
A ‘Defective’ Conjugated Porous Poly-Azo as Dual Photocatalyst




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
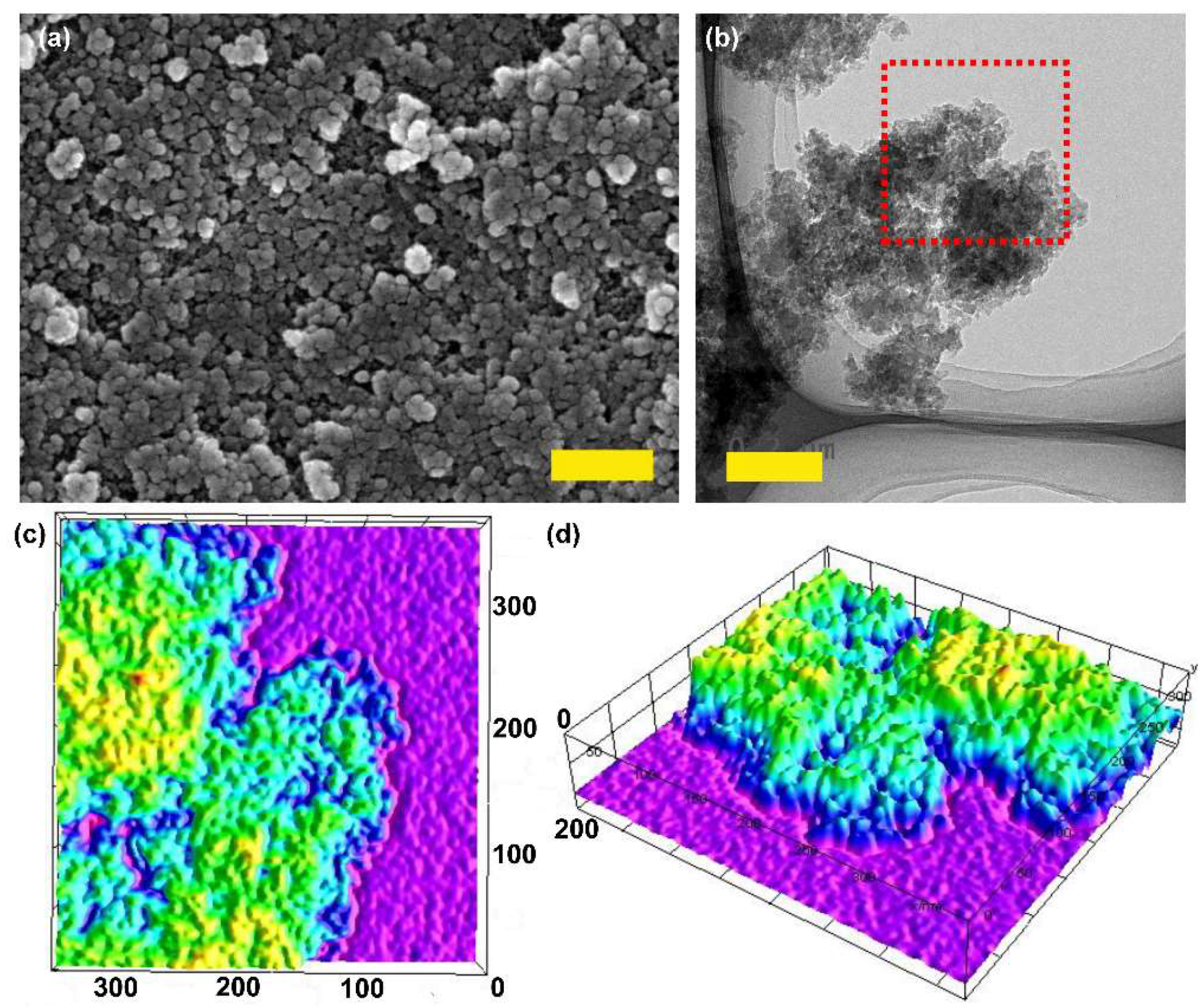
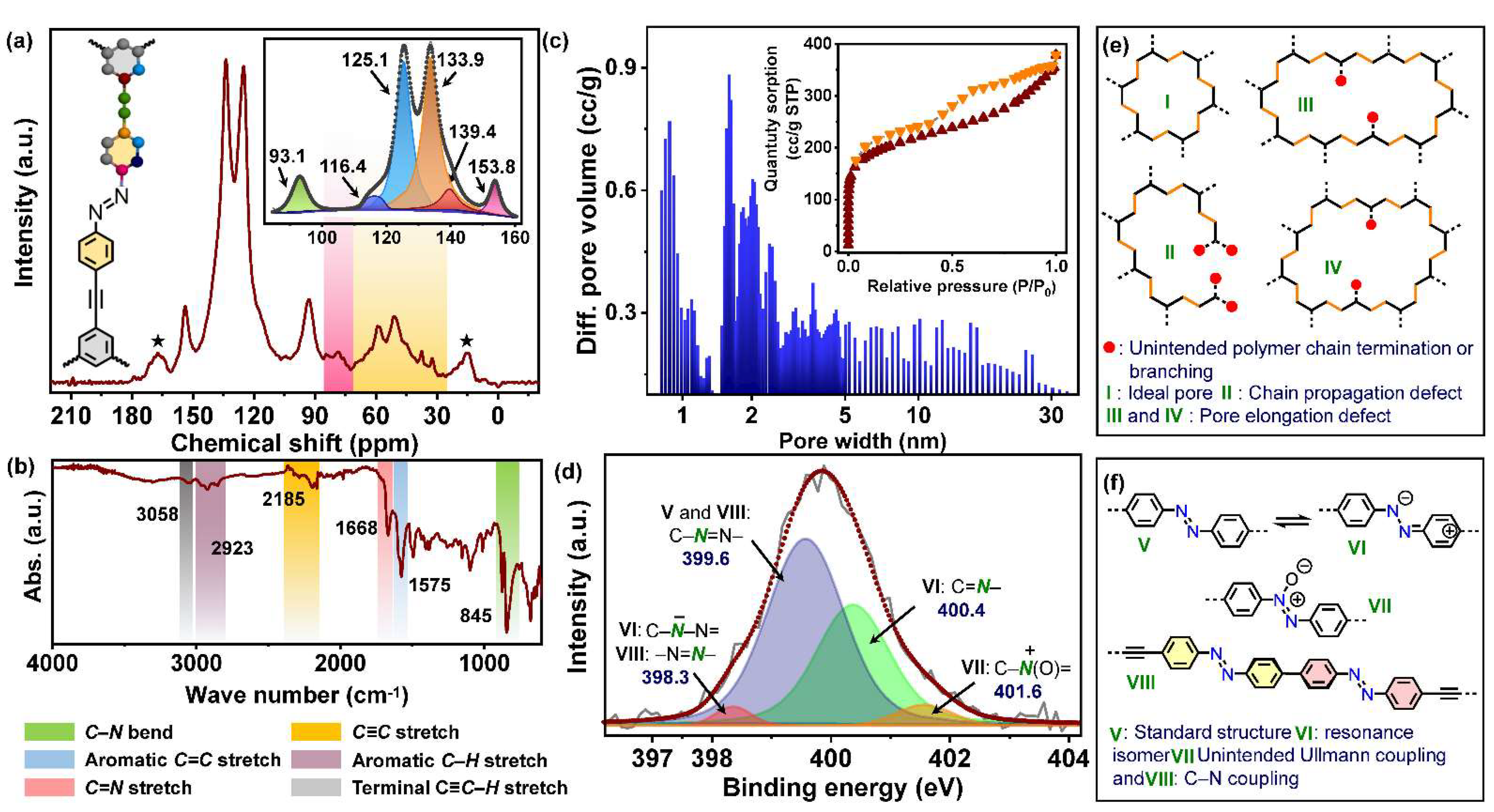
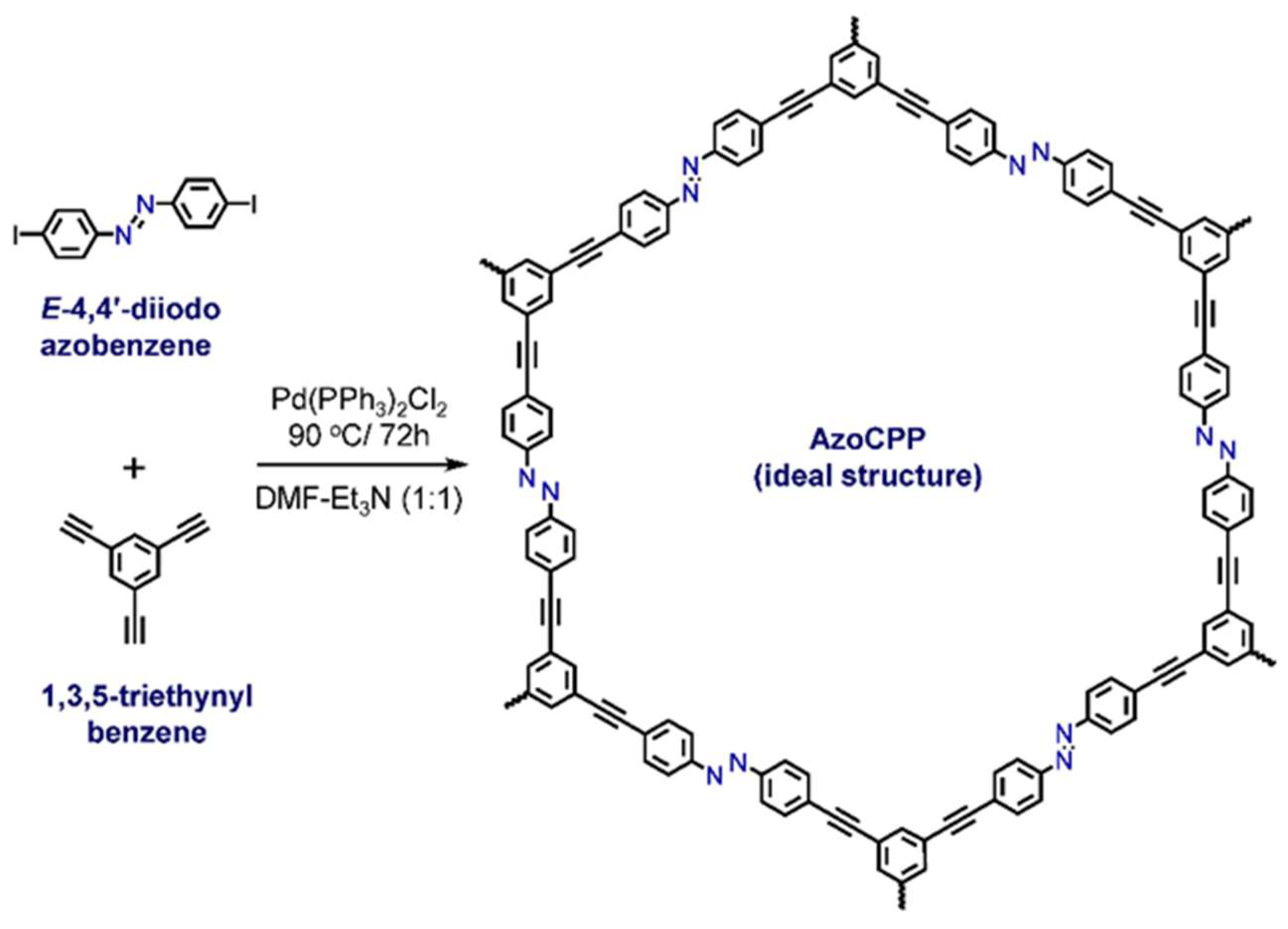
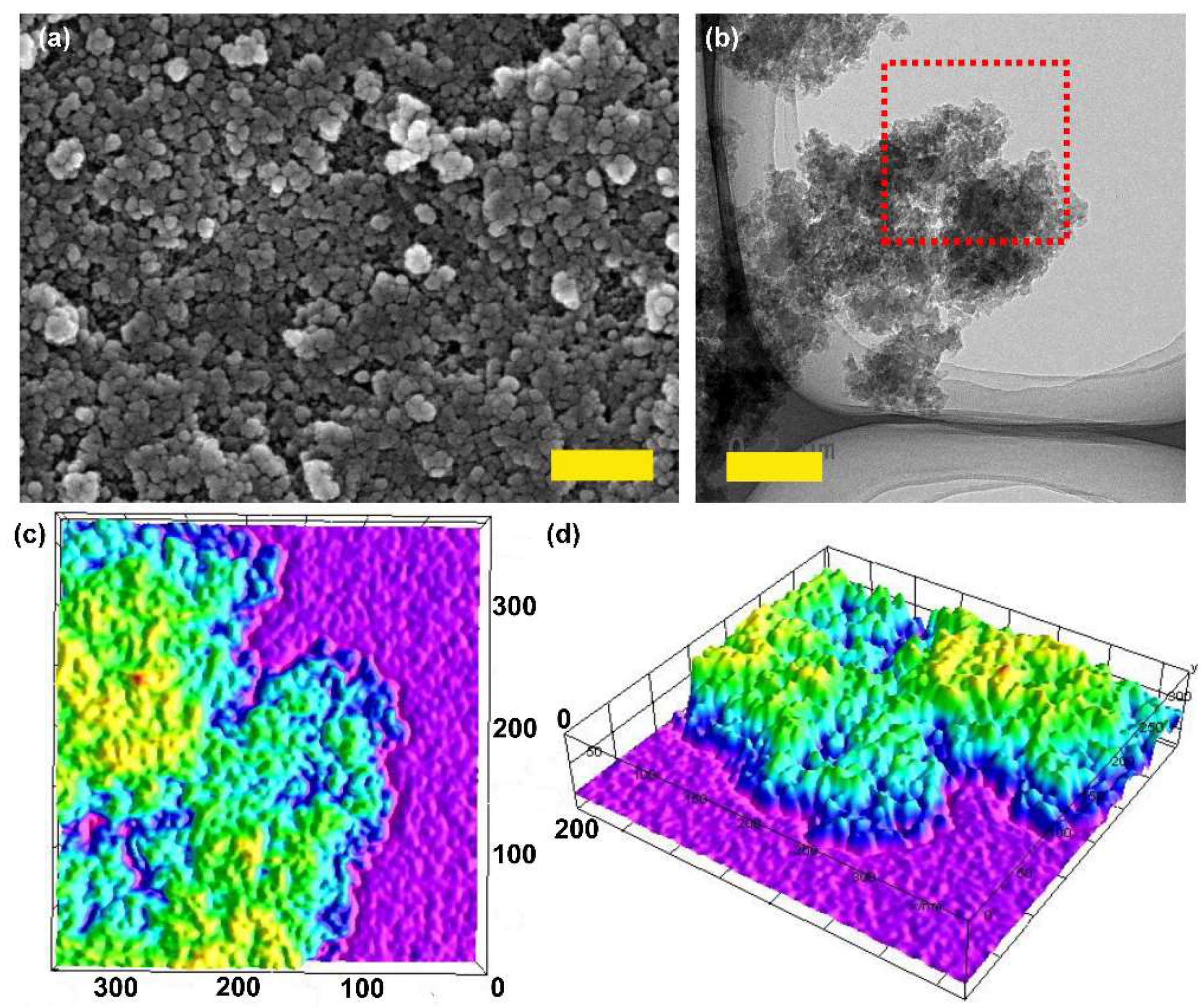
2.1. Synthesis and Characterizations
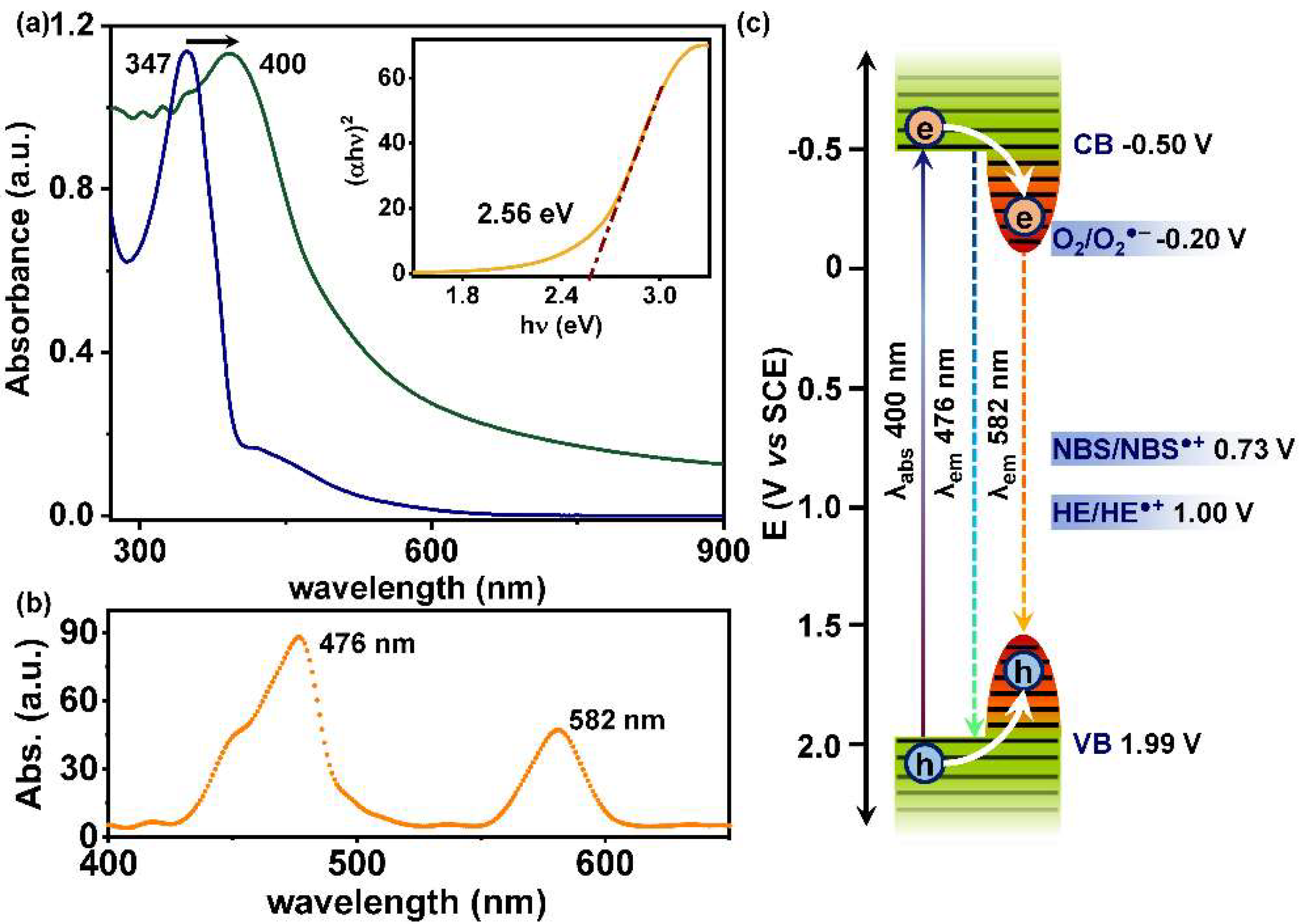
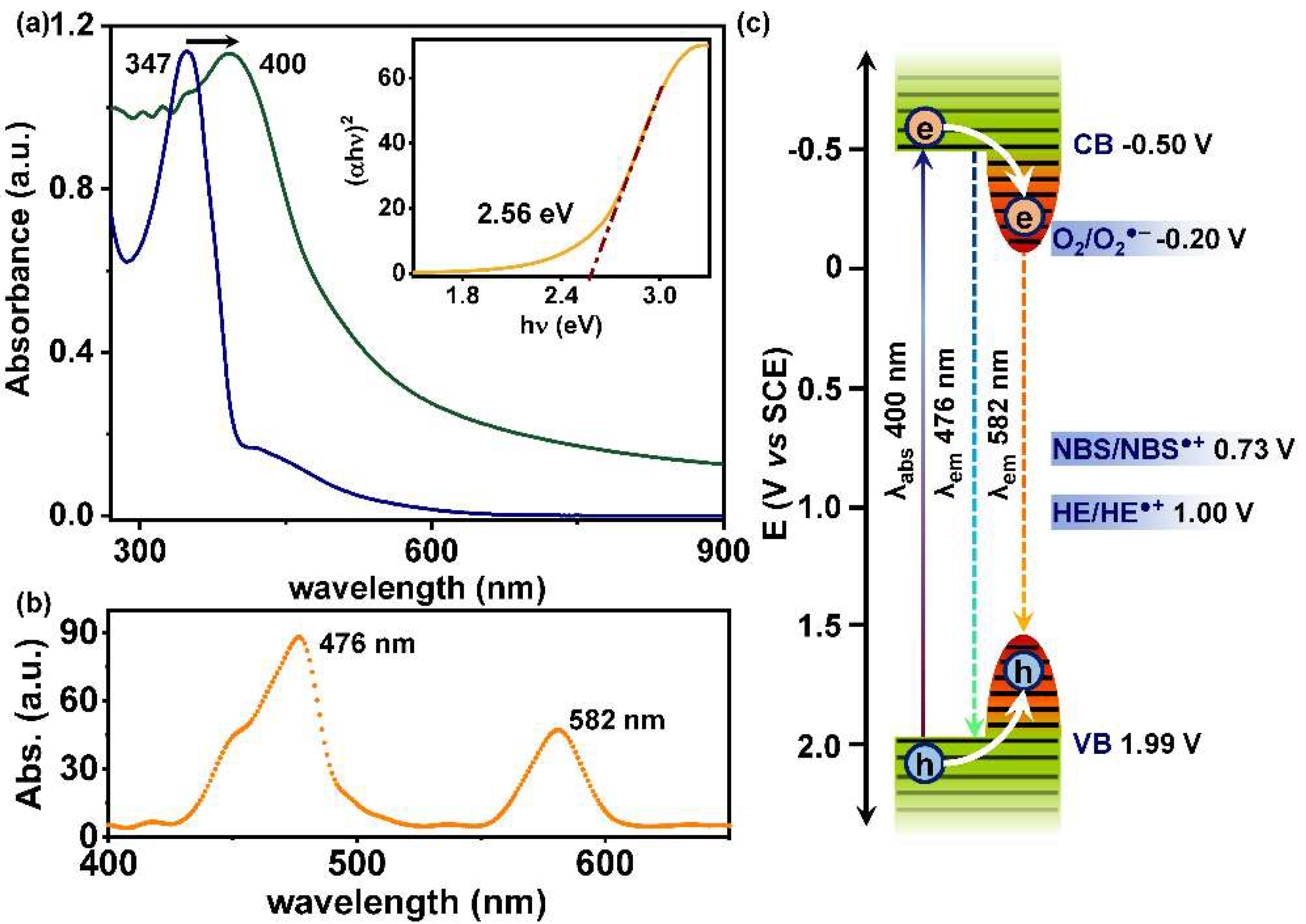
2.2. Photophysical and Electrochemical Properties
2.3. Photocatalysis
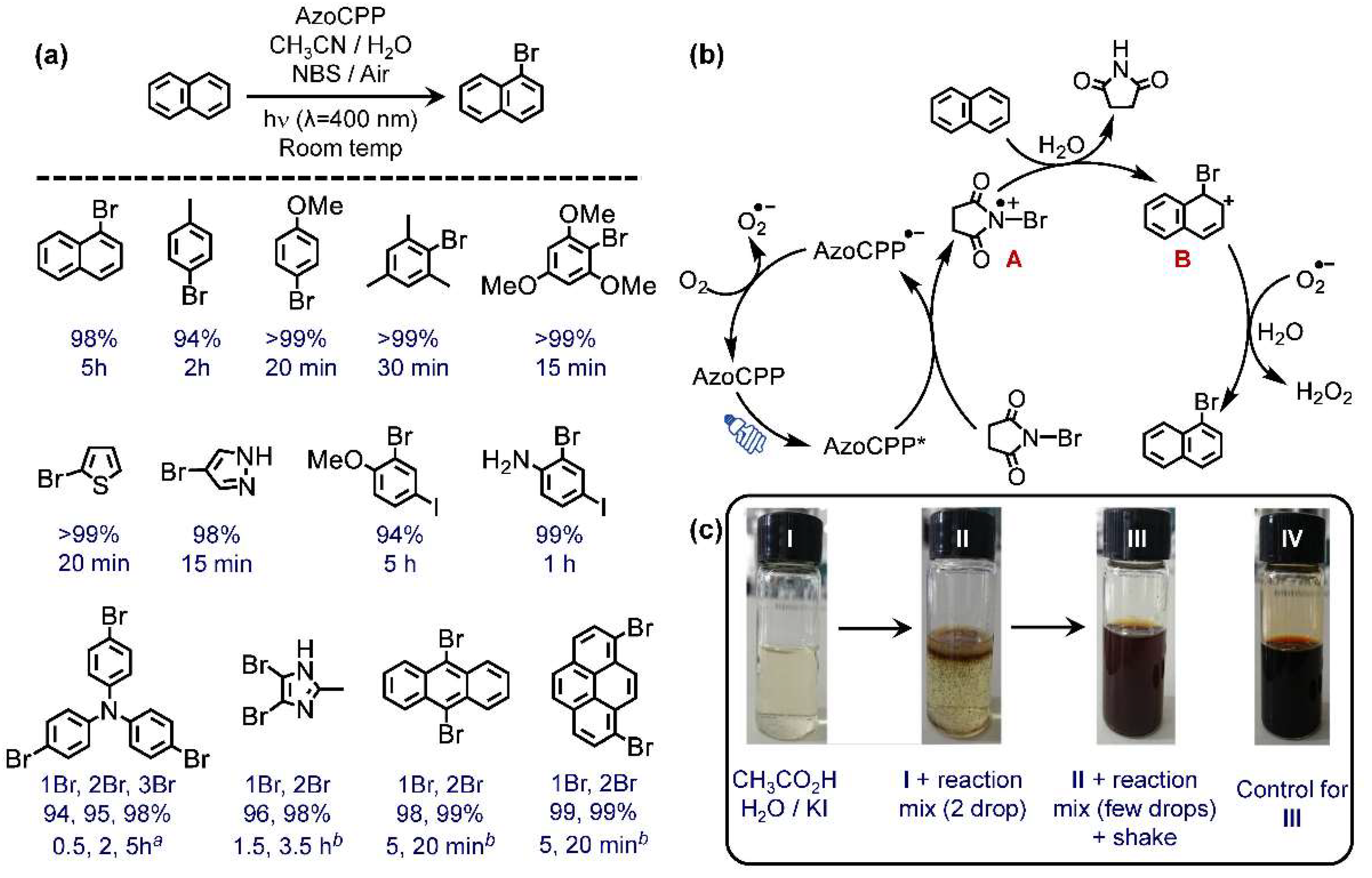
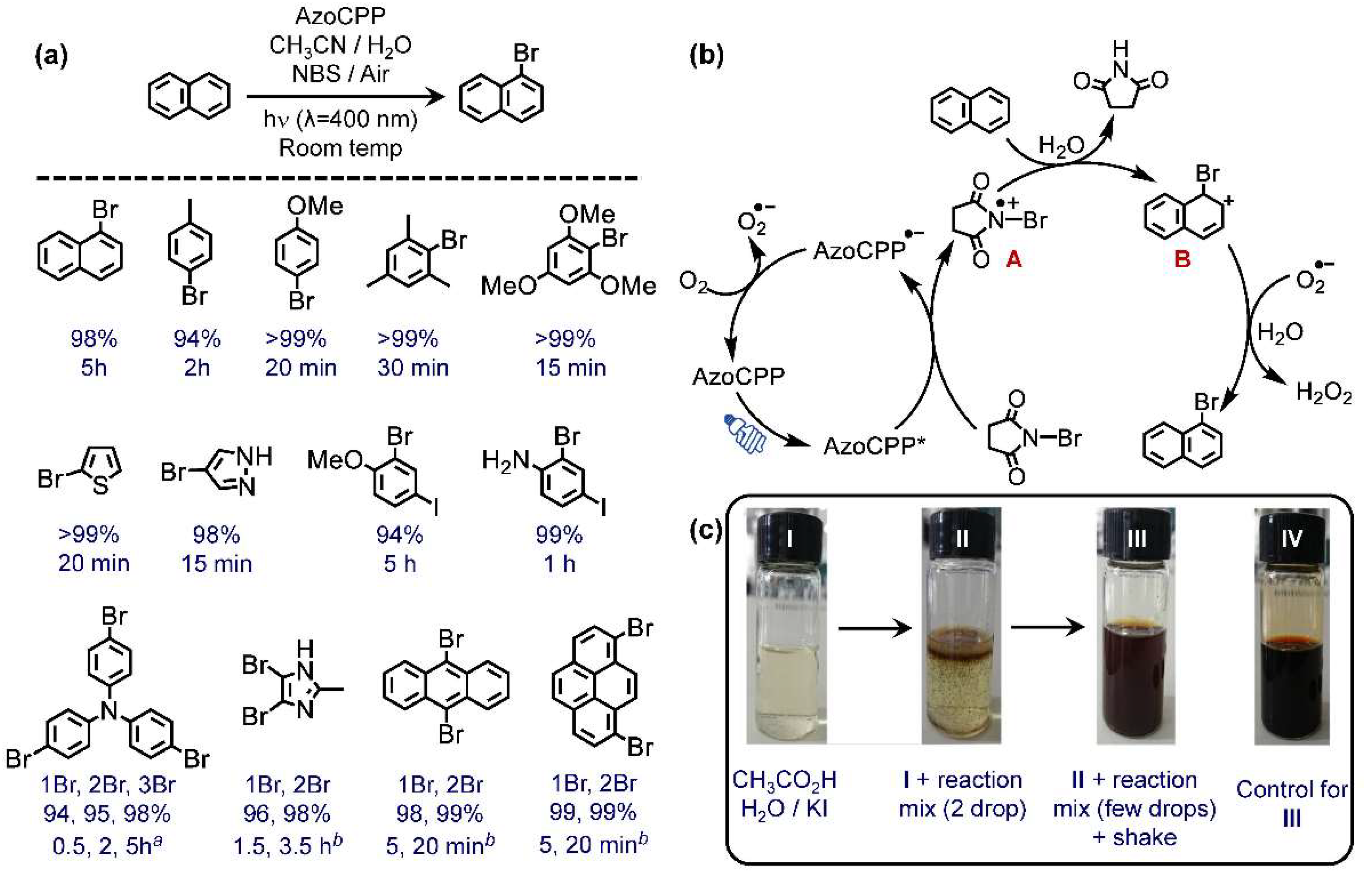
2.3.1. Oxidative Bromination
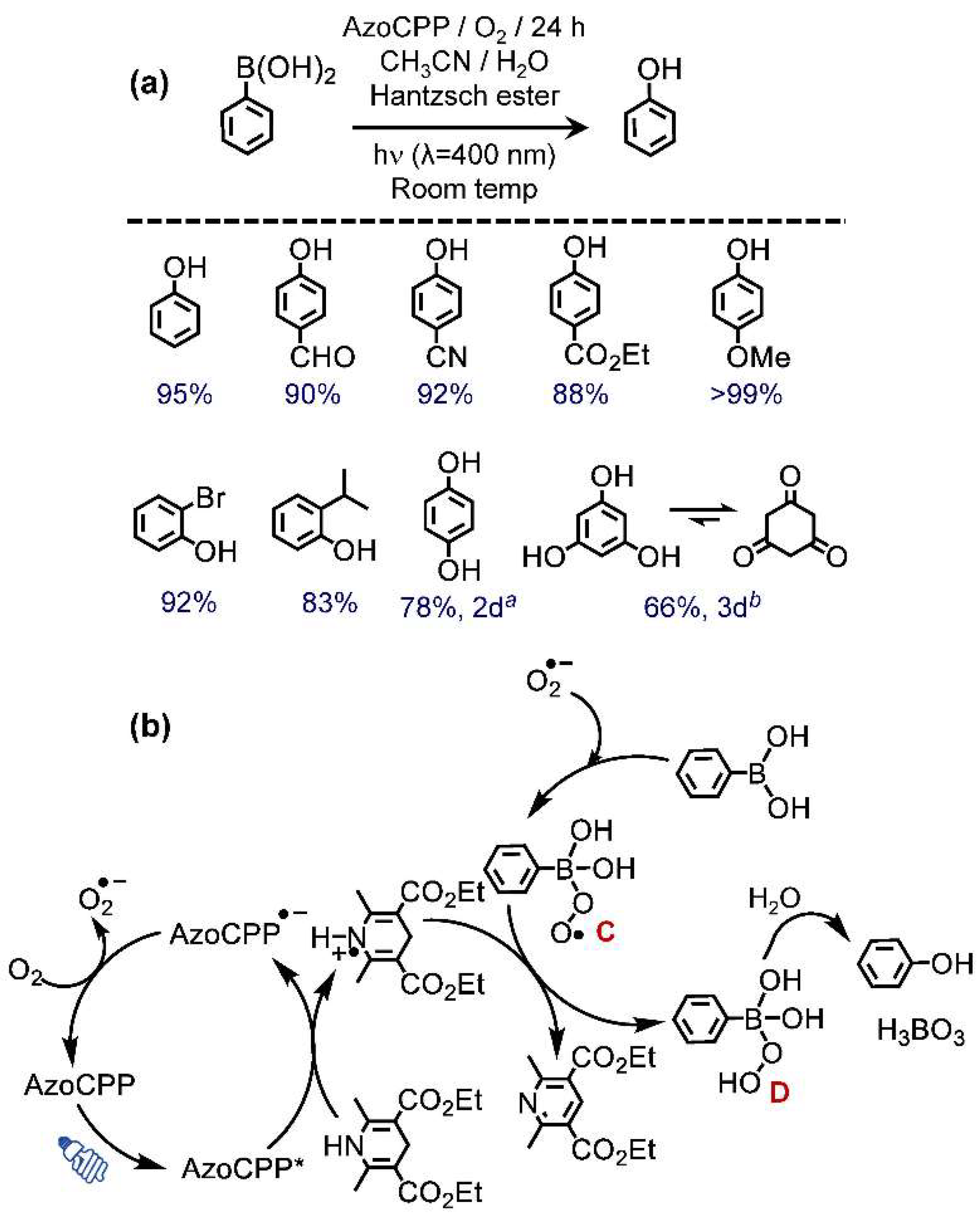
2.3.2. Oxidative Dehydroxylation of Boronic Acids
2.3.3. Structure–Property–Catalysis Interplay at AzoCPP
3. Materials and Methods
3.1. General Methods
3.2. Synthesis of AzoCPP
3.3. General Procedure for Photocatalytic Oxidative Bromination
3.4. General Procedure for Photocatalytic Dehydroxylation of Boronic Acid
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Cooper, A.I. Conjugated microporous polymers. Adv. Mater. 2009, 21, 1291–1295. [Google Scholar] [CrossRef]
- Xu, Y.; Jin, S.; Xu, H.; Nagai, A.; Jiang, D. Conjugated microporous polymers: Design, synthesis and application. Chem. Soc. Rev. 2013, 42, 8012–8031. [Google Scholar] [CrossRef] [PubMed]
- Vilela, F.; Zhang, K.A.I.; Antonietti, M. Conjugated porous polymers for energy applications. Energy Environ. Sci. 2012, 5, 7819–7832. [Google Scholar] [CrossRef]
