Donors in Upgrading Heavy Oil and Bitumen. Sustain. Energy Fuels 2022, 6, 1866–1890. [Google Scholar] [CrossRef]
- Stepacheva, A.; Gavrilenko, A.; Markova, M.; Semenova, A.; Monzharenko, M.; Sulman, M. The Use of Supercritical Solvents in Crude Oil Fraction Conversion. J. Phys. Conf. Ser. 2020, 1658, 12057. [Google Scholar] [CrossRef]
- Hart, A.; Lewis, C.; White, T.; Greaves, M.; Wood, J. Effect of Cyclohexane as Hydrogen-Donor in Ultradispersed Catalytic Upgrading of Heavy Oil. Fuel Process. Technol. 2015, 138, 724–733. [Google Scholar] [CrossRef]
- Ovalles, C.; Vallejos, C.; Vasquez, T.; Rojas, I.; Ehrman, U.; Benitez, J.L.; Martinez, R. Downhole Upgrading of Extra-Heavy Crude Oil Using Hydrogen Donors and Methane under Steam Injection Conditions. Pet. Sci. Technol. 2003, 21, 255–274. [Google Scholar] [CrossRef]
- Alemán-Vázquez, L.O.; Cano-Domínguez, J.L.; García-Gutiérrez, J.L. Effect of Tetralin, Decalin and Naphthalene as Hydrogen Donors in the Upgrading of Heavy Oils. Procedia Eng. 2012, 42, 532–539. [Google Scholar] [CrossRef]
- Fang, D.; Wang, G.; Sheng, Q.; Ge, S.; Gao, C.; Gao, J. Preparation of Hydrogen Donor Solvent for Asphaltenes Efficient Liquid-Phase Conversion via Heavy Cycle Oil Selective Hydrogenation. Fuel 2019, 257, 115886. [Google Scholar] [CrossRef]
- Ovalles, C.; Rivero, V.; Salazar, A. Downhole Upgrading of Orinoco Basin Extra-Heavy Crude Oil Using Hydrogen Donors under Steam Injection Conditions. Effect of the Presence of Iron Nanocatalysts. Catalysts 2015, 5, 286–297. [Google Scholar] [CrossRef]
- Al-Muntaser, A.A.; Varfolomeev, M.A.; Suwaid, M.A.; Saleh, M.M.; Djimasbe, R.; Yuan, C.; Zairov, R.R.; Ancheyta, J. Effect of Decalin as Hydrogen-Donor for in-Situ Upgrading of Heavy Crude Oil in Presence of Nickel-Based Catalyst. Fuel 2022, 313, 122652. [Google Scholar] [CrossRef]
- Hart, A.; Adam, M.; Robinson, J.P.; Rigby, S.P.; Wood, J. Tetralin and Decalin H-Donor Effect on Catalytic Upgrading of Heavy Oil Inductively Heated with Steel Balls. Catalysts 2020, 10, 393. [Google Scholar] [CrossRef]
- Kim, S.H.; Kim, K.D.; Lee, H.; Lee, Y.K. Beneficial Roles of H-Donors as Diluent and H-Shuttle for Asphaltenes in Catalytic Upgrading of Vacuum Residue. Chem. Eng. J. 2017, 314, 1–10. [Google Scholar] [CrossRef]
- Bai, J.K.; Zhang, X.B.; Li, W.; Wang, X.B.; Du, Z.Y.; Li, W.Y. Rate Constant of Hydrogen Transfer from H-Donor Solvents to Coal Radicals. Fuel 2022, 318, 123621. [Google Scholar] [CrossRef]
