
Role of Reaction Intermediate Diffusion on the Performance of Platinum Electrodes in Solid Acid Fuel Cells

 , , , , , ,
, , , , , ,  , and
, and
Abstract
:
1. Introduction
2. Results
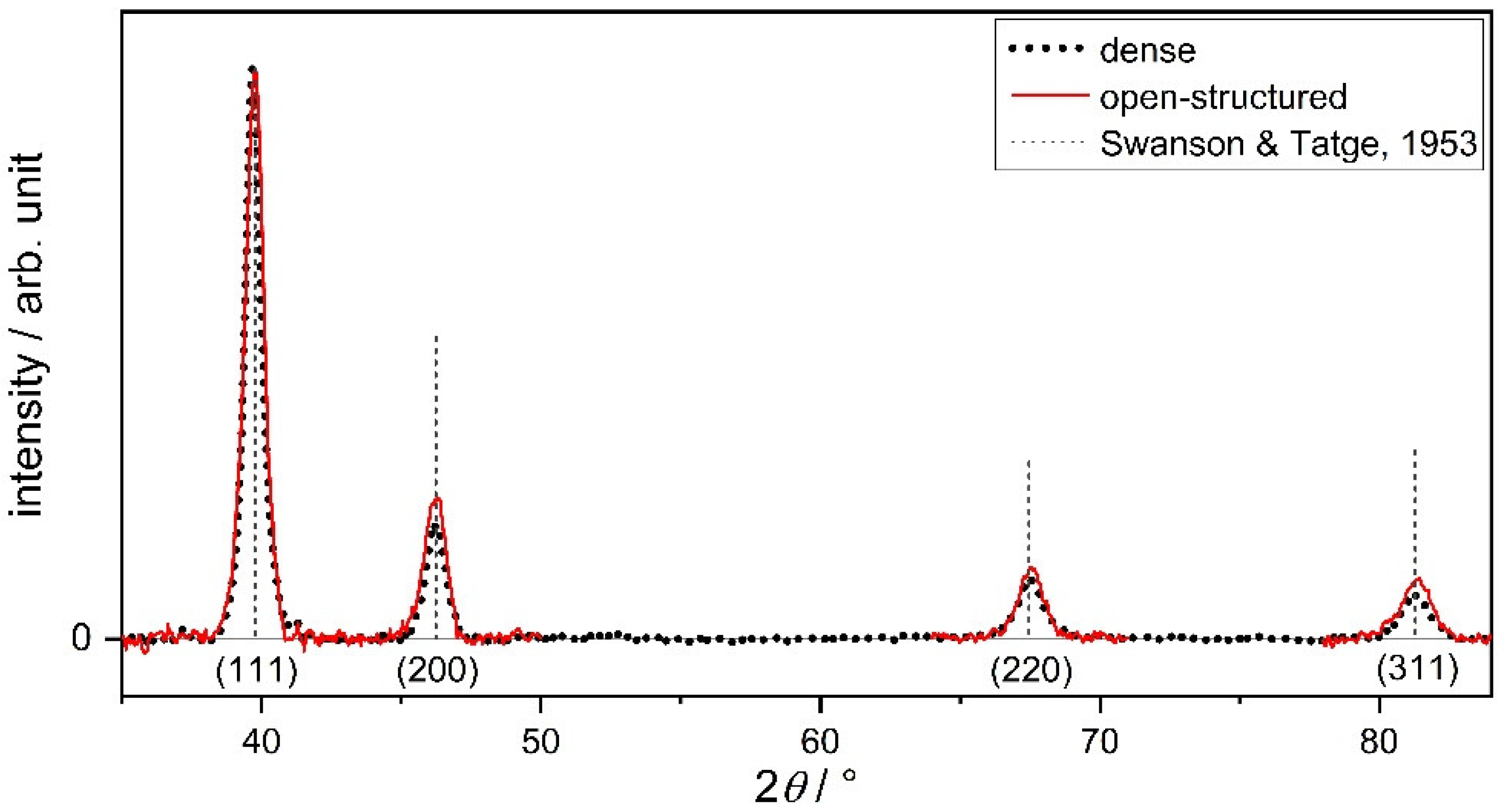
2.1. Structural Characterization
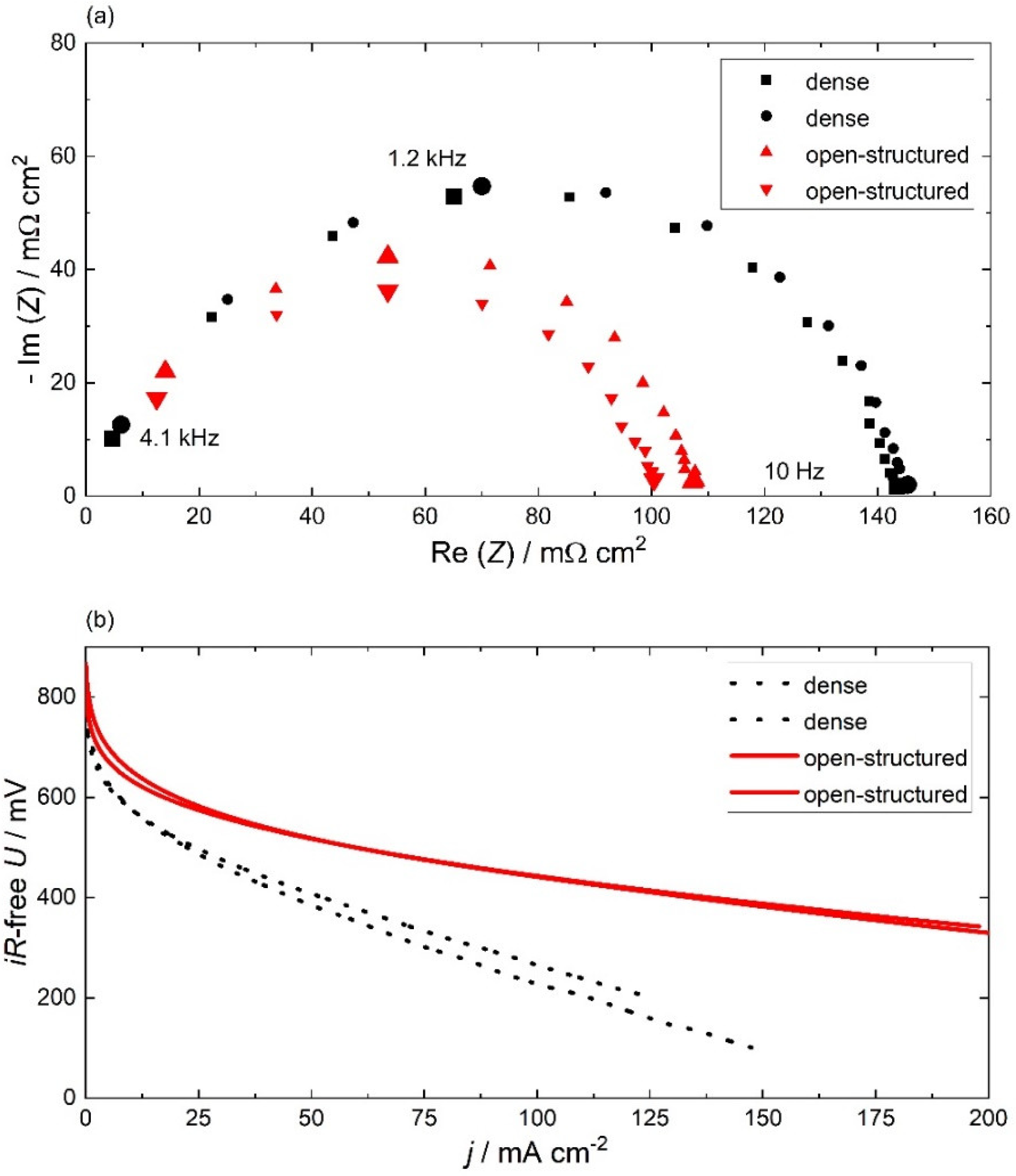
2.2. Electrochemical Characterization
2.2.1. Hydrogen Oxidation Reaction (HOR)
2.2.2. Oxygen Reduction Reaction (ORR)
2.2.3. Electrochemical Stability
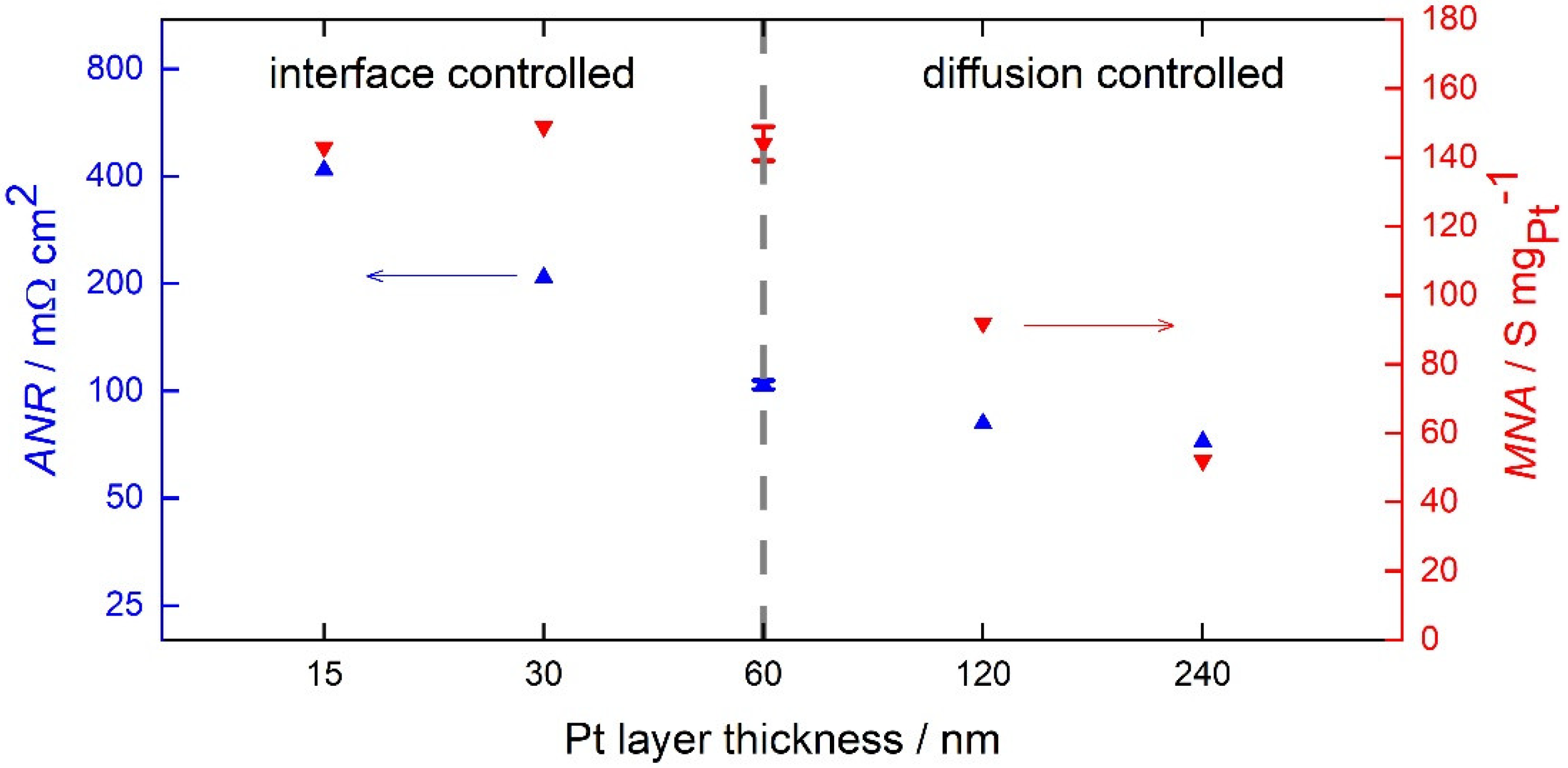
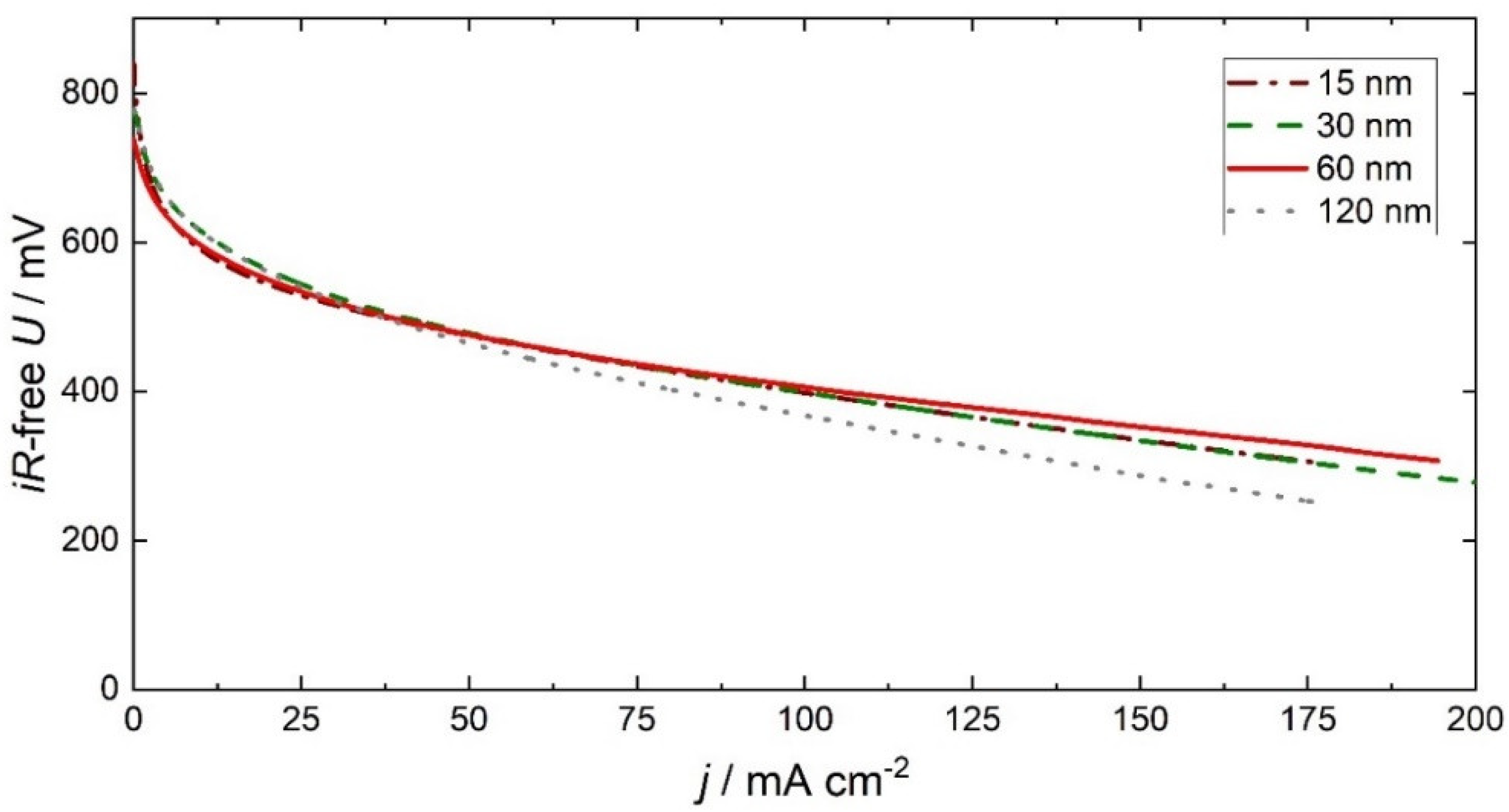
3. Discussion
| Tafel step: H2 ⇄ 2 Had | (1) |
| Heyrovsky step: H2 ⇄ Had + e− + H+ | (2) |
| Volmer step: Had ⇄ H+ + e− | (3) |
4. Materials and Methods
4.1. Electrode and Cell Fabrication
4.2. Physical and Electrochemical Characterization