- Taylor, D.; Dalgarno, S.J.; Xu, Z.; Vilela, F. Conjugated porous polymers: Incredibly versatile materials with far-reaching applications. Chem. Soc. Rev. 2020, 49, 3981–4042. [Google Scholar] [CrossRef]
- Chakraborty, J.; Nath, I.; Song, S.; Mohamed, S.; Khan, A.; Heynderickx, P.M.; Verpoort, F. Porous organic polymer composites as surging catalysts for visible-light-driven chemical transformations and pollutant degradation. J. Photochem. Photobiol. C Photochem. Rev. 2019, 41, 100319. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, Q. Recent progress in covalent organic frameworks as light-emitting materials. Mater. Today Energy 2021, 20, 100635. [Google Scholar] [CrossRef]
- She, P.; Qin, Y.; Wang, X.; Zhang, Q. Recent progress in external-stimulus-responsive 2D covalent organic frameworks. Adv. Mater. 2021, 2101175. [Google Scholar] [CrossRef]
- Zhi, Y.; Wang, Z.; Zhang, H.-L.; Zhang, Q. Recent progress in metal-free covalent organic frameworks as heterogeneous catalysts. Small 2020, 16, e2001070. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.-J.; Wu, Z.; Xie, J.; Yu, F.; Guo, W.; Xu, Z.J.; Li, D.-S.; Zhang, S.; Zhang, Q. Two-dimensional (2D) covalent organic framework as efficient cathode for binder-free lithium-ion battery. ChemSusChem 2019, 13, 2457–2463. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, T.; Podjaski, F.; Kröger, J.; Biswal, B.P.; Lotsch, B.V. Polymer photocatalysts for solar-to-chemical energy conversion. Nat. Rev. Mater. 2021, 6, 168–190. [Google Scholar] [CrossRef]
- Wisser, F.M.; Berruyer, P.; Cardenas, L.; Mohr, Y.; Quadrelli, E.A.; Lesage, A.; Farrusseng, D.; Canivet, J. Hammett parameter in microporous solids as macroligands for heterogenized photocatalysts. ACS Catal. 2018, 8, 1653–1661. [Google Scholar] [CrossRef]
- Kim, S.; Ogata, T.; Kurihara, S. Azobenzene-containing polymers for photonic crystal materials. Polym. J. 2017, 49, 407–412. [Google Scholar] [CrossRef]
- Zhu, C.; Lu, Y.; Sun, J.; Yu, Y. Dynamic interfacial regulation by photodeformable azobenzene-containing liquid crystal polymer micro/nanostructures. Langmuir 2020, 36, 6611–6625. [Google Scholar] [CrossRef]
- Nath, I.; Chakraborty, J.; Khan, A.; Arshad, M.N.; Azum, N.; Rab, M.A.; Asiri, A.M.; Alamry, K.A.; Verpoort, F. Conjugated mesoporous polyazobenzene–Pd(II) composite: A potential catalyst for visible-light-induced Sonogashira coupling. J. Catal. 2019, 377, 183–189. [Google Scholar] [CrossRef]
- Nath, I.; Chakraborty, J.; Zhang, G.; Chen, C.; Chaemchuen, S.; Park, J.; Zhuiykov, S.; Han, T.; Verpoort, F. Understanding the roles of variable Pd(II)/Pd(0) ratio supported on conjugated poly-azobenzene network: From characteristic alteration in properties to their cooperation towards visible-light-induced selective hydrogenation. J. Catal. 2020, 385, 120–128. [Google Scholar] [CrossRef] [Green Version]