- Al-Muntaser, A.A.; Varfolomeev, M.A.; Suwaid, M.A.; Yuan, C.; Chemodanov, A.E.; Feoktistov, D.A.; Rakhmatullin, I.Z.; Abbas, M.; Domínguez-Álvarez, E.; Akhmadiyarov, A.A.; et al. Hydrothermal Upgrading of Heavy Oil in the Presence of Water at Sub-Critical, near-Critical and Supercritical Conditions. J. Pet. Sci. Eng. 2020, 184, 106592. [Google Scholar] [CrossRef]
- Muraza, O.; Galadima, A. Aquathermolysis of Heavy Oil: A Review and Perspective on Catalyst Development. Fuel 2015, 157, 219–231. [Google Scholar] [CrossRef]
- Liu, H.; Wang, J.; Wang, Z.; Fan, S.; Chen, K. Hydrogenation of Nickel Octaethylporphyrin over Dispersed MoS2 Catalysts Formed In Situ. ChemistrySelect 2018, 3, 4292–4297. [Google Scholar] [CrossRef]
- Alkhaldi, S.; Husein, M.M. Hydrocracking of Heavy Oil by Means of in Situ Prepared Ultradispersed Nickel Nanocatalyst. Energy Fuels 2014, 28, 643–649. [Google Scholar] [CrossRef]
- Chianelli, R. Catalysis Reviews: Science and Engineering Fundamental Studies of Transition Metal Sulfide Hydrodesulfurization Catalysts. Catal. Rev. 1984, 26, 361–393. [Google Scholar] [CrossRef]
- Urazov, K.K.; Sviridenko, N.N. NiO Based Catalysts Obtained “in-Situ” for Heavy Crude Oil Upgrading: Effect of NiO Precursor on the Catalytic Cracking Products Composition. J. Taiwan Inst. Chem. Eng. 2021, 127, 151–156. [Google Scholar] [CrossRef]
- Avbenake, O.P.; Al-Hajri, R.S.; Jibril, B.Y. Saturates and Aromatics Characterization in Heavy Crude Oil Upgrading Using Ni–Co/γ-Al2O3 Catalysts. Pet. Sci. Technol. 2020, 38, 800–807. [Google Scholar] [CrossRef]
- Sviridenko, N.N.; Golovko, A.K.; Kirik, N.P.; Anshits, A.G. Upgrading of Heavy Crude Oil by Thermal and Catalytic Cracking in the Presence of NiCr/WC Catalyst. J. Taiwan Inst. Chem. Eng. 2020, 112, 97–105. [Google Scholar] [CrossRef]
- Morelos-Santos, O.; Reyes de la Torre, A.I.; Melo-Banda, J.A.; Mendoza-Martínez, A.M.; Schacht-Hernández, P.; Portales-Martínez, B.; Soto-Escalante, I.; Domínguez-Esquivel, J.M.; José-Yacamán, M. Dispersed Nickel-Based Catalyst for Enhanced Oil Recovery (EOR) Under Limited Hydrogen Conditions. Top. Catal. 2020, 63, 504–510. [Google Scholar] [CrossRef]
- Okunev, A.G.; Parkhomchuk, E.V.; Lysikov, A.I.; Parunin, P.D.; Semeikina, V.S.; Parmon, V.N. Catalytic Hydroprocessing of Heavy Oil Feedstocks. Russ. Chem. Rev. 2015, 84, 981–999. [Google Scholar] [CrossRef]
- Wu, C.; Lei, G.L.; Yao, C.J.; Gai, P.Y.; Cao, Y.B.; Li, X.N. Mechanism for Reducing the Viscosity of Extra-Heavy Oil by Aquathermolysis with an Amphiphilic Catalyst. Ranliao Huaxue Xuebao/J. Fuel Chem. Technol. 2010, 38, 684–690. [Google Scholar] [CrossRef]
