
Carbon Monoxide Tolerant Pt-Based Electrocatalysts for H2-PEMFC Applications: Current Progress and Challenges



Abstract
1. Introduction
2. Fundamentals of CO Tolerance
2.1. HOR and CO Poisoning Kinetics
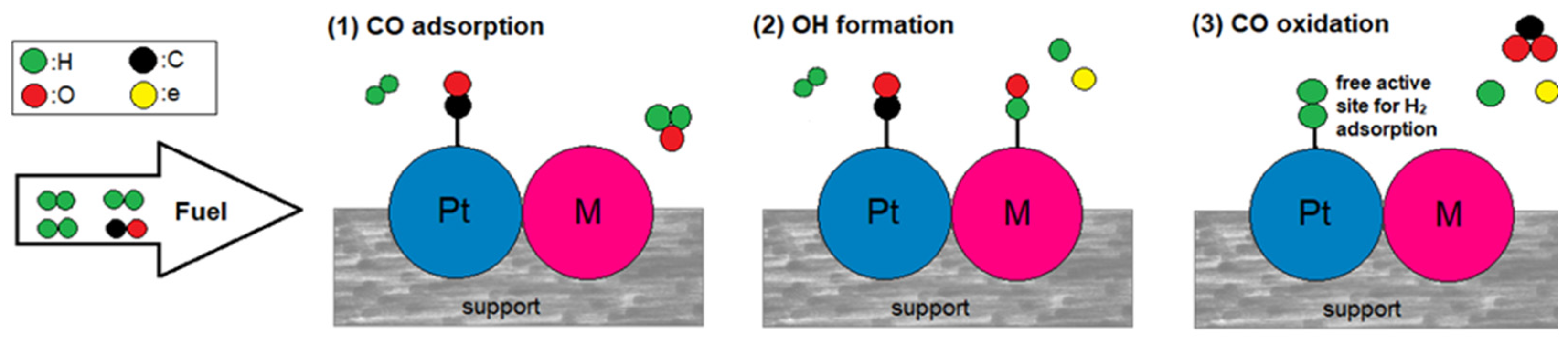
2.2. CO Tolerance Mechanisms
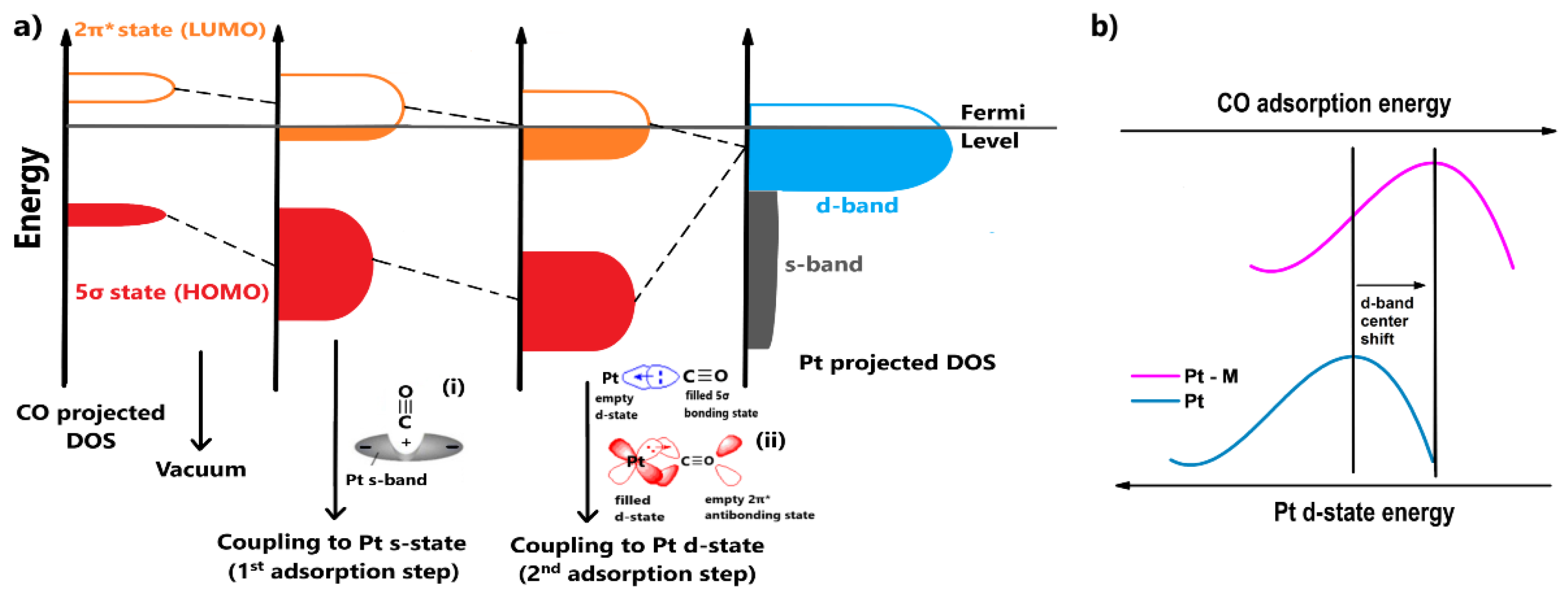
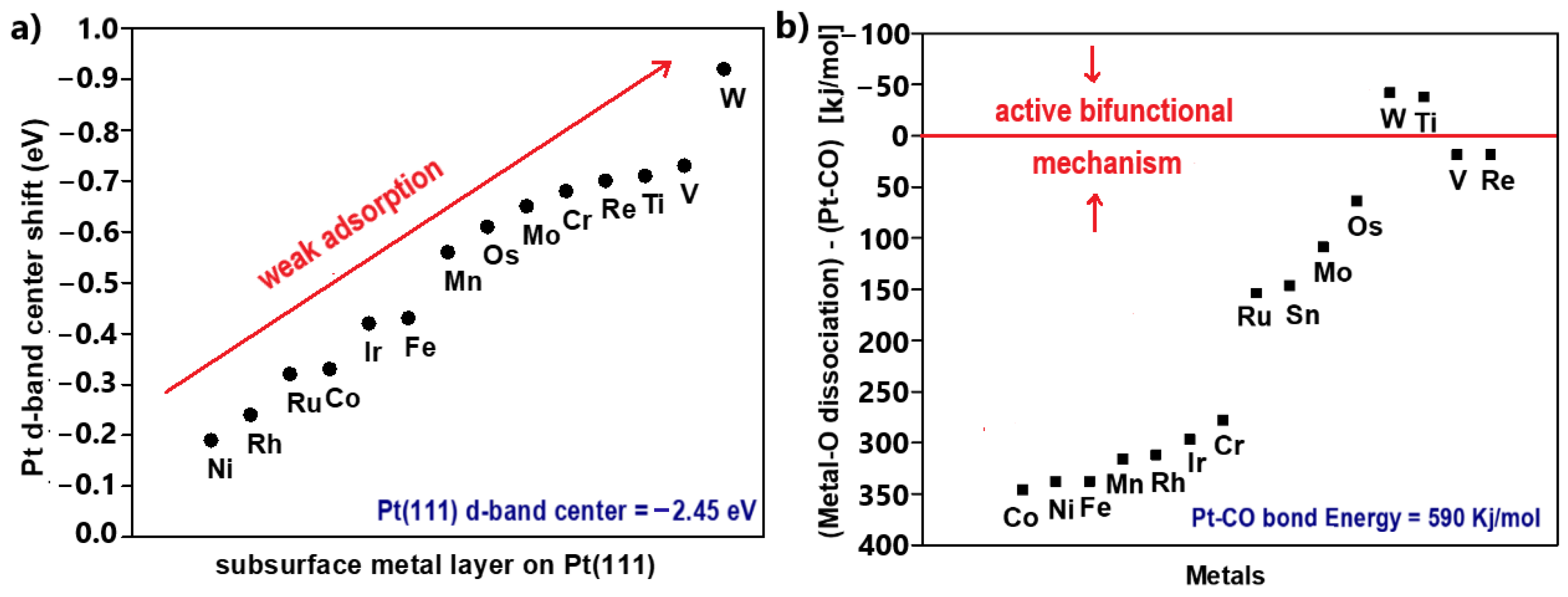
- The adsorbate orbitals are hybridized with the s-state of Pt. This interaction results in the energy downshifting and broadening of both 2π* and 5σ CO states, as shown in Figure 2a. Schiros et al. [42] suggested that this type of hybridization positively polarizes the metal s-state due to an electron distribution (Figure 2a(i)); therefore, the orbital of the adsorbate is coordinated through an attractive electrostatic interaction, promoting to some extent the bond formation;
- Two stabilization effects occur synergistically between the renormalized CO states and Pt d-state. Through this interaction, the respective bonding and antibonding states of 5σ and 2π* states are formed. The contribution of the 5σ antibonding state is considered minimal. First, an electron transfer arises from the highest occupied molecular orbital (HOMO) of CO, the filled 5σ bonding state, to the Pt d-state, increasing its electron density. To compensate for this effect, electrons are back-donated from the Pt d-state to the lowest occupied molecular orbital (LUMO) of CO, the empty 2π* antibonding state (Figure 2a(ii)); therefore, total charge stabilization between the interacted DOS is achieved, leading to the Pt-CO binding [43,44,45].