- Gon, M.; Wakabayashi, J.; Nakamura, M.; Tanaka, K.; Chujo, Y. Preparation of near-infrared emissive π-conjugated polymer films based on boron-fused azobenzene complexes with perpendicularly protruded aryl substituents. Macromol. Rapid Commun. 2020, 42, 2000566. [Google Scholar] [CrossRef]
- Tobin, J.M.; McCabe, T.J.D.; Prentice, A.W.; Holzer, S.; Lloyd, G.O.; Paterson, M.J.; Arrighi, V.; Cormack, P.A.G.; Vilela, F. Polymer-supported photosensitizers for oxidative organic transformations in flow and under visible light irradiation. ACS Catal. 2017, 7, 4602–4612. [Google Scholar] [CrossRef]
- Luo, J.; Zhang, X.; Zhang, J. Carbazolic porous organic framework as an efficient, metal-free visible-light photocatalyst for organic synthesis. ACS Catal. 2015, 5, 2250–2254. [Google Scholar] [CrossRef]
- Su, C.; Tandiana, R.; Tian, B.; Sengupta, A.; Tang, W.; Su, J.; Loh, K.P. Visible-light photocatalysis of aerobic oxidation reactions using carbazolic conjugated microporous polymers. ACS Catal. 2016, 6, 3594–3599. [Google Scholar] [CrossRef]
- Takanabe, K. Photocatalytic water splitting: Quantitative approaches toward photocatalyst by design. ACS Catal. 2017, 7, 8006–8022. [Google Scholar] [CrossRef]