- Hart, A.; Leeke, G.; Greaves, M.; Wood, J. Downhole heavy crude oil upgrading using CAPRI: Effect of steam upon upgrading and coke formation. Energy Fuels 2014, 28, 1811–1819. [Google Scholar] [CrossRef]
- Urazov, K.K.; Sviridenko, N.N. Structural Transformations of Heavy Oil Asphaltenes from the Zyuzeevskoye Field upon Thermocatalytic Cracking. Solid Fuel Chem. 2022, 56, 128–132. [Google Scholar] [CrossRef]
- Waldner, P. Fe-Solubility of Ni7S6 and Ni9S 8: Thermodynamic Analysis. J. Chem. Thermodyn. 2011, 43, 315–318. [Google Scholar] [CrossRef]
- Li, Q.; Wang, X.; Zhang, R.; Mi, J.; Wu, M. Insights into the Effects of Metal-Ion Doping on the Structure and Hot-Coal-Gas Desulfurization Properties of Zn-Based Sorbents Supported on SBA-15. Fuel 2022, 315, 123198. [Google Scholar] [CrossRef]
- Picón-Hernández, H.J.; Centeno-Hurtado, A.; Pantoja-Agreda, E.F. Morphological Classification of Coke Formed from the Castilla and Jazmín Crude Oils. CTyF-Cienc. Tecnol. Future 2008, 3, 169–183. [Google Scholar] [CrossRef]
- Crelling, J.C. Coal Carbonization. Appl. Coal Petrol. 2008, 173–192. [Google Scholar] [CrossRef]
- Nguyen, M.T.; Nguyen, D.L.T.; Xia, C.; Nguyen, T.B.; Shokouhimehr, M.; Sana, S.S.; Grace, A.N.; Aghbashlo, M.; Tabatabaei, M.; Sonne, C.; et al. Recent Advances in Asphaltene Transformation in Heavy Oil Hydroprocessing: Progress, Challenges, and Future Perspectives. Fuel Process. Technol. 2021, 213, 106681. [Google Scholar] [CrossRef]
- Gao, Q.; Luo, W.; Ma, X.; Ma, Z.; Li, S.; Gou, F.; Shen, W.; Jiang, Y.; He, R.; Li, M. Electronic Modulation and Vacancy Engineering of Ni9S8 to Synergistically Boost Efficient Water Splitting: Active Vacancy-Metal Pairs. Appl. Catal. B Environ. 2022, 310, 121356. [Google Scholar] [CrossRef]
- Chen, L.; Deng, W.; Chen, Z.; Wang, X. Hetero-Architectured Core-Shell NiMoO4@Ni9S8/MoS2 Nanorods Enabling High-Performance Supercapacitors. J. Mater. Res. 2022, 37, 284–293. [Google Scholar] [CrossRef]
- Hussain, S.; Ullah, N.; Zhang, Y.; Shaheen, A.; Javed, M.S.; Lin, L.; Zulfiqar; Shah, S.B.; Liu, G.; Qiao, G. One-Step Synthesis of Unique Catalyst Ni9S8@C for Excellent MOR Performances. Int. J. Hydrog. Energy 2019, 44, 24525–24533. [Google Scholar] [CrossRef]
- Fergoug, T.; Bouhadda, Y. Determination of Hassi Messaoud Asphaltene Aromatic Structure from 1H & 13C NMR Analysis. Fuel 2014, 115, 521–526. [Google Scholar] [CrossRef]
- Sun, Y.D.; Yang, C.H.; Zhao, H.; Shan, H.H.; Shen, B.X. Influence of Asphaltene on the Residue Hydrotreating Reaction. Energy Fuels 2010, 24, 5008–5011. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Indices | Initial Oil | Experiment | |||||