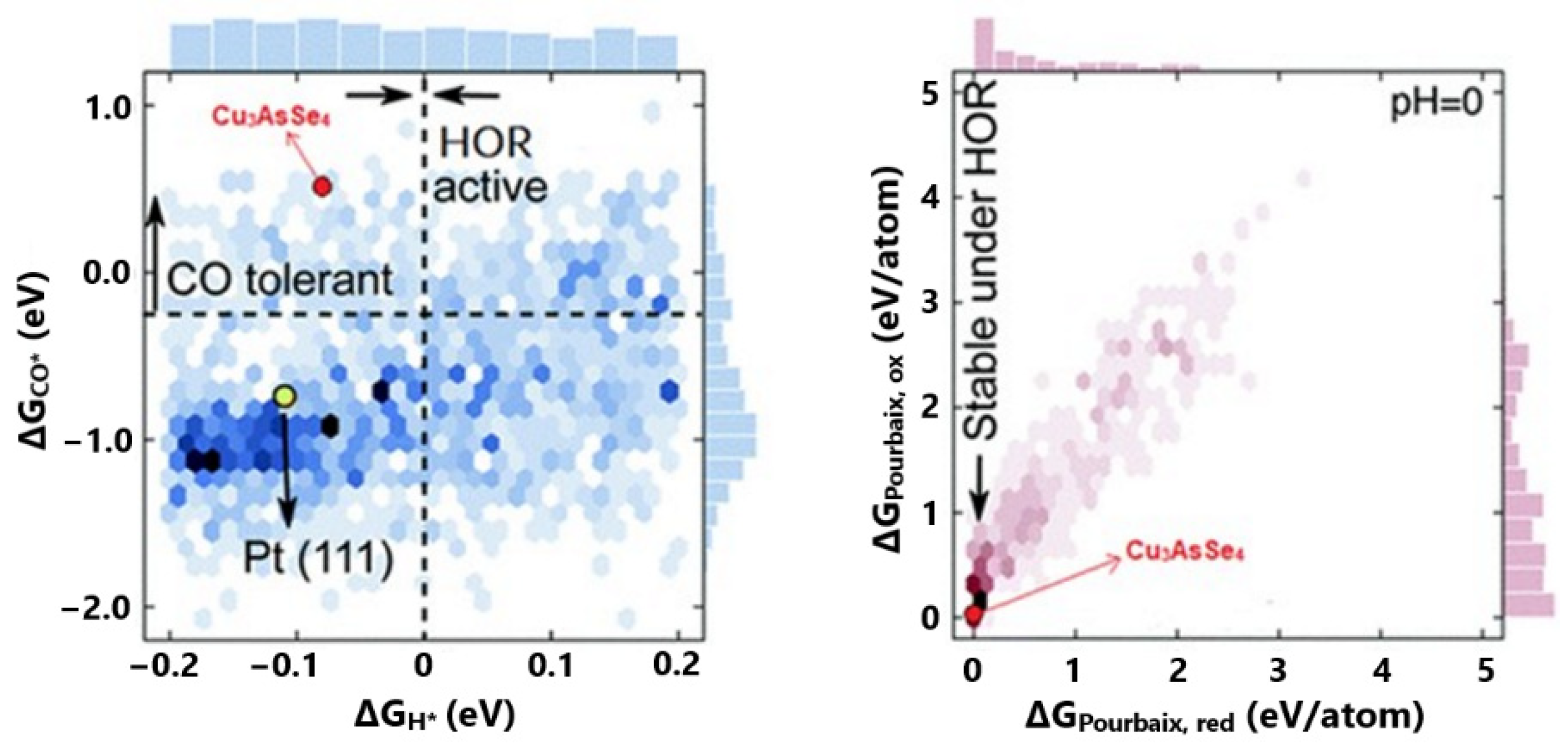
2.3. Energetic View of CO Tolerance
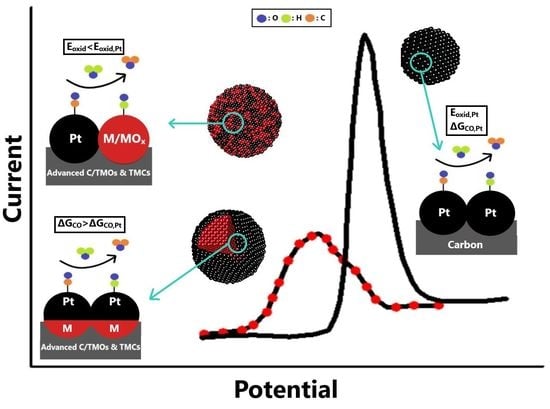
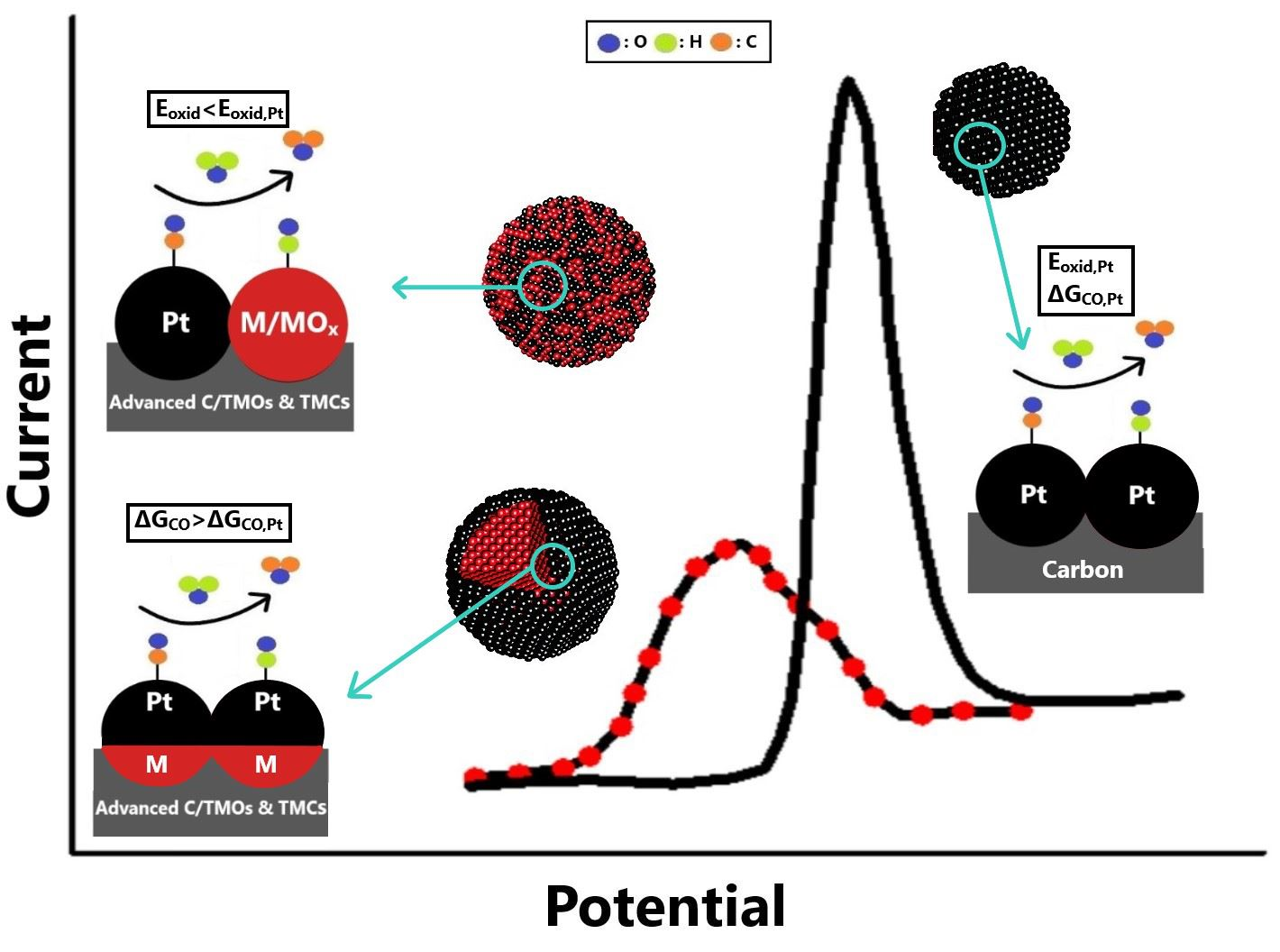
3. Overview of State-of-the-Art CO-Tolerant Electrocatalysts
3.1. Pt-Based Electrocatalysts Supported on Carbon
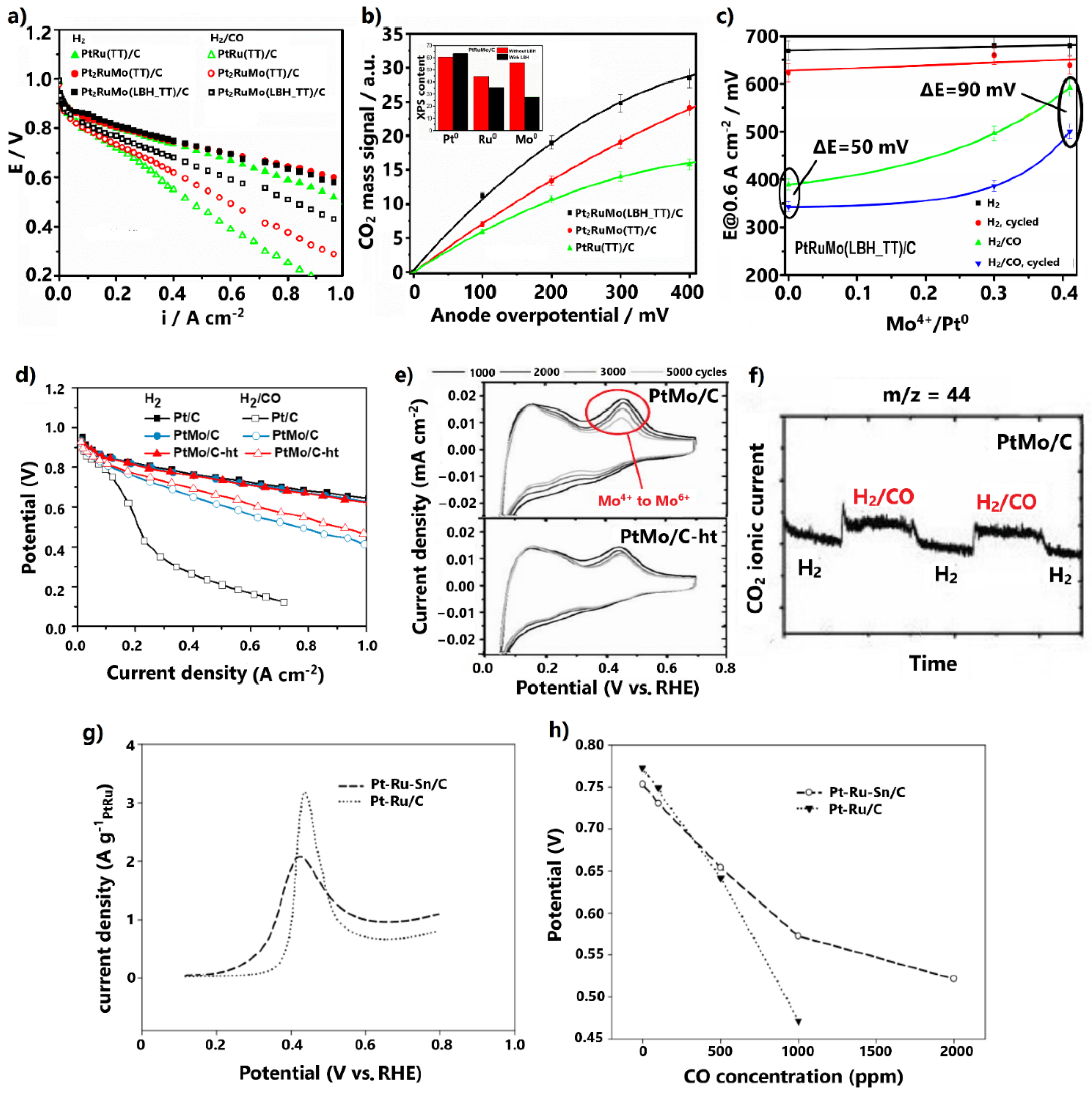
3.1.1. Alloys
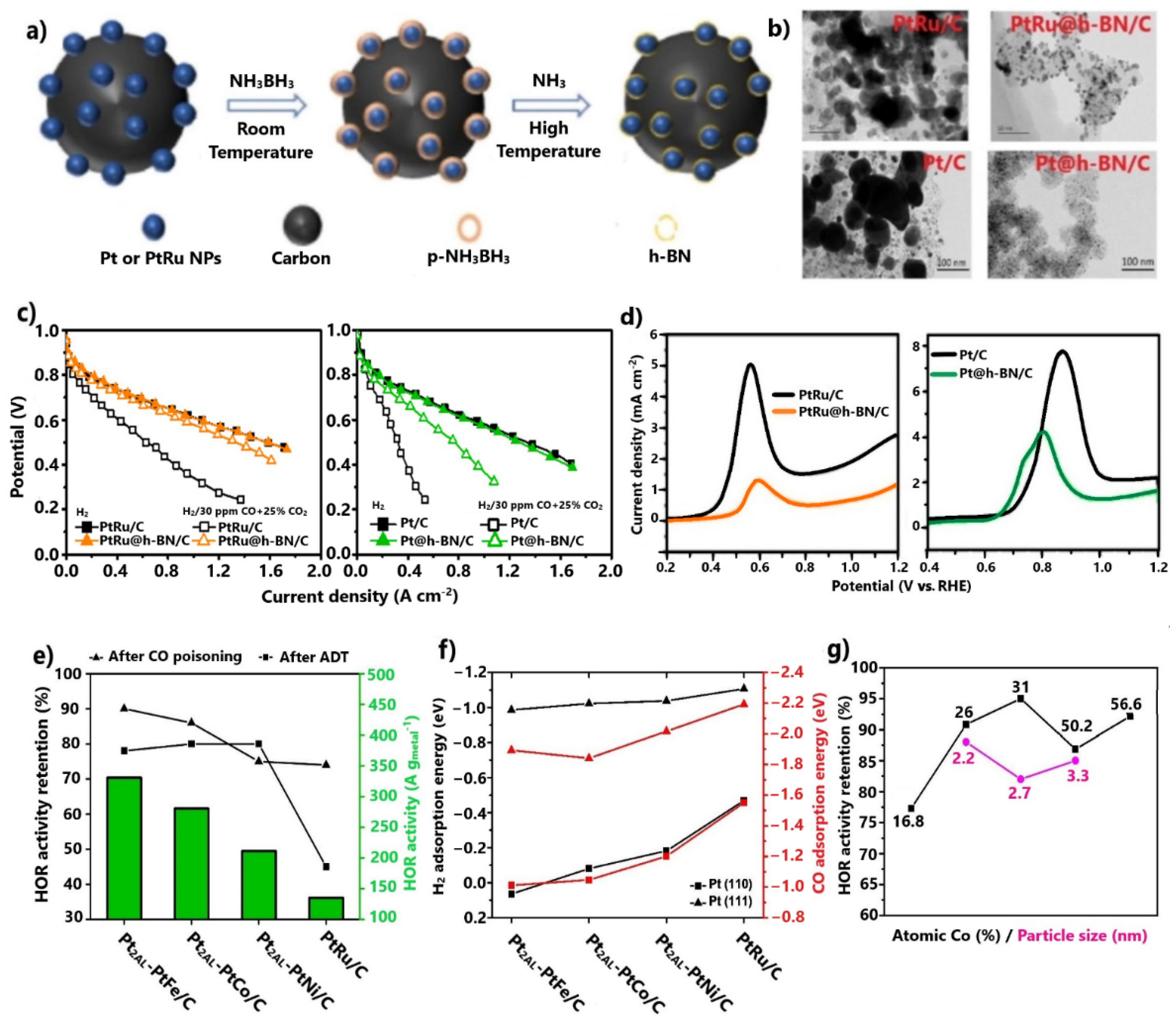
3.1.2. Core–Shell Structures
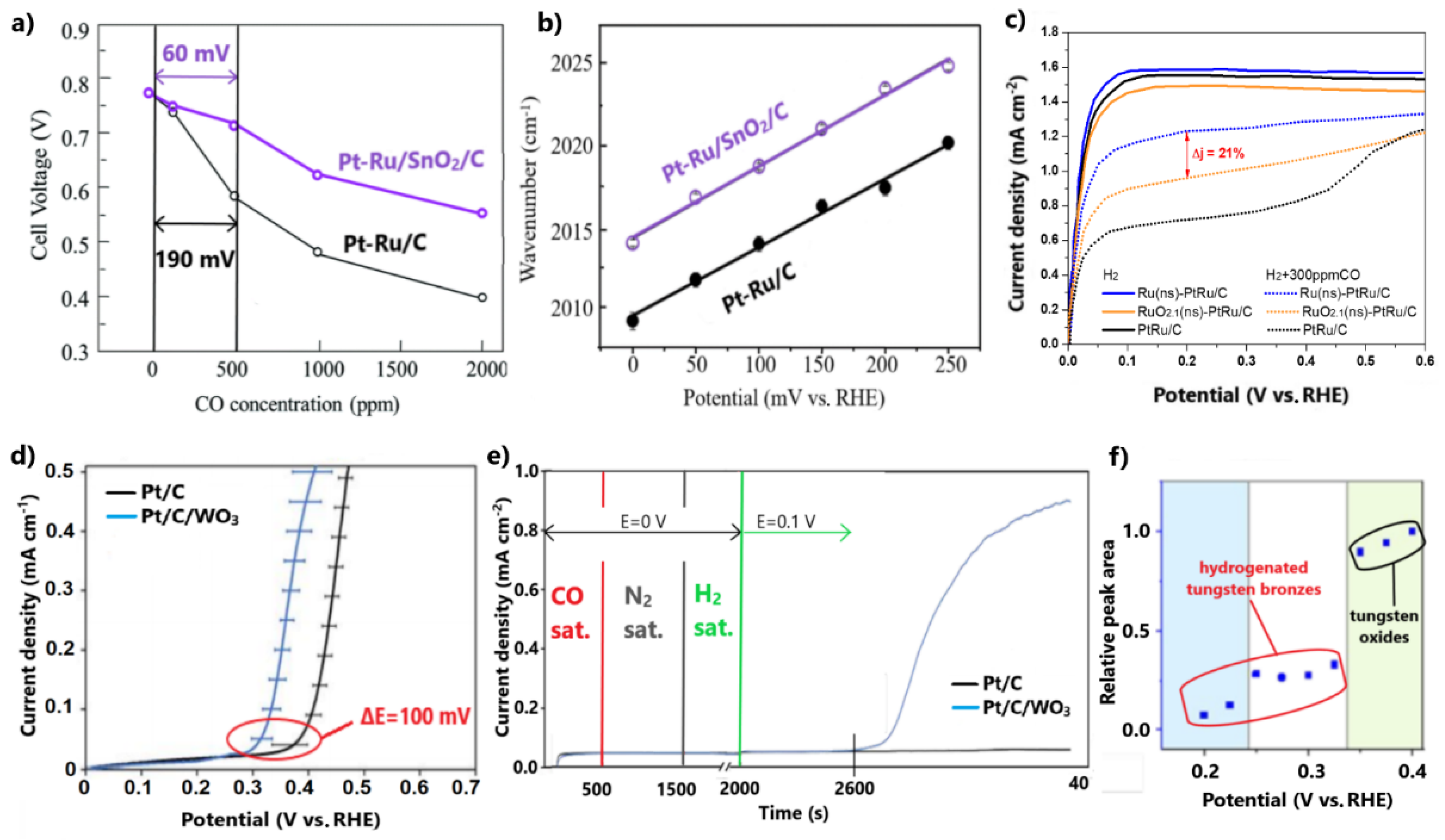
3.1.3. Combinations with Metal Oxides
3.1.4. Surface-Modified Pt/C
3.2. Pt-Based Electrocatalysts Supported on Alternative Materials
3.2.1. Advanced Carbonaceous Supports
3.2.2. Transition Metal Oxides and Carbides
4. Outlook
5. Concluding Remarks
- The optimal regulation of the alloying degree of Pt alloys via appropriate synthesis pathways can considerably improve CO tolerance and stability; however, since the added non-noble metals are exposed on the catalyst surface in the alloyed structures, Pt alloys do not generally present adequate stability for practical utilization. Nevertheless, examination of the Pt alloys is essential to reveal the correlations between the physicochemical properties, stability, and CO tolerance mechanisms;
- The combination of Pt-based electrocatalysts with TMOs can enhance the CO tolerance through the promotion of the bifunctional mechanism via the oxygenated species on their surfaces; however, to avoid the blockage of the Pt’s active surface and the dissolution of metal oxides, the structure of the developed electrocatalysts should be suitably controlled;
- The surface-sensitive nature of CO electrooxidation enables the enhancement of the CO tolerance of Pt/C (susceptible to CO poisoning) by controlling its morphology and structure through suitable synthesis routes. The regulation of the lattice strain, electronic structure, metal NP size, terraces adlayers, surface defects, and exposed crystalline facets can positively affect the CO adsorption and CO oxidation;
- The substitution of conventional the carbon black support with advanced carbonaceous materials can improve the CO tolerance through better electronic interactions between the deposited metal NPs and the support. Additionally, the functionalization of the carbon surface via chemical pre-treatment or doping with elements promotes the bifunctional mechanism, accelerating CO oxidation;
- The utilization of Mo and W oxides and carbides as alternative catalyst supports can help avoid the use of the corrosive carbon and enhance CO tolerance, mainly through the bifunctional characteristics of Mo and W; however, to ensure adequate stability, Mo and W atoms must be incorporated sufficiently in the composite;
- Core–shell-structured CO-tolerant electrocatalysts are the most promising for practical applications. Generally, the encapsulation of non-noble-metal-based core within the Pt shell prevents its leaching, resulting in excellent stability improvements. Concurrently, through the electronic modification introduced by the core to the Pt shell, the CO adsorption is significantly suppressed. Novel approaches related to 2D material shells, such as the use of hexagonal boron nitride, present exceptional potential for further research. Similar behavior can be obtained with 2D material shells by modifying the surfaces of conventional electrocatalysts with organic compounds.
Author Contributions
Funding
Acknowledgments

Conflicts of Interest
References
- Sopian, K.; Daud, W.R.W. Challenges and future developments in proton exchange membrane fuel cells. Renew. Energy 2006, 31, 719–727. [Google Scholar] [CrossRef]
- Zhao, J.; Li, X. A review of polymer electrolyte membrane fuel cell durability for vehicular applications: Degradation modes and experimental techniques. Energy Convers. Manag. 2019, 199, 112022. [Google Scholar] [CrossRef]
- Bernardo, G.; Araújo, T.; da Silva Lopes, T.; Sousa, J.; Mendes, A. Recent advances in membrane technologies for hydrogen purification. Int. J. Hydrog. Energy 2020, 45, 7313–7338. [Google Scholar] [CrossRef]
- Nikolaidis, P.; Poullikkas, A. A comparative overview of hydrogen production processes. Renew. Sustain. Energy Rev. 2017, 67, 597–611. [Google Scholar] [CrossRef]
- Kaiwen, L.; Bin, Y.; Tao, Z. Economic analysis of hydrogen production from steam reforming process: A literature review. Energy Sources Part B Econ. Plan. Policy 2018, 13, 109–115. [Google Scholar] [CrossRef]
- Cheng, X.; Shi, Z.; Glass, N.; Zhang, L.; Zhang, J.; Song, D.; Liu, Z.-S.; Wang, H.; Shen, J. A review of PEM hydrogen fuel cell contamination: Impacts, mechanisms, and mitigation. J. Power Sources 2007, 165, 739–756. [Google Scholar] [CrossRef]
- Zamel, N.; Li, X. Effect of contaminants on polymer electrolyte membrane fuel cells. Prog. Energy Combust. Sci. 2011, 37, 292–329. [Google Scholar] [CrossRef]
- Garbis, P.; Kern, C.; Jess, A. Kinetics and reactor design aspects of selective methanation of CO over a Ru/γ-Al2O3 catalyst in CO2/H2 rich gases. Energies 2019, 12, 469. [Google Scholar] [CrossRef]
- Matsuda, Y.; Shimizu, T.; Mitsushima, S. Adsorption behavior of low concentration carbon monoxide on polymer electrolyte fuel cell anodes for automotive applications. J. Power Sources 2016, 318, 1–8. [Google Scholar] [CrossRef]
- Li, Y.; Wang, X.; Mei, B.; Wang, Y.; Luo, Z.; Luo, E.; Yang, X.; Shi, Z.; Liang, L.; Jin, Z. Carbon monoxide powered fuel cell towards H2-onboard purification. Sci. Bull. 2021, 66, 1305–1311. [Google Scholar] [CrossRef]
- St-Pierre, J. PEMFC contaminant tolerance limit—CO in H2. Electrochim. Acta 2010, 55, 4208–4211. [Google Scholar] [CrossRef]
- Delgado, S.; Lagarteira, T.; Mendes, A. Air bleeding strategies to increase the efficiency of proton exchange membrane fuel cell stationary applications fuelled with CO ppm-levels. Int. J. Electrochem. Sci 2020, 15, 613–627. [Google Scholar] [CrossRef]
- Baschuk, J.; Li, X. Carbon monoxide poisoning of proton exchange membrane fuel cells. Int. J. Energy Res. 2001, 25, 695–713. [Google Scholar] [CrossRef]
- Zhang, C.; Shen, X.; Pan, Y.; Peng, Z. A review of Pt-based electrocatalysts for oxygen reduction reaction. Front. Energy 2017, 11, 268–285. [Google Scholar] [CrossRef]
- Igarashi, H.; Fujino, T.; Zhu, Y.; Uchida, H.; Watanabe, M. CO tolerance of Pt alloy electrocatalysts for polymer electrolyte fuel cells and the detoxification mechanism. Phys. Chem. Chem. Phys. 2001, 3, 306–314. [Google Scholar] [CrossRef]
- Lopes, P.P.; Freitas, K.S.; Ticianelli, E.A. CO tolerance of PEMFC anodes: Mechanisms and electrode designs. Electrocatalysis 2010, 1, 200–212. [Google Scholar] [CrossRef]
- Ren, X.; Lv, Q.; Liu, L.; Liu, B.; Wang, Y.; Liu, A.; Wu, G. Current progress of Pt and Pt-based electrocatalysts used for fuel cells. Sustain. Energy Fuels 2020, 4, 15–30. [Google Scholar] [CrossRef]
- Watanabe, M.; Tryk, D.A.; Wakisaka, M.; Yano, H.; Uchida, H. Overview of recent developments in oxygen reduction electrocatalysis. Electrochim. Acta 2012, 84, 187–201. [Google Scholar] [CrossRef]
- Zhao, Y.; Mao, Y.; Zhang, W.; Tang, Y.; Wang, P. Reviews on the effects of contaminations and research methodologies for PEMFC. Int. J. Hydrog. Energy 2020, 45, 23174–23200. [Google Scholar] [CrossRef]
- Shabani, B.; Hafttananian, M.; Khamani, S.; Ramiar, A.; Ranjbar, A. Poisoning of proton exchange membrane fuel cells by contaminants and impurities: Review of mechanisms, effects, and mitigation strategies. J. Power Sources 2019, 427, 21–48. [Google Scholar] [CrossRef]
- Valdés-López, V.F.; Mason, T.; Shearing, P.R.; Brett, D.J. Carbon monoxide poisoning and mitigation strategies for polymer electrolyte membrane fuel cells–A review. Prog. Energy Combust. Sci. 2020, 79, 100842. [Google Scholar] [CrossRef]
- Ehteshami, S.M.M.; Chan, S.H. A review of electrocatalysts with enhanced CO tolerance and stability for polymer electrolyte membarane fuel cells. Electrochim. Acta 2013, 93, 334–345. [Google Scholar] [CrossRef]
- Durst, J.; Siebel, A.; Simon, C.; Hasche, F.; Herranz, J.; Gasteiger, H. New insights into the electrochemical hydrogen oxidation and evolution reaction mechanism. Energy Environ. Sci. 2014, 7, 2255–2260. [Google Scholar] [CrossRef]