- Noda, Y.; Merschjann, C.; Tarábek, J.; Amsalem, P.; Koch, N.; Bojdys, M.J. Directional charge transport in layered two-dimensional triazine-based graphitic carbon nitride. Angew. Chem. Int. Ed. 2019, 58, 9394–9398. [Google Scholar] [CrossRef]
- Bässler, H.; Köhler, A. Unimolecular and Supramolecular Electronics I: Chemistry and Physics Meet at Metal-Molecule Interfaces; Metzger, R.M., Ed.; Springer: Berlin/Heidelberg, Germany, 2011; Volume 312, pp. 1–65. [Google Scholar]
- Wang, Y.; Vogel, A.; Sachs, M.; Sprick, R.S.; Wilbraham, L.; Moniz, S.J.A.; Godin, R.; Zwijnenburg, M.A.; Durrant, J.R.; Cooper, A.I.; et al. Current understanding and challenges of solar-driven hydrogen generation using polymeric photocatalysts. Nat. Energy 2019, 4, 746–760. [Google Scholar] [CrossRef]
- Pelzer, K.M.; Darling, S.B. Charge generation in organic photovoltaics: A review of theory and computation. Mol. Syst. Des. Eng. 2016, 1, 10–24. [Google Scholar] [CrossRef]
- Tamai, Y.; Ohkita, H.; Benten, H.; Ito, S. Exciton diffusion in conjugated polymers: From fundamental understanding to improvement in photovoltaic conversion efficiency. J. Phys. Chem. Lett. 2015, 6, 3417–3428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregg, B.A. Charged defects in soft semiconductors and their influence on organic photovoltaics. Soft Matter 2009, 5, 2985–2989. [Google Scholar] [CrossRef]