|---|---|---|---|---|---|---|---|
| C | CA | CA+T | C+T | CA+D | C+D | ||
| Gas yield, wt.% | 5.0 | 5.8 | 5.6 | 6.9 | 7.9 | 8.2 | |
| - Sulfur content in gas, wt.% | 0.64 | 1.41 | 1.91 | 1.18 | 1.15 | 1.20 | |
| Coke yield, wt.% | 3.4 | 1.8 | 0.8 | 1.2 | 5.2 | 4.9 | |
| - Sulfur content in coke, wt.% | 0.14 | 0.11 | 0.03 | 0.09 | 0.39 | 0.28 | |
| Liquid products (LP), wt.% | 100 | 91.6 | 92.4 | 93.6 | 91.9 | 86.9 | 86.9 |
| - Sulfur content in LP, wt.% | 4.53 | 3.75 | 3.01 | 2.59 | 3.26 | 2.99 | 3.05 |
| SARA, wt.% | |||||||
| Saturates+aromatics | 68.1 | 74.7 | 75.2 | 74.9 | 70.5 | 70.4 | 71.7 |
| Resins | 21.5 | 11.0 | 10.2 | 13.1 | 14.4 | 12.5 | 11.5 |
| Asphaltenes | 10.4 | 5.9 | 7.0 | 5.6 | 7.0 | 4.0 | 3.7 |
| Asphaltenes | Content, wt.% | |||||
|---|---|---|---|---|---|---|
| C | H | N | S | O | H/C | |
| Initial oil | 80.01 | 7.63 | 1.81 | 5.51 | 5.04 | 1.144 |
| C | 79.56 | 5.78 | 2.31 | 5.46 | 6.89 | 0.872 |
| CA | 79.78 | 5.75 | 2.21 | 7.25 | 5.01 | 0.865 |
| CA+T | 81.49 | 6.10 | 2.25 | 7.16 | 3.00 | 0.898 |
| C+T | 81.06 | 5.79 | 1.95 | 7.54 | 3.66 | 0.857 |
| CA+D | 80.15 | 5.54 | 2.04 | 7.67 | 4.60 | 0.829 |
| C+D | 80.15 | 5.64 | 1.92 | 7.51 | 4.78 | 0.844 |
| Parameters | Initial Oil | C | CA | CA+T | C+T |
|---|---|---|---|---|---|
| MM, a.m.u. | 1920 | 782 | 851 | 863 | 828 |
| Number of atoms in a mean molecule | |||||
| C | 128.0 | 51.8 | 56.6 | 58.6 | 55.9 |
| H | 146.5 | 45.2 | 48.9 | 52.6 | 47.9 |
| N | 2.5 | 1.3 | 1.3 | 1.4 | 1.2 |
| S | 3.3 | 1.3 | 1.9 | 1.9 | 2.0 |
| O | 6.0 | 3.4 | 2.7 | 1.6 | 1.9 |
| Number of rings | |||||
| RT | 24.8 | 13.1 | 14.9 | 14.6 | 14.3 |
| RAr | 15.5 | 9.6 | 11.3 | 10.3 | 10.8 |
| RN | 9.3 | 3.5 | 3.6 | 4.3 | 3.5 |
| ma | 3.81 | 1.89 | 1.10 | 1.47 | 1.35 |
| σa | 0.56 | 0.41 | 0.45 | 0.46 | 0.40 |
| Number of carbon atoms of different types in a mean molecule | |||||
| CAr | 62.0 | 34.3 | 36.5 | 37.3 | 37.3 |
| CN | 32.4 | 12.4 | 12.6 | 16.2 | 10.6 |
| CS | 33.6 | 5.2 | 7.5 | 5.1 | 8.0 |
| n | 3.56 | 2.49 | 2.79 | 2.50 | 2.63 |
| ƒa | 48.5 | 66.1 | 64.5 | 63.6 | 66.7 |
| Characteristics | Zyuzeevskoye Field |
|---|---|
| Density at 20 °C, kg/m3 | 940.0 |
| Dynamic viscosity, mPa s—at 20 °C | 743 |
| Elemental composition, wt.% | |
| - Carbon | 81.01 |
| - Hydrogen | 11.45 |
| - Sulfur | 4.53 |
| - Nitrogen | 0.89 |
| - Oxygen | 2.12 |
| - H/C | 1.69 |
| Component composition, wt.% | |
| - Saturated hydrocarbons | 24.6 |
| - Aromatic hydrocarbons | 43.5 |
| - Resins | 21.5 |
| - Asphaltenes | 10.4 |
| Fractional composition, wt.% | |
| - ibp-200 °C | 13.5 |
| - 200–360 °C | 17.3 |