- Tzorbatzoglou, F.; Brouzgou, A.; Jing, S.; Wang, Y.; Song, S.; Tsiakaras, P. Oxygen reduction and hydrogen oxidation reaction on novel carbon supported PdxIry electrocatalysts. Int. J. Hydrog. Energy 2018, 43, 11766–11777. [Google Scholar] [CrossRef]
- Lei, H.-Y.; Piao, J.-H.; Brouzgou, A.; Gorbova, E.; Tsiakaras, P.; Liang, Z.-X. Synthesis of nitrogen-doped mesoporous carbon nanosheets for oxygen reduction electrocatalytic activity enhancement in acid and alkaline media. Int. J. Hydrog. Energy 2019, 44, 4423–4431. [Google Scholar] [CrossRef]
- Wang, Y.; Song, S.; Maragou, V.; Shen, P.K.; Tsiakaras, P. High surface area tungsten carbide microspheres as effective Pt catalyst support for oxygen reduction reaction. Appl. Catal. B Environ. 2009, 89, 223–228. [Google Scholar] [CrossRef]
- Durst, J.; Simon, C.; Hasché, F.; Gasteiger, H.A. Hydrogen oxidation and evolution reaction kinetics on carbon supported Pt, Ir, Rh, and Pd electrocatalysts in acidic media. J. Electrochem. Soc. 2014, 162, F190. [Google Scholar] [CrossRef]
- Tzorbatzoglou, F.; Brouzgou, A.; Tsiakaras, P. Electrocatalytic activity of Vulcan-XC-72 supported Pd, Rh and PdxRhy toward HOR and ORR. Appl. Catal. B Environ. 2015, 174, 203–211. [Google Scholar] [CrossRef]
- Sheng, W.; Gasteiger, H.A.; Shao-Horn, Y. Hydrogen oxidation and evolution reaction kinetics on platinum: Acid vs. alkaline electrolytes. J. Electrochem. Soc. 2010, 157, B1529. [Google Scholar] [CrossRef]
- Jiménez, S.; Soler, J.; Valenzuela, R.; Daza, L. Assessment of the performance of a PEMFC in the presence of CO. J. Power Sources 2005, 151, 69–73. [Google Scholar] [CrossRef]
- Brouzgou, A.; Podias, A.; Tsiakaras, P. PEMFCs and AEMFCs directly fed with ethanol: A current status comparative review. J. Appl. Electrochem. 2013, 43, 119–136. [Google Scholar] [CrossRef]
- Camara, G.; Ticianelli, E.; Mukerjee, S.; Lee, S.; McBreen, J. The CO poisoning mechanism of the hydrogen oxidation reaction in proton exchange membrane fuel cells. J. Electrochem. Soc. 2002, 149, A748. [Google Scholar] [CrossRef]
- Grgur, B.N.; Marković, N.M.; Lucas, C.A.; Ross, P.N., Jr. Electrochemical oxidation of carbon monoxide: From platinum single crystals to low temperature fuel cells catalysts. Part I: Carbon monoxide oxidation onto low index platinum single crystals. J. Serbian Chem. Soc. 2001, 66, 785–797. [Google Scholar] [CrossRef]
- Bilondi, A.M.; Abdollahzadeh, M.; Kermani, M.; Heidary, H.; Havaej, P. Numerical study of anode side CO contamination effects on PEM fuel cell performance; and mitigation methods. Energy Convers. Manag. 2018, 177, 519–534. [Google Scholar] [CrossRef]
- Wee, J.-H.; Lee, K.-Y. Overview of the development of CO-tolerant anode electrocatalysts for proton-exchange membrane fuel cells. J. Power Sources 2006, 157, 128–135. [Google Scholar] [CrossRef]
- Ehteshami, S.M.M.; Jia, Q.; Halder, A.; Chan, S.; Mukerjee, S. The role of electronic properties of Pt and Pt alloys for enhanced reformate electro-oxidation in polymer electrolyte membrane fuel cells. Electrochim. Acta 2013, 107, 155–163. [Google Scholar] [CrossRef]
- Gasteiger, H.A.; Markovic, N.M.; Ross, P.N., Jr. H2 and CO electrooxidation on well-characterized Pt, Ru, and Pt-Ru. 1. Rotating disk electrode studies of the pure gases including temperature effects. J. Phys. Chem. 1995, 99, 8290–8301. [Google Scholar] [CrossRef]
- Grgur, B.N.; Marković, N.M.; Ross, P.N., Jr. Electrochemical oxidation of carbon monoxide: From platinum single crystals to low temperature fuel cells catalysts–Part II: Electrooxidation of H2, CO and H2/CO mixtures on well characterized PtMo. J. Serbian Chem. Soc. 2003, 68, 191–205. [Google Scholar] [CrossRef]
- Mukerjee, S.; Urian, R.; Lee, S.; Ticianelli, E.A.; McBreen, J. Electrocatalysis of CO tolerance by carbon-supported PtMo electrocatalysts in PEMFCs. J. Electrochem. Soc. 2004, 151, A1094. [Google Scholar] [CrossRef][Green Version]
- Hammer, B.; Morikawa, Y.; Nørskov, J.K. CO chemisorption at metal surfaces and overlayers. Phys. Rev. Lett. 1996, 76, 2141. [Google Scholar] [CrossRef]
- Blyholder, G. Molecular orbital view of chemisorbed carbon monoxide. J. Phys. Chem. 1964, 68, 2772–2777. [Google Scholar] [CrossRef]
- Schiros, T.; Takahashi, O.; Andersson, K.J.; Öström, H.; Pettersson, L.G.; Nilsson, A.; Ogasawara, H. The role of substrate electrons in the wetting of a metal surface. J. Chem. Phys. 2010, 132, 094701. [Google Scholar] [CrossRef] [PubMed]
- Ray, N.K.; Anderson, A.B. Molecular orbital study of CO chemisorption and oxidation on a Pt (111) surface. Surf. Sci. 1982, 119, 35–45. [Google Scholar] [CrossRef]
- Gajdoš, M.; Eichler, A.; Hafner, J. CO adsorption on close-packed transition and noble metal surfaces: Trends from ab initio calculations. J. Phys. Condens. Matter 2004, 16, 1141. [Google Scholar] [CrossRef]
- Pettersson, L.G.M.; Nilsson, A. A molecular perspective on the d-band model: Synergy between experiment and theory. Top. Catal. 2014, 57, 2–13. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Abild-Pedersen, F.; Studt, F.; Bligaard, T. Density functional theory in surface chemistry and catalysis. Proc. Natl. Acad. Sci. USA 2011, 108, 937–943. [Google Scholar] [CrossRef]
- Christoffersen, E.; Liu, P.; Ruban, A.; Skriver, H.L.; Nørskov, J.K. Anode materials for low-temperature fuel cells: A density functional theory study. J. Catal. 2001, 199, 123–131. [Google Scholar] [CrossRef]
- Wakisaka, M.; Mitsui, S.; Hirose, Y.; Kawashima, K.; Uchida, H.; Watanabe, M. Electronic structures of Pt−Co and Pt−Ru alloys for CO-tolerant anode catalysts in polymer electrolyte fuel cells studied by EC−XPS. J. Phys. Chem. B 2006, 110, 23489–23496. [Google Scholar] [CrossRef]
- Lopes, P.P.; Ticianelli, E.A. The CO tolerance pathways on the Pt–Ru electrocatalytic system. J. Electroanal. Chem. 2010, 644, 110–116. [Google Scholar] [CrossRef]
- Liu, Z.; Jackson, G.S.; Eichhorn, B.W. PtSn Intermetallic, Core–Shell, and Alloy Nanoparticles as CO-Tolerant Electrocatalysts for H2 Oxidation. Angew. Chem. 2010, 122, 3241–3244. [Google Scholar] [CrossRef]
- Andreadis, G.; Podias, A.; Tsiakaras, P. The effect of the parasitic current on the direct ethanol PEM fuel cell operation. J. Power Sources 2008, 181, 214–227. [Google Scholar] [CrossRef]
- Liu, P.; Logadottir, A.; Nørskov, J.K. Modeling the electro-oxidation of CO and H2/CO on Pt, Ru, PtRu and Pt3Sn. Electrochim. Acta 2003, 48, 3731–3742. [Google Scholar] [CrossRef]
- Kitchin, J.; Nørskov, J.K.; Barteau, M.; Chen, J. Modification of the surface electronic and chemical properties of Pt (111) by subsurface 3d transition metals. J. Chem. Phys. 2004, 120, 10240–10246. [Google Scholar] [CrossRef]
- Shubina, T.; Koper, M. Quantum-chemical calculations of CO and OH interacting with bimetallic surfaces. Electrochim. Acta 2002, 47, 3621–3628. [Google Scholar] [CrossRef]
- Liu, Y. Is the free energy change of adsorption correctly calculated? J. Chem. Eng. Data 2009, 54, 1981–1985. [Google Scholar] [CrossRef]
- Koper, M.T.; Shubina, T.E.; van Santen, R.A. Periodic density functional study of CO and OH adsorption on Pt−Ru alloy surfaces: Implications for CO tolerant fuel cell catalysts. J. Phys. Chem. B 2002, 106, 686–692. [Google Scholar] [CrossRef]
- Liu, N.; Mao, J.; Yang, Z. Mitigation of CO poisoning on functionalized palladium monolayer supported on titanium carbide. Surf. Coat. Technol. 2020, 402, 125925. [Google Scholar] [CrossRef]
- Arab, A. DFT study of the interaction between carbon monoxide and Rh-Cu bimetallic nanoclusters. Mater. Today Commun. 2021, 26, 102013. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Bligaard, T.; Rossmeisl, J.; Christensen, C.H. Towards the computational design of solid catalysts. Nat. Chem. 2009, 1, 37–46. [Google Scholar] [CrossRef]
- Back, S.; Na, J.; Tran, K.; Ulissi, Z.W. In silico discovery of active, stable, CO-tolerant and cost-effective electrocatalysts for hydrogen evolution and oxidation. Phys. Chem. Chem. Phys. 2020, 22, 19454–19458. [Google Scholar] [CrossRef]