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goñi-Urtiaga, A.; Presvytes, D.; Scott, K. Solid Acids as Electrolyte Materials for Proton Exchange Membrane (PEM) Electrolysis: Review. Int. J. Hydrogen Energy 2012, 37, 3358–3372. [Google Scholar] [CrossRef]
- Paschos, O.; Kunze, J.; Stimming, U.; Maglia, F. A Review on Phosphate Based, Solid State, Protonic Conductors for Intermediate Temperature Fuel Cells. J. Phys. Condens. Matter 2011, 23, 234110. [Google Scholar] [CrossRef]
- Scott, K.; Xu, C.; Wu, X. Intermediate Temperature Proton-Conducting Membrane Electrolytes for Fuel Cells: Intermediate Temperature Proton-Conducting Membrane Electrolytes for Fuel Cells. WIREs Energy Environ. 2014, 3, 24–41. [Google Scholar] [CrossRef]
- Haile, S.M.; Boysen, D.A.; Chisholm, C.R.I.; Merle, R.B. Solid Acids as Fuel Cell Electrolytes. Nature 2001, 410, 910–913. [Google Scholar] [CrossRef] [Green Version]
- Taninouchi, Y.; Uda, T.; Awakura, Y.; Ikeda, A.; Haile, S.M. Dehydration Behavior of the Superprotonic Conductor CsH2PO4 at Moderate Temperatures: 230 to 260 °C. J. Mater. Chem. 2007, 17, 3182. [Google Scholar] [CrossRef]
- Uda, T.; Boysen, D.A.; Chisholm, C.R.I.; Haile, S.M. Alcohol Fuel Cells at Optimal Temperatures. Electrochem. Solid State Lett. 2006, 9, A261. [Google Scholar] [CrossRef]
- Chisholm, C.R.I.; Boysen, D.A.; Papandrew, A.B.; Zecevic, S.; Cha, S.; Sasaki, K.A.; Varga, Á.; Giapis, K.P.; Haile, S.M. From Laboratory Breakthrough to Technological Realization: The Development Path for Solid Acid Fuel Cells. Electrochem. Soc. Interface 2009, 18, 53. [Google Scholar]