- Godin, R.; Wang, Y.; Zwijnenburg, M.A.; Tang, J.; Durrant, J.R. Time-resolved spectroscopic investigation of charge trapping in carbon nitrides photocatalysts for hydrogen generation. J. Am. Chem. Soc. 2017, 139, 5216–5224. [Google Scholar] [CrossRef] [PubMed]
- Steinmann, S.N.; Melissen, S.T.A.G.; Le Bahers, T.; Sautet, P. Challenges in calculating the bandgap of triazine-based carbon nitride structures. J. Mater. Chem. A 2017, 5, 5115–5122. [Google Scholar] [CrossRef] [Green Version]
- Lau, V.W.-H.; Yu, V.W.-Z.; Ehrat, F.; Botari, T.; Moudrakovski, I.; Simon, T.; Duppel, V.; Medina, E.; Stolarczyk, J.; Feldmann, J.; et al. Urea-modified carbon nitrides: Enhancing photocatalytic hydrogen evolution by rational defect engineering. Adv. Energy Mater. 2017, 7, 1602251. [Google Scholar] [CrossRef]
- Noriega, R.; Rivnay, J.; Vandewal, K.; Koch, F.P.V.; Stingelin, N.; Smith, P.; Toney, M.F.; Salleo, A. A general relationship between disorder, aggregation and charge transport in conjugated polymers. Nat. Mater. 2013, 12, 1038–1044. [Google Scholar] [CrossRef]
- Zhi, Y.; Li, K.; Xia, H.; Xue, M.; Mu, Y.; Liu, X. Robust porous organic polymers as efficient heterogeneous organo-photocatalysts for aerobic oxidation reactions. J. Mater. Chem. A 2017, 5, 8697–8704. [Google Scholar] [CrossRef]