- Kwon, S.; Ham, D.J.; Kim, T.; Kwon, Y.; Lee, S.G.; Cho, M. Active methanol oxidation reaction by enhanced CO tolerance on bimetallic Pt/Ir electrocatalysts using electronic and bifunctional effects. ACS Appl. Mater. Interfaces 2018, 10, 39581–39589. [Google Scholar] [CrossRef]
- Mahata, A.; Nair, A.S.; Pathak, B. Recent advancements in Pt-nanostructure-based electrocatalysts for the oxygen reduction reaction. Catal. Sci. Technol. 2019, 9, 4835–4863. [Google Scholar] [CrossRef]
- Sui, S.; Wang, X.; Zhou, X.; Su, Y.; Riffat, S.; Liu, C.-J. A comprehensive review of Pt electrocatalysts for the oxygen reduction reaction: Nanostructure, activity, mechanism and carbon support in PEM fuel cells. J. Mater. Chem. A 2017, 5, 1808–1825. [Google Scholar] [CrossRef]
- Wang, C.; Markovic, N.M.; Stamenkovic, V.R. Advanced platinum alloy electrocatalysts for the oxygen reduction reaction. ACS Catal. 2012, 2, 891–898. [Google Scholar] [CrossRef]
- Liu, Z.; Ma, L.; Zhang, J.; Hongsirikarn, K.; Goodwin, J.G., Jr. Pt alloy electrocatalysts for proton exchange membrane fuel cells: A review. Catal. Rev. 2013, 55, 255–288. [Google Scholar] [CrossRef]
- Anićijević, D.D.V.; Nikolić, V.M.; Marčeta-Kaninski, M.P.; Pašti, I.A. Is platinum necessary for efficient hydrogen evolution?–DFT study of metal monolayers on tungsten carbide. Int. J. Hydrog. Energy 2013, 38, 16071–16079. [Google Scholar] [CrossRef]
- Taufany, F.; Pan, C.J.; Lai, F.J.; Chou, H.L.; Sarma, L.S.; Rick, J.; Lin, J.M.; Lee, J.F.; Tang, M.T.; Hwang, B.J. Relating the composition of PtxRu100−x/C nanoparticles to their structural aspects and electrocatalytic activities in the methanol oxidation reaction. Chem.–A Eur. J. 2013, 19, 905–915. [Google Scholar] [CrossRef]
- Brouzgou, A.; Seretis, A.; Song, S.; Shen, P.K.; Tsiakaras, P. CO tolerance and durability study of PtMe(Me = Ir or Pd) electrocatalysts for H2-PEMFC application. Int. J. Hydrog. Energy 2021, 46, 13865–13877. [Google Scholar] [CrossRef]
- Watanabe, M.; Igarashi, H.; Fujino, T. Design of CO tolerant anode catalysts for polymer electrolyte fuel cell. Electrochemistry 1999, 67, 1194–1196. [Google Scholar] [CrossRef]
- González-Hernández, M.; Antolini, E.; Perez, J. Synthesis, characterization and CO tolerance evaluation in PEMFCs of Pt2RuMo electrocatalysts. Catalysts 2019, 9, 61. [Google Scholar] [CrossRef]
- Lee, K.-S.; Jeon, T.-Y.; Yoo, S.J.; Park, I.-S.; Cho, Y.-H.; Kang, S.H.; Choi, K.H.; Sung, Y.-E. Effect of PtRu alloying degree on electrocatalytic activities and stabilities. Appl. Catal. B Environ. 2011, 102, 334–342. [Google Scholar] [CrossRef]
- Seo, A.; Lee, J.; Han, K.; Kim, H. Performance and stability of Pt-based ternary alloy catalysts for PEMFC. Electrochim. Acta 2006, 52, 1603–1611. [Google Scholar] [CrossRef]
- Hassan, A.; Ticianelli, E.A. Activity and Stability of Dispersed Multi Metallic Pt-based Catalysts for CO Tolerance in Proton Exchange Membrane Fuel Cell Anodes. Anais Acad. Bras. Ciênc. 2018, 90, 697–718. [Google Scholar] [CrossRef] [PubMed]
- Bezerra, C.W.; Zhang, L.; Liu, H.; Lee, K.; Marques, A.L.; Marques, E.P.; Wang, H.; Zhang, J. A review of heat-treatment effects on activity and stability of PEM fuel cell catalysts for oxygen reduction reaction. J. Power Sources 2007, 173, 891–908. [Google Scholar] [CrossRef]
- Mukerjee, S.; Lee, S.; Ticianelli, E.; McBreen, J.; Grgur, B.; Markovic, N.; Ross, P.; Giallombardo, J.; De Castro, E. Investigation of enhanced CO tolerance in proton exchange membrane fuel cells by carbon supported PtMo alloy catalyst. Electrochem. Solid State Lett. 1998, 2, 12. [Google Scholar] [CrossRef]
- Santiago, E.I.; Camara, G.A.; Ticianelli, E.A. CO tolerance on PtMo/C electrocatalysts prepared by the formic acid method. Electrochim. Acta 2003, 48, 3527–3534. [Google Scholar] [CrossRef]
- Takeguchi, T.; Sirinutsomboon, B.; Wongsawa, T.; Ui, K. Effect of Valency of Sn in Sn-Modified Pt-Ru Catalyst on CO Tolerance and HOR Activity for PEFC. ECS Trans. 2017, 77, 1375. [Google Scholar] [CrossRef]
- Santiago, E.I.; Batista, M.S.; Assaf, E.M.; Ticianelli, E.A. Mechanism of CO tolerance on molybdenum-based electrocatalysts for PEMFC. J. Electrochem. Soc. 2004, 151, A944. [Google Scholar] [CrossRef]
- Huang, H.; Nassr, A.B.A.A.; Celorrio, V.; Taylor, S.R.; Puthiyapura, V.K.; Hardacre, C.; Brett, D.J.; Russell, A.E. Effects of heat treatment atmosphere on the structure and activity of Pt3Sn nanoparticle electrocatalysts: A characterisation case study. Faraday Discuss. 2018, 208, 555–573. [Google Scholar] [CrossRef]
- Zhu, M.; Sun, G.; Xin, Q. Effect of alloying degree in PtSn catalyst on the catalytic behavior for ethanol electro-oxidation. Electrochim. Acta 2009, 54, 1511–1518. [Google Scholar] [CrossRef]
- Venkataraman, R.; Kunz, H.; Fenton, J. Development of new CO tolerant ternary anode catalysts for proton exchange membrane fuel cells. J. Electrochem. Soc. 2003, 150, A278. [Google Scholar] [CrossRef]
- Liang, Y.; Zhang, H.; Zhong, H.; Zhu, X.; Tian, Z.; Xu, D.; Yi, B. Preparation and characterization of carbon-supported PtRuIr catalyst with excellent CO-tolerant performance for proton-exchange membrane fuel cells. J. Catal. 2006, 238, 468–476. [Google Scholar] [CrossRef]
- Papageorgopoulos, D.; Keijzer, M.; De Bruijn, F. The inclusion of Mo, Nb and Ta in Pt and PtRu carbon supported electrocatalysts in the quest for improved CO tolerant PEMFC anodes. Electrochim. Acta 2002, 48, 197–204. [Google Scholar] [CrossRef]
- Garcia, A.C.; Paganin, V.A.; Ticianelli, E.A. CO tolerance of PdPt/C and PdPtRu/C anodes for PEMFC. Electrochim. Acta 2008, 53, 4309–4315. [Google Scholar] [CrossRef]
- Bortoloti, F.; Garcia, A.; Angelo, A. Electronic effect in intermetallic electrocatalysts with low susceptibility to CO poisoning during hydrogen oxidation. Int. J. Hydrog. Energy 2015, 40, 10816–10824. [Google Scholar] [CrossRef]
- Lim, D.-H.; Choi, D.-H.; Lee, W.-D.; Lee, H.-I. A new synthesis of a highly dispersed and CO tolerant PtSn/C electrocatalyst for low-temperature fuel cell; its electrocatalytic activity and long-term durability. Appl. Catal. B Environ. 2009, 89, 484–493. [Google Scholar] [CrossRef]
- Wang, K.; Wang, Y.; Liang, Z.; Liang, Y.; Wu, D.; Song, S.; Tsiakaras, P. Ordered mesoporous tungsten carbide/carbon composites promoted Pt catalyst with high activity and stability for methanol electrooxidation. Appl. Catal. B Environ. 2014, 147, 518–525. [Google Scholar] [CrossRef]
- Zhang, L.; Shi, W.; Zhang, B. A review of electrocatalyst characterization by transmission electron microscopy. J. Energy Chem. 2017, 26, 1117–1135. [Google Scholar] [CrossRef]
- Gong, W.; Jiang, Z.; Wu, R.; Liu, Y.; Huang, L.; Hu, N.; Tsiakaras, P.; Shen, P.K. Cross-double dumbbell-like Pt–Ni nanostructures with enhanced catalytic performance toward the reactions of oxygen reduction and methanol oxidation. Appl. Catal. B Environ. 2019, 246, 277–283. [Google Scholar] [CrossRef]
- Jiang, R.; Tung, S.; Tang, Z.; Li, L.; Ding, L.; Xi, X.; Liu, Y.; Zhang, L.; Zhang, J. A review of core-shell nanostructured electrocatalysts for oxygen reduction reaction. Energy Storage Mater. 2018, 12, 260–276. [Google Scholar] [CrossRef]
- Ochal, P.; de la Fuente, J.L.G.; Tsypkin, M.; Seland, F.; Sunde, S.; Muthuswamy, N.; Rønning, M.; Chen, D.; Garcia, S.; Alayoglu, S. CO stripping as an electrochemical tool for characterization of Ru@Pt core-shell catalysts. J. Electroanal. Chem. 2011, 655, 140–146. [Google Scholar] [CrossRef]
- Zhang, L.; Kim, J.; Chen, H.M.; Nan, F.; Dudeck, K.; Liu, R.-S.; Botton, G.A.; Zhang, J. A novel CO-tolerant PtRu core–shell structured electrocatalyst with Ru rich in core and Pt rich in shell for hydrogen oxidation reaction and its implication in proton exchange membrane fuel cell. J. Power Sources 2011, 196, 9117–9123. [Google Scholar] [CrossRef]