| - 360–500 °C | 25.1 |
| - >500 °C | 44.1 |
| Type of Hydrogen | Chemical Shift (ppm) | Assignments |
|---|---|---|
| HAr | 6.5–9.5 | aromatic hydrogen |
| Hα | 2.0–4.5 | α hydrogen of aliphatic chains |
| Hβ | 1.0–2.0 | β hydrogen of aliphatic chains |
| Hγ | 0.5–1.0 | γ hydrogen of aliphatic chains |
| Symbol | Structure Parameters | Calculated Formulas |
|---|---|---|
| HT | Total hydrogen numbers | Mw × H% |
| CT | Total carbon numbers | (Mw × C%)/12 |
| n | Average alkyl chain length | (Hα + Hβ + Hγ)/Hα |
| fa | Aromaticity factor | (C/H − Hα/2 − Hβ/2 − Hγ/2)/(C/H) |
| HAU/CAr | Condensation degree parameter of the aromatic ring | (Hα/2 + HAr)/(C/H − Hα/2 − Hβ/2 − Hγ/2) |
| σ | Replacement rate of periphery hydrogen in the aromatic ring system | (Hα/2)/(Hα/2 + HAr) |
| CAr | Aromatic carbon numbers | fa × CT |
| Cs | Saturated carbon numbers | CT − CAr |
| Cap | Peripheral carbon in a fused aromatic ring | CAr × HAU/CAr |
| Ci | Internal carbon in a fused aromatic ring | CAr − Cap |
| CAr(us) | Aromatic carbon number per unit structure | (2.503/HAU/CAr)2 |
| u | Blocks number in molecule | CAr / CAr(us) |
| RT | Total rings | CT − HT/2 + 1 − CAr/2 |
| RAr | Aromatic rings | CAr/2 − (Hα × HT)/4 − (HAr × HT)/2 + 1 |
| RN | Naphthenic rings | RT − RAr |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Urazov, K.K.; Sviridenko, N.N.; Iovik, Y.A.; Kolobova, E.N.; Grabchenko, M.V.; Kurzina, I.A.; Mukhamatdinov, I.I. Effect of Hydrogen-Donor of Heavy Crude Oil Catalytic Aquathermolysis in the Presence of a Nickel-Based Catalyst. Catalysts 2022, 12, 1154. https://doi.org/10.3390/catal12101154
Urazov KK, Sviridenko NN, Iovik YA, Kolobova EN, Grabchenko MV, Kurzina IA, Mukhamatdinov II. Effect of Hydrogen-Donor of Heavy Crude Oil Catalytic Aquathermolysis in the Presence of a Nickel-Based Catalyst. Catalysts. 2022; 12(10):1154. https://doi.org/10.3390/catal12101154
Chicago/Turabian StyleUrazov, Khoshim Kh., Nikita N. Sviridenko, Yuliya A. Iovik, Ekaterina N. Kolobova, Maria V. Grabchenko, Irina A. Kurzina, and Irek I. Mukhamatdinov. 2022. "Effect of Hydrogen-Donor of Heavy Crude Oil Catalytic Aquathermolysis in the Presence of a Nickel-Based Catalyst" Catalysts 12, no. 10: 1154. https://doi.org/10.3390/catal12101154
APA StyleUrazov, K. K., Sviridenko, N. N., Iovik, Y. A., Kolobova, E. N., Grabchenko, M. V., Kurzina, I. A., & Mukhamatdinov, I. I. (2022). Effect of Hydrogen-Donor of Heavy Crude Oil Catalytic Aquathermolysis in the Presence of a Nickel-Based Catalyst. Catalysts, 12(10), 1154. https://doi.org/10.3390/catal12101154