- Chakraborty, J.; Nath, I.; Verpoort, F. Pd-nanoparticle decorated azobenzene based colloidal porous organic polymer for visible and natural sunlight induced Mott-Schottky junction mediated instantaneous Suzuki coupling. Chem. Eng. J. 2019, 358, 580–588. [Google Scholar] [CrossRef]
- Zhou, J.; Nomenyo, K.; Cesar, C.C.; Lusson, A.; Schwartzberg, A.; Yen, C.-C.; Woon, W.-Y.; Lerondel, G. Giant defect emission enhancement from ZnO nanowires through desulfurization process. Sci. Rep. 2020, 10, 4237. [Google Scholar] [CrossRef]
- Wang, H.; Jiang, S.; Chen, S.; Zhang, X.; Shao, W.; Sun, X.; Zhao, Z.; Zhang, Q.; Luo, Y.; Xie, Y. Insights into the excitonic processes in polymeric photocatalysts. Chem. Sci. 2017, 8, 4087–4092. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, R.; Paesani, F. Unraveling the effect of defects, domain size, and chemical doping on photophysics and charge transport in covalent organic frameworks. Chem. Sci. 2021, 12, 8373–8384. [Google Scholar] [CrossRef] [PubMed]
- Roessler, M.M.; Salvadori, E. Principles and applications of EPR spectroscopy in the chemical sciences. Chem. Soc. Rev. 2018, 47, 2534–2553. [Google Scholar] [CrossRef] [PubMed]
- Saikia, I.; Borah, A.J.; Phukan, P. Use of bromine and bromo-organic compounds in organic synthesis. Chem. Rev. 2016, 116, 6837–7042. [Google Scholar] [CrossRef]
- Dastan, A.; Tahir, M.N.; Ülkü, D.; Balci, M. Bromination of naphthalene and derivatives: High temperature bromination XI. Tetrahedron 1999, 55, 12853–12864. [Google Scholar] [CrossRef]