- Aricò, A.S.; Stassi, A.; D’Urso, C.; Sebastián, D.; Baglio, V. Synthesis of Pd3Co1@Pt/C Core-Shell Catalysts for Methanol-Tolerant Cathodes of Direct Methanol Fuel Cells. Chem.—A Eur. J. 2014, 20, 10679–10684. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Park, S.; Kim, H. Preparation of CO-tolerant PtRuNi/C ternary electrocatalyst having a composition gradient shell. Chem. Eng. J. 2021, 414, 128792. [Google Scholar] [CrossRef]
- Sun, M.; Lv, Y.; Song, Y.; Wu, H.; Wang, G.; Zhang, H.; Chen, M.; Fu, Q.; Bao, X. CO-tolerant PtRu@h-BN/C core–shell electrocatalysts for proton exchange membrane fuel cells. Appl. Surf. Sci. 2018, 450, 244–250. [Google Scholar] [CrossRef]
- Sun, M.; Dong, J.; Lv, Y.; Zhao, S.; Meng, C.; Song, Y.; Wang, G.; Li, J.; Fu, Q.; Tian, Z. Pt@h-BN core–shell fuel cell electrocatalysts with electrocatalysis confined under outer shells. Nano Res. 2018, 11, 3490–3498. [Google Scholar] [CrossRef]
- Zhang, H.; Xu, L.; Tian, Y.; Jiao, A.; Li, S.; Liu, X.; Chen, M.; Chen, F. Convenient synthesis of 3D fluffy PtPd nanocorals loaded on 2D h-BN supports as highly efficient and stable electrocatalysts for alcohol oxidation reaction. ACS Omega 2019, 4, 11163–11172. [Google Scholar] [CrossRef]
- Shi, G.; Yano, H.; Tryk, D.; Watanabe, M.; Iiyama, A.; Uchida, H. A novel Pt–Co alloy hydrogen anode catalyst with superlative activity, CO-tolerance and robustness. Nanoscale 2016, 8, 13893–13897. [Google Scholar] [CrossRef]
- Shi, G.; Yano, H.; Tryk, D.A.; Iiyama, A.; Uchida, H. Highly active, CO-tolerant, and robust hydrogen anode catalysts: Pt–M(M = Fe, Co, Ni) alloys with stabilized Pt-skin layers. ACS Catal. 2017, 7, 267–274. [Google Scholar] [CrossRef]
- Shi, G.; Yano, H.; Tryk, D.A.; Iiyama, A.; Uchida, H. Effect of core-alloy composition and particle size of stabilized Pt Skin/PtCo alloy nanocatalysts on the CO-Tolerant hydrogen oxidation electrocatalysis. Electrochim. Acta 2019, 328, 135056. [Google Scholar] [CrossRef]
- Tran, D.T.; Kshetri, T.; Chuong, N.D.; Gautam, J.; Van Hien, H.; Kim, N.H.; Lee, J.H. Emerging core-shell nanostructured catalysts of transition metal encapsulated by two-dimensional carbon materials for electrochemical applications. Nano Today 2018, 22, 100–131. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, J.; Gu, J.; Su, L.; Cheng, L. An overview of metal oxide materials as electrocatalysts and supports for polymer electrolyte fuel cells. Energy Environ. Sci. 2014, 7, 2535–2558. [Google Scholar] [CrossRef]
- Zhu, Y.; Lin, Q.; Zhong, Y.; Tahini, H.A.; Shao, Z.; Wang, H. Metal oxide-based materials as an emerging family of hydrogen evolution electrocatalysts. Energy Environ. Sci. 2020, 13, 3361–3392. [Google Scholar] [CrossRef]
- Takeguchi, T.; Kunifuji, A.; Narischat, N.; Ito, M.; Noguchi, H.; Uosaki, K.; Mukai, S.R. Ligand effect of SnO2 on a Pt–Ru catalyst and the relationship between bond strength and CO tolerance. Catal. Sci. Technol. 2016, 6, 3214–3219. [Google Scholar] [CrossRef]
- Takimoto, D.; Ohnishi, T.; Ayato, Y.; Mochizuki, D.; Sugimoto, W. Suppression of CO adsorption on PtRu/C catalysts modified with metallic ruthenium nanosheets. J. Electrochem. Soc. 2016, 163, F367. [Google Scholar] [CrossRef]
- Stewart, D.W.; Scott, K.; Wain, A.J.; Rosser, T.E.; Brightman, E.; Macphee, D.; Mamlouk, M. The role of tungsten oxide in enhancing the carbon monoxide tolerance of platinum-based hydrogen oxidation catalysts. ACS Appl. Mater. Interfaces 2020, 12, 37079–37091. [Google Scholar] [CrossRef]
- Ciapina, E.G.; Santos, S.F.; Gonzalez, E.R. Electrochemical CO stripping on nanosized Pt surfaces in acid media: A review on the issue of peak multiplicity. J. Electroanal. Chem. 2018, 815, 47–60. [Google Scholar] [CrossRef]
- Koper, M.T. Structure sensitivity and nanoscale effects in electrocatalysis. Nanoscale 2011, 3, 2054–2073. [Google Scholar] [CrossRef]
- Ramos, A.; Santos, M.; Godoi, C.; de Queiroz, L.; Nandenha, J.; Fontes, E.; Brito, W.; Machado, M.; Neto, A.; de Souza, R. High CO tolerance of Pt nanoparticles synthesized by sodium borohydride in a time-domain NMR spectrometer. Int. J. Hydrog. Energy 2020, 45, 22973–22978. [Google Scholar] [CrossRef]
- Wang, T.; Chen, Z.-X.; Yu, S.; Sheng, T.; Ma, H.-B.; Chen, L.-N.; Rauf, M.; Xia, H.-P.; Zhou, Z.-Y.; Sun, S.-G. Constructing canopy-shaped molecular architectures to create local Pt surface sites with high tolerance to H2S and CO for hydrogen electrooxidation. Energy Environ. Sci. 2018, 11, 166–171. [Google Scholar] [CrossRef]
- Yaldagard, M.; Jahanshahi, M.; Seghatoleslami, N. Carbonaceous nanostructured support materials for low temperature fuel cell electrocatalysts—A review. World J. Nano Sci. Eng. 2013, 2013, 40507. [Google Scholar] [CrossRef]
- Sharma, S.; Pollet, B.G. Support materials for PEMFC and DMFC electrocatalysts—A review. J. Power Sources 2012, 208, 96–119. [Google Scholar] [CrossRef]
- Shrestha, S.; Liu, Y.; Mustain, W.E. Electrocatalytic activity and stability of Pt clusters on state-of-the-art supports: A review. Catal. Rev. 2011, 53, 256–336. [Google Scholar] [CrossRef]
- Narischat, N.; Takeguchi, T.; Mori, T.; Iwamura, S.; Ogino, I.; Mukai, S.R.; Ueda, W. Effect of the mesopores of carbon supports on the CO tolerance of Pt2Ru3 polymer electrolyte fuel cell anode catalyst. Int. J. Hydrog. Energy 2016, 41, 13697–13704. [Google Scholar] [CrossRef]
- He, C.; Song, S.; Liu, J.; Maragou, V.; Tsiakaras, P. KOH-activated multi-walled carbon nanotubes as platinum supports for oxygen reduction reaction. J. Power Sources 2010, 195, 7409–7414. [Google Scholar] [CrossRef]
- Tan, A.-D.; Wang, Y.-F.; Fu, Z.-Y.; Tsiakaras, P.; Liang, Z.-X. Highly effective oxygen reduction reaction electrocatalysis: Nitrogen-doped hierarchically mesoporous carbon derived from interpenetrated nonporous metal-organic frameworks. Appl. Catal. B Environ. 2017, 218, 260–266. [Google Scholar] [CrossRef]
- Sebastián, D.; Suelves, I.; Moliner, R.; Lázaro, M. The effect of the functionalization of carbon nanofibers on their electronic conductivity. Carbon 2010, 48, 4421–4431. [Google Scholar] [CrossRef]
- Wang, S.; Cochell, T.; Manthiram, A. Boron-doped carbon nanotube-supported Pt nanoparticles with improved CO tolerance for methanol electro-oxidation. Phys. Chem. Chem. Phys. 2012, 14, 13910–13913. [Google Scholar] [CrossRef] [PubMed]
- Tsiouvaras, N.; Martínez-Huerta, M.; Moliner, R.; Lázaro, M.; Rodríguez, J.; Pastor, E.; Pena, M.; Fierro, J. CO tolerant PtRu–MoOx nanoparticles supported on carbon nanofibers for direct methanol fuel cells. J. Power Sources 2009, 186, 299–304. [Google Scholar] [CrossRef]
- González-Hernández, M.; Antolini, E.; Perez, J. CO tolerance and stability of PtRu and PtRuMo electrocatalysts supported on N-doped graphene nanoplatelets for polymer electrolyte membrane fuel cells. Int. J. Hydrog. Energy 2020, 45, 5276–5284. [Google Scholar] [CrossRef]
- González-Hernández, M.; Antolini, E.; Perez, J. CO Tolerance and Stability of Graphene and N-Doped Graphene Supported Pt Anode Electrocatalysts for Polymer Electrolyte Membrane Fuel Cells. Catalysts 2020, 10, 597. [Google Scholar] [CrossRef]
- Escudero-Cid, R.; Varela, A.; Hernández-Fernández, P.; Fatás, E.; Ocón, P. The effect of functionalised multi-walled carbon nanotubes in the hydrogen electrooxidation reaction in reactive currents impurified with CO. Int. J. Hydrog. Energy 2014, 39, 5063–5073. [Google Scholar] [CrossRef]
- Liu, Y.; Kelly, T.G.; Chen, J.G.; Mustain, W.E. Metal carbides as alternative electrocatalyst supports. ACS Catal. 2013, 3, 1184–1194. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Shao, Y.; Yin, G.; Lin, Y. Recent progress in nanostructured electrocatalysts for PEM fuel cells. J. Mater. Chem. A 2013, 1, 4631–4641. [Google Scholar] [CrossRef]
- Hassan, A.; Paganin, V.A.; Ticianelli, E.A. Pt modified tungsten carbide as anode electrocatalyst for hydrogen oxidation in proton exchange membrane fuel cell: CO tolerance and stability. Appl. Catal. B Environ. 2015, 165, 611–619. [Google Scholar] [CrossRef]
- Wang, D.; Subban, C.V.; Wang, H.; Rus, E.; DiSalvo, F.J.; Abruña, H.D. Highly stable and CO-tolerant Pt/Ti0.7W0.3O2 electrocatalyst for proton-exchange membrane fuel cells. J. Am. Chem. Soc. 2010, 132, 10218–10220. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.; Paganin, V.A.; Ticianelli, E.A. Investigation of carbon supported PtW catalysts as CO tolerant anodes at high temperature in proton exchange membrane fuel cell. J. Power Sources 2016, 325, 375–382. [Google Scholar] [CrossRef]
- Brkovic, S.M.; Kaninski, M.P.M.; Lausevic, P.Z.; Saponjic, A.B.; Radulovic, A.M.; Rakic, A.A.; Pasti, I.A.; Nikolic, V.M. Non-stoichiometric tungsten-carbide-oxide-supported Pt–Ru anode catalysts for PEM fuel cells–From basic electrochemistry to fuel cell performance. Int. J. Hydrog. Energy 2020, 45, 13929–13938. [Google Scholar] [CrossRef]
- Vass, Á.; Borbáth, I.; Bakos, I.; Pászti, Z.; Sajó, I.; Tompos, A. Novel Pt electrocatalysts: Multifunctional composite supports for enhanced corrosion resistance and improved CO tolerance. Top. Catal. 2018, 61, 1300–1312. [Google Scholar] [CrossRef]
- Vass, Á.; Borbáth, I.; Bakos, I.; Pászti, Z.; Sáfrán, G.; Tompos, A. Stability issues of CO tolerant Pt-based electrocatalysts for polymer electrolyte membrane fuel cells: Comparison of Pt/Ti0.8Mo0.2O2–C with PtRu/C. React. Kinet. Mech. Catal. 2019, 126, 679–699. [Google Scholar] [CrossRef]
- Liu, J.; Jiang, L. Electrostatic self-assembly of Pt nanoparticles on hexagonal tungsten oxide as an active CO-tolerant hydrogen oxidation electrocatalyst. Int. J. Hydrog. Energy 2018, 43, 8944–8952. [Google Scholar] [CrossRef]
- Jackson, C.; Raymakers, L.; Mulder, M.; Kucernak, A. Assessing electrocatalyst hydrogen activity and CO tolerance: Comparison of performance obtained using the high mass transport ‘floating electrode’technique and in electrochemical hydrogen pumps. Appl. Catal. B Environ. 2020, 268, 118734. [Google Scholar] [CrossRef]
- Bakos, I.; Borbáth, I.; Vass, Á.; Pászti, Z.; Tompos, A. Design and investigation of molybdenum modified platinum surfaces for modeling of CO tolerant electrocatalysts. Top. Catal. 2018, 61, 1385–1395. [Google Scholar] [CrossRef]
- Li, H.; Knights, S.; Shi, Z.; Van Zee, J.W.; Zhang, J. Proton Exchange Membrane Fuel Cells: Contamination and Mitigation Strategies; CRC Press: Boca Raton, FL, USA, 2010. [Google Scholar]
- Schmidt, T.; Noeske, M.; Gasteiger, H.A.; Behm, R.; Britz, P.; Brijoux, W.; Bönnemann, H. Electrocatalytic activity of PtRu alloy colloids for CO and CO/H2 electrooxidation: Stripping voltammetry and rotating disk measurements. Langmuir 1997, 13, 2591–2595. [Google Scholar] [CrossRef]
- Olu, P.-Y.; Ohnishi, T.; Ayato, Y.; Mochizuki, D.; Sugimoto, W. Insights into the enhanced tolerance to carbon monoxide on model tungsten trioxide-decorated polycrystalline platinum electrode. Electrochem. Commun. 2016, 71, 69–72. [Google Scholar] [CrossRef]
- Kwon, K.; Jung, Y.; Ku, H.; Lee, K.H.; Kim, S.; Sohn, J.; Pak, C. CO-tolerant Pt–BeO as a novel anode electrocatalyst in proton exchange membrane fuel cells. Catalysts 2016, 6, 68. [Google Scholar] [CrossRef]
- Brković, S.M.; Nikolić, V.M.; Kaninski, M.P.M.; Pašti, I.A. Pt/C catalyst impregnated with tungsten-oxide–Hydrogen oxidation reaction vs. CO tolerance. Int. J. Hydrog. Energy 2019, 44, 13364–13372. [Google Scholar] [CrossRef]
- Lee, M.J.; Kang, J.S.; Kang, Y.S.; Chung, D.Y.; Shin, H.; Ahn, C.-Y.; Park, S.; Kim, M.-J.; Kim, S.; Lee, K.-S. Understanding the bifunctional effect for removal of CO poisoning: Blend of a platinum nanocatalyst and hydrous ruthenium oxide as a model system. ACS Catal. 2016, 6, 2398–2407. [Google Scholar] [CrossRef]
- Garg, A.; Milina, M.; Ball, M.; Zanchet, D.; Hunt, S.T.; Dumesic, J.A.; Román-Leshkov, Y. Transition-metal nitride core@noble-metal shell nanoparticles as highly CO tolerant catalysts. Angew. Chem. Int. Ed. 2017, 56, 8828–8833. [Google Scholar] [CrossRef]
- Obradović, M.D.; Gojković, S.L. CO tolerant Pt/Ru0.7Ti0.3O2 nanocatalyst for hydrogen oxidation reaction. Zašt. Mater. 2018, 59, 265–272. [Google Scholar] [CrossRef]
- Kurdin, K.A.; Kuznetsov, V.V.; Sinitsyn, V.V.; Galitskaya, E.A.; Filatova, E.A.; Belina, C.A.; Stevenson, K.J. Synthesis and characterization of Pt-HxMoO3 catalysts for CO-tolerant PEMFCs. Catal. Today 2020. In press. [Google Scholar] [CrossRef]
- Ogihara, Y.; Yano, H.; Watanabe, M.; Iiyama, A.; Uchida, H. Effect of an Sb-Doped SnO2 Support on the CO-Tolerance of Pt2Ru3 Nanocatalysts for Residential Fuel Cells. Catalysts 2016, 6, 139. [Google Scholar] [CrossRef]
- Takimoto, D.; Ohnishi, T.; Nutariya, J.; Shen, Z.; Ayato, Y.; Mochizuki, D.; Demortière, A.; Boulineau, A.; Sugimoto, W. Ru-core@Pt-shell nanosheet for fuel cell electrocatalysts with high activity and durability. J. Catal. 2017, 345, 207–215. [Google Scholar] [CrossRef]
- Gubán, D.; Tompos, A.; Bakos, I.; Vass, Á.; Pászti, Z.; Szabó, E.G.; Sajó, I.; Borbáth, I. Preparation of CO-tolerant anode electrocatalysts for polymer electrolyte membrane fuel cells. Int. J. Hydrog. Energy 2017, 42, 13741–13753. [Google Scholar] [CrossRef]
- Roh, G.; Lee, H.; Jeong, Y.; Kim, J.H.; Kim, H. Preparation of carbon-supported Pt–Ru core-shell nanoparticles using carbonized polydopamine and ozone for a CO tolerant electrocatalyst. Int. J. Hydrog. Energy 2019, 44, 21588–21596. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, J.; Han, G.; Du, C.; Sun, Y.; Du, L.; An, M.; Yin, G.; Gao, Y.; Song, Y. Superior catalytic performance and CO tolerance of Ru@Pt/C-TiO2 electrocatalyst toward methanol oxidation reaction. Appl. Surf. Sci. 2019, 473, 943–950. [Google Scholar] [CrossRef]
- Na, H.; Choi, H.; Oh, J.-W.; Jung, Y.S.; Cho, Y.S. Enhanced CO oxidation and cyclic activities in three-dimensional platinum/indium tin oxide/carbon black electrocatalysts processed by cathodic arc deposition. ACS Appl. Mater. Interfaces 2019, 11, 25179–25185. [Google Scholar] [CrossRef]
- Olu, P.-Y.; Ohnishi, T.; Mochizuki, D.; Sugimoto, W. Uncovering the real active sites of ruthenium oxide for the carbon monoxide electro-oxidation reaction on platinum: The catalyst acts as a co-catalyst. J. Electroanal. Chem. 2018, 810, 109–118. [Google Scholar] [CrossRef]
- Antoniassi, R.M.; Quiroz, J.; Barbosa, E.; Parreira, L.S.; Isidoro, R.A.; Spinace, E.; Silva, J.; Camargo, P.H. Improving the Electrocatalytic Activities and CO Tolerance of Pt NPs by Incorporating TiO2 Nanocubes onto Carbon Supports. ChemCatChem 2021, 13, 1931–1939. [Google Scholar] [CrossRef]
- Wang, H.; Yuan, X.-Z.; Li, H. PEM Fuel Cell Diagnostic Tools; CRC Press: Boca Raton, FL, USA, 2019. [Google Scholar]
- Anderson, A.B.; Grantscharova, E.; Seong, S. Systematic theoretical study of alloys of platinum for enhanced methanol fuel cell performance. J. Electrochem. Soc. 1996, 143, 2075. [Google Scholar] [CrossRef]
- Xin, H.; Linic, S. Communications: Exceptions to the d-band model of chemisorption on metal surfaces: The dominant role of repulsion between adsorbate states and metal d-states. J. Chem. Phys. 2010, 132, 221101. [Google Scholar] [CrossRef]
- Wang, Q.; Tao, H.; Li, Z.; Chen, C.; Liu, S.; Han, L.; Lu, X. Improving electrochemical activity of PtRu/SnO2/C catalyst by reduction treatment and alkaline etching. J. Energy Chem. 2016, 25, 811–816. [Google Scholar] [CrossRef]
- Ganesan, A.; Narayanasamy, M. Ultra-low loading of platinum in proton exchange membrane-based fuel cells: A brief review. Mater. Renew. Sustain. Energy 2019, 8, 18. [Google Scholar] [CrossRef]
- Iezzi, R.C.; Santos, R.D.; da Silva, G.C.; Paganin, V.A.; Ticianelli, E.A. CO tolerance and stability of proton exchange membrane fuel cells with Nafion® and Aquivion® membranes and Mo-based anode electrocatalysts. J. Braz. Chem. Soc. 2018, 29, 1094–1104. [Google Scholar] [CrossRef]
- Yazici, M.S.; Dursun, S.; Borbáth, I.; Tompos, A. Reformate gas composition and pressure effect on CO tolerant Pt/Ti0.8Mo0.2O2–C electrocatalyst for PEM fuel cells. Int. J. Hydrog. Energy 2021, 46, 13524–13533. [Google Scholar] [CrossRef]
- Vielstich, W.; Lamm, A.; Gasteiger, H. Handbook of Fuel Cells. Fundamentals, Technology, Applications; ETDEWEB World Energy Base: Oak Ridge, TN, USA, 2003.