- Rogers, D.; Brown, R.G.; Brandeburg, Z.C.; Ko, E.Y.; Hopkins, M.D.; Leblanc, G.; Lamar, A.A. Organic dye-catalyzed, visible-light photoredox bromination of arenes and heteroarenes using n-bromosuccinimide. ACS Omega 2018, 3, 12868–12877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, C.; Tu, W.-C.; Levers, O.; Bröhl, A.; Hallett, J.P. Green and sustainable solvents in chemical processes. Chem. Rev. 2018, 118, 747–800. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nath, I.; Chakraborty, J.; Abednatanzi, S.; Van Der Voort, P. A ‘Defective’ Conjugated Porous Poly-Azo as Dual Photocatalyst. Catalysts 2021, 11, 1064. https://doi.org/10.3390/catal11091064
Nath I, Chakraborty J, Abednatanzi S, Van Der Voort P. A ‘Defective’ Conjugated Porous Poly-Azo as Dual Photocatalyst. Catalysts. 2021; 11(9):1064. https://doi.org/10.3390/catal11091064
Chicago/Turabian StyleNath, Ipsita, Jeet Chakraborty, Sara Abednatanzi, and Pascal Van Der Voort. 2021. "A ‘Defective’ Conjugated Porous Poly-Azo as Dual Photocatalyst" Catalysts 11, no. 9: 1064. https://doi.org/10.3390/catal11091064
APA StyleNath, I., Chakraborty, J., Abednatanzi, S., & Van Der Voort, P. (2021). A ‘Defective’ Conjugated Porous Poly-Azo as Dual Photocatalyst. Catalysts, 11(9), 1064. https://doi.org/10.3390/catal11091064