- Zhang, T.; Wang, P.; Chen, H.; Pei, P. A review of automotive proton exchange membrane fuel cell degradation under start-stop operating condition. Appl. Energy 2018, 223, 249–262. [Google Scholar] [CrossRef]
- Brouzgou, A.; Song, S.; Tsiakaras, P. Low and non-platinum electrocatalysts for PEMFCs: Current status, challenges and prospects. Appl. Catal. B Environ. 2012, 127, 371–388. [Google Scholar] [CrossRef]
- Antolini, E.; Salgado, J.R.; Gonzalez, E.R. The stability of Pt–M(M = first row transition metal) alloy catalysts and its effect on the activity in low temperature fuel cells: A literature review and tests on a Pt–Co catalyst. J. Power Sources 2006, 160, 957–968. [Google Scholar] [CrossRef]
- Du, L.; Prabhakaran, V.; Xie, X.; Park, S.; Wang, Y.; Shao, Y. Low-PGM and PGM-free catalysts for proton exchange membrane fuel cells: Stability challenges and material solutions. Adv. Mater. 2021, 33, 1908232. [Google Scholar] [CrossRef] [PubMed]
- Prass, S.; Friedrich, K.A.; Zamel, N. Tolerance and recovery of ultralow-loaded platinum anode electrodes upon carbon monoxide and hydrogen sulfide exposure. Molecules 2019, 24, 3514. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
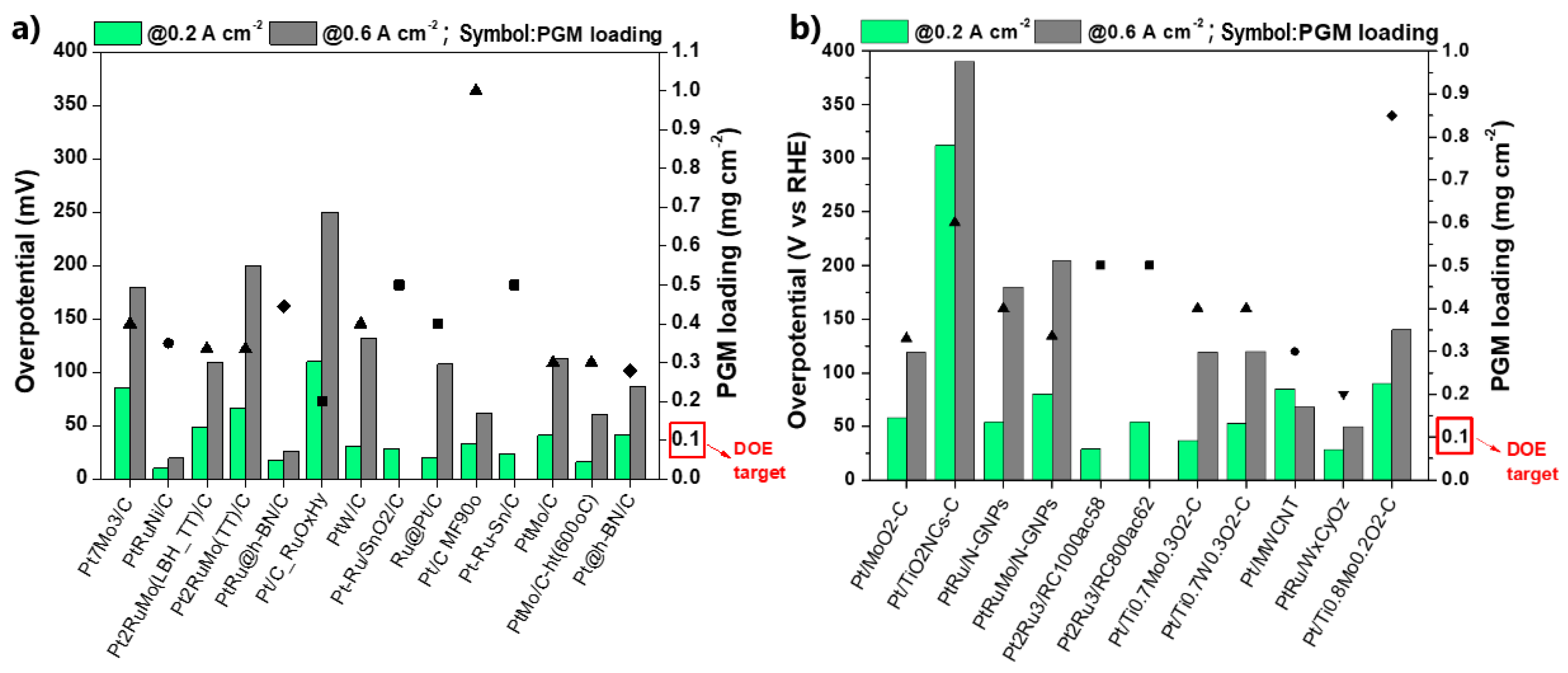{kind=link}
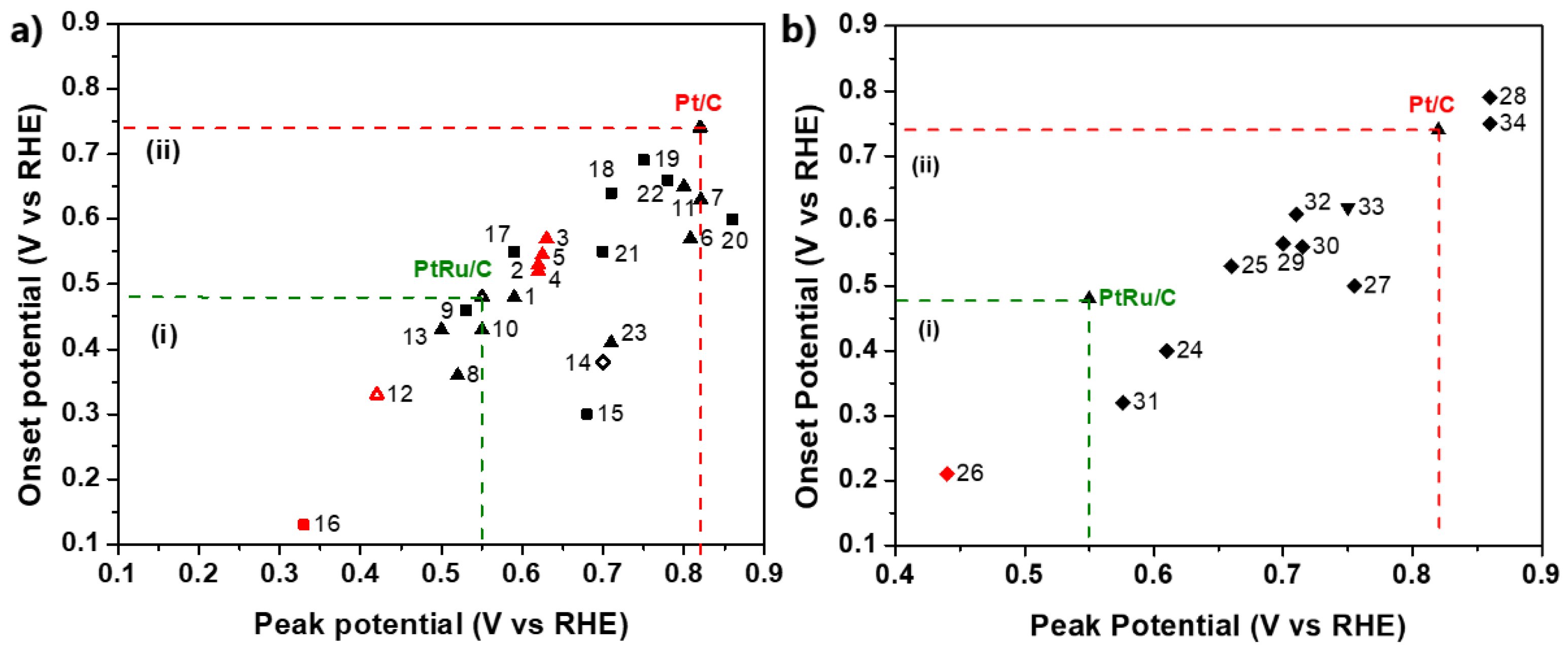
| Electrocatalyst | Experimental Conditions | Refs (Data) | Electrocatalyst | Experimental Conditions | Refs (Data) |
|---|---|---|---|---|---|
| PtRu@h-BN/C | 25 °C, 0.1 M HClO4, 20 mV/s | [95] (1) | WO3/Ptpc | 25 °C, 0.1 M HClO4, 50 mV/s | [136] (18) |
| PtxAL‒Pt69Co31/C | 70 °C, 0.1 M HClO4, 20 mV/s | [100] (2) | Pt-BeO/C | 25 °C, 0.1 M HClO4, 20 mV/s | [137] (19) |
| Pt2AL −PtFe/C | 70 °C, 0.1 M HClO4, 20 mV/s | [99] (3) | Pt/C+WOx | 25 °C, 0.5 M HClO4, 100 mV/s | [138] (20) |
| Pt2AL −PtCo/C | 70 °C, 0.1 M HClO4, 20 mV/s | [99] (4) | Pt/C-RuOxHy_ | 25 °C, 0.5 M H2SO4, 20 mV/s | [139] (21) |
| Pt2AL −PtNi/C | 70 °C, 0.1 M HClO4, 20 mV/s | [99] (5) | Pt/C/WO3 | 25 °C, 0.1 M H2SO4, 10 mV/s | [106] (22) |
| Pt/TiWN/C | 25 °C, 0.1 M HClO4, 50 mV/s | [140] (6) | PtRuNi/C | 25 °C, 0.5 M H2SO4, 10 mV/s | [94] (23) |
| Pt/TiWC/C | 25 °C, 0.1 M HClO4, 50 mV/s | [140] (7) | Pt/Ru0.7Ti0.3O2 | 25 °C, 0.1 M HClO4, 20 mV/s | [141] (24) |
| Ru(ns)-PtRu/C | 25 °C, 0.1 M HClO4, 10 mV/s | [105] (8) | Pt-HxMoO3 | 25 °C, 0.5 M H2SO4, 2 mV/s | [142](25) |
| RuO2.1ns-PtRu/C | 25 °C, 0.1 M HClO4, 10 mV/s | [105] (9) | Pt2Ru3/Sb-SnO2 | 70 °C, 0.1 M HClO4, 20 mV/s | [143] (26) |
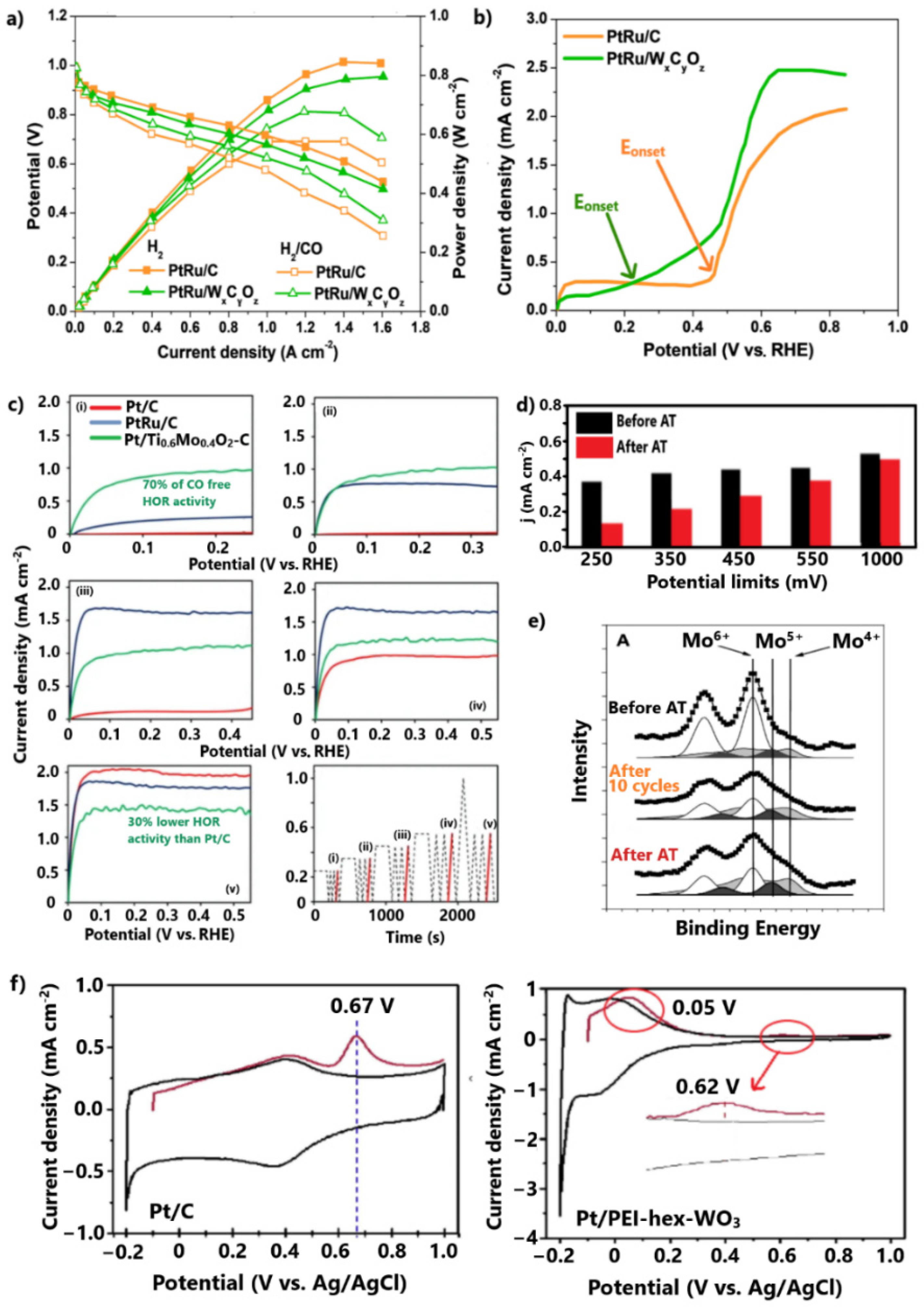
| Ru@Pt-1.5(ns)/C | 25 °C, 0.1 M HClO4, 10 mV/s | [144] (10) | PtRu/WxCyOz | 25 °C, 0.5 M HClO4, 100 mV/s | [128] (27) |
| Pt@h-BN/C | 25 °C, 0.1 M HClO4, 20 mV/s | [96] (11) | Pt/PEI-hex-WO3 | 25 °C, 0.5 M H2SO4, 20 mV/s | [131] (28) |
| Pt-Ru-Sn/C | 60 °C, 0.06 M HClO4, 10 mV/s | [77] (12) | Pt/Ti0.7W0.3O2-C | 25 °C, 0.5 M H2SO4, 10 mV/s | [145] (29) |
| Ru@Pt/C | 25 °C, 0.5 M H2SO4, 10 mV/s | [146] (13) | Pt/Ti0.7Mo0.3O2-C | 25 °C, 0.5 M H2SO4, 10 mV/s | [145] (30) |
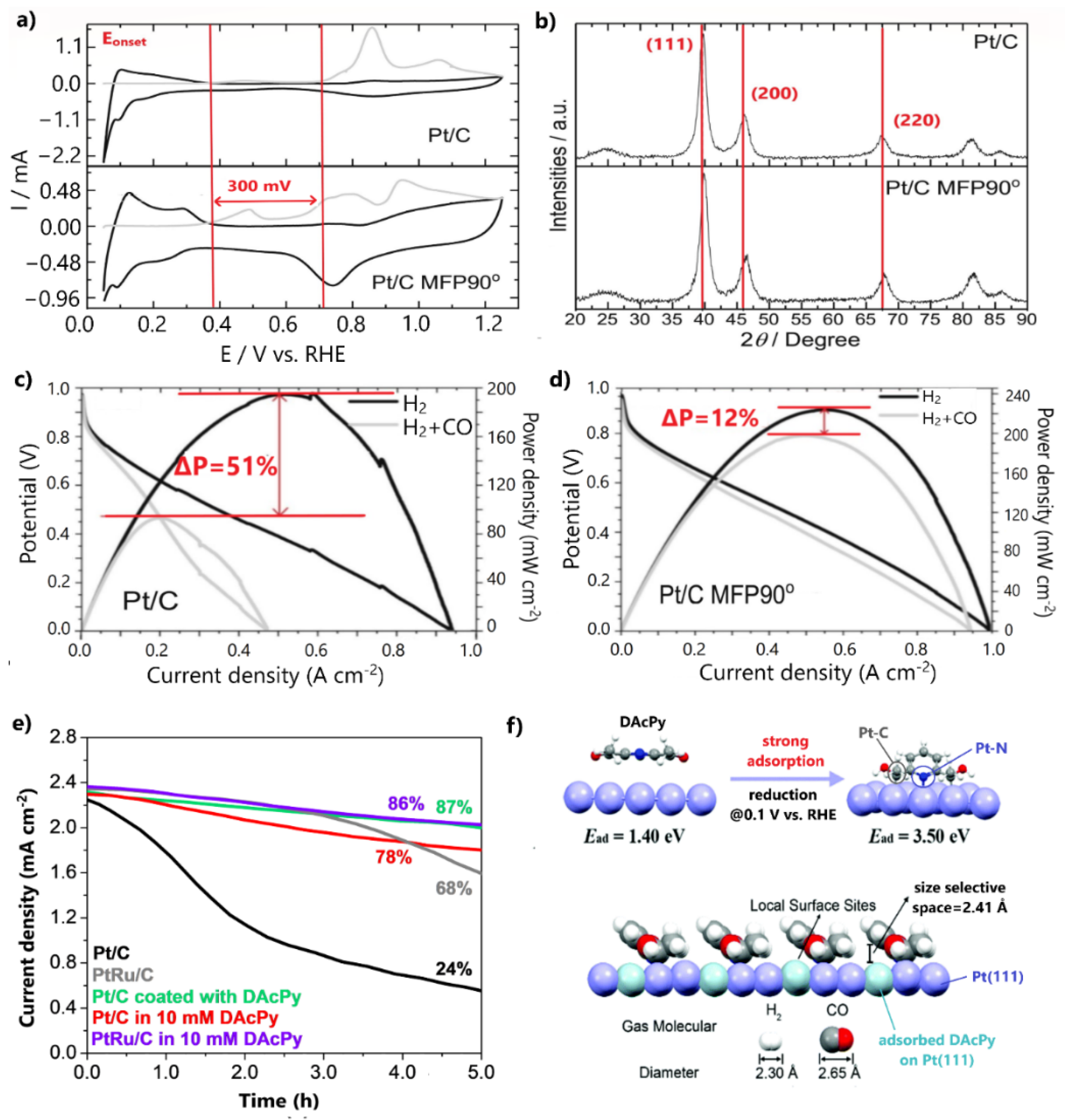
| Pt/C MFP90° | 25 °C, 0.5 M H2SO4, 10 mV/s | [109] (14) | Ru@Pt/C-TiO2 | 25 °C, 0.5 M H2SO4, 10 mV/s | [147] (31) |
| Pt/ITO/CB | 25 °C, 0.1 M HClO4, 20 mV/s | [148] (15) | Pt/Ti0.6Mo0.4O2-C | 25 °C, 0.5 M H2SO4, 10 mV/s | [130] (32) |
| PtRu/SnO2/C | 60 °C, 0.1 M HClO4, 10 mV/s | [104] (16) | Pt/N-GNP | 25 °C, 0.5 M H2SO4, 10 mV/s | [122] (33) |
| RuO2(ns)/Pt | 25 °C, 0.1 M HClO4, 50 mV/s | [149] (17) | Pt/TiO2NCs-C | 25 °C, 0.5 M H2SO4, 50 mV/s | [150] (34) |
| Electrocatalyst | Advantages | Challenges | Refs |
|---|---|---|---|
| PtRuNi/C (composition gradient shell) | - Only 11% performance decay after 10 ppm CO poisoning - Complete stability after 100 h of operation for 10 ppm CO/H2 feed | - Unknown CO tolerance under poisoning with greater than 10 ppm CO concentrations - Complicated synthesis method | [94] |
| PtxAL-Pt69Co31/C | - 95% HOR activity retention after 1000 ppm CO poisoning at 70 °C - 78% HOR activity retention after 1000 ppm CO poisoning and after a durability test of 4000 cycles | - Unknown CO tolerance behavior under practical H2-PEMFC operation | [100] |
| PtRu@h-BN/C | - Only 18 and 26 mV potential loss at 0.2 and 0.6 A cm−2, respectively, after 30 ppm CO + 25% CO2 poisoning - Thermochemically and electrochemically stable | - Unknown stability for long-term H2-PEMFC operation under CO poisoning - Complicated synthesis method | [95] |
| Pt/TixMo1-xO2-C (x = 0.8–0.6) | - CO oxidation at potentials smaller than 250 mV - 130 mV overpotential at 1 A cm−2 after 100 ppm CO poisoning | - Lower HOR activity in the absence of CO than Pt/C, due to lower support conductivity - Inadequate stability due to dissolution of non-incorporated Mo | [129,130,145] |
| Pt/C MFP90° | - Only 12% loss in maximum power density after 100 ppm CO poisoning | - High PGM loading | [109] |
| Pt/C coated with DAcPy | - 87% HOR activity retention after 5 h of continuous poisoning with 100 ppm CO | - High PGM loading - Unknown CO tolerance behavior under practical H2-PEMFC operation | [110] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Molochas, C.; Tsiakaras, P. Carbon Monoxide Tolerant Pt-Based Electrocatalysts for H2-PEMFC Applications: Current Progress and Challenges. Catalysts 2021, 11, 1127. https://doi.org/10.3390/catal11091127
Molochas C, Tsiakaras P. Carbon Monoxide Tolerant Pt-Based Electrocatalysts for H2-PEMFC Applications: Current Progress and Challenges. Catalysts. 2021; 11(9):1127. https://doi.org/10.3390/catal11091127
Chicago/Turabian StyleMolochas, Costas, and Panagiotis Tsiakaras. 2021. "Carbon Monoxide Tolerant Pt-Based Electrocatalysts for H2-PEMFC Applications: Current Progress and Challenges" Catalysts 11, no. 9: 1127. https://doi.org/10.3390/catal11091127
APA StyleMolochas, C., & Tsiakaras, P. (2021). Carbon Monoxide Tolerant Pt-Based Electrocatalysts for H2-PEMFC Applications: Current Progress and Challenges. Catalysts, 11(9), 1127. https://doi.org/10.3390/catal11091127