- Lohmann, F.P.; Schulze, P.S.C.; Wagner, M.; Naumov, O.; Lotnyk, A.; Abel, B.; Varga, Á. The next Generation Solid Acid Fuel Cell Electrodes: Stable, High Performance with Minimized Catalyst Loading. J. Mater. Chem. A 2017, 5, 15021–15025. [Google Scholar] [CrossRef]
- Suryaprakash, R.C.; Lohmann, F.P.; Wagner, M.; Abel, B.; Varga, A. Spray Drying as a Novel and Scalable Fabrication Method for Nanostructured CsH 2 PO 4, Pt-Thin-Film Composite Electrodes for Solid Acid Fuel Cells. RSC Adv. 2014, 4, 60429–60436. [Google Scholar] [CrossRef]
- Lohmann-Richters, F.P.; Abel, B.; Varga, Á. In Situ Determination of the Electrochemically Active Platinum Surface Area: Key to Improvement of Solid Acid Fuel Cells. J. Mater. Chem. A 2018, 6, 2700–2707. [Google Scholar] [CrossRef]
- Thoi, V.S.; Usiskin, R.E.; Haile, S.M. Platinum-Decorated Carbon Nanotubes for Hydrogen Oxidation and Proton Reduction in Solid Acid Electrochemical Cells. Chem. Sci. 2015, 6, 1570–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uda, T.; Haile, S.M. Thin-Membrane Solid-Acid Fuel Cell. Electrochem. Solid State Lett. 2005, 8, A245–A246. [Google Scholar] [CrossRef] [Green Version]
- Papandrew, A.B.; Chisholm, C.R.I.; Elgammal, R.A.; Özer, M.M.; Zecevic, S.K. Advanced Electrodes for Solid Acid Fuel Cells by Platinum Deposition on CsH 2 PO 4. Chem. Mater. 2011, 23, 1659–1667. [Google Scholar] [CrossRef] [Green Version]
- Lim, D.-K.; Liu, J.; Pandey, S.A.; Paik, H.; Chisholm, C.R.I.; Hupp, J.T.; Haile, S.M. Atomic Layer Deposition of Pt@CsH2PO4 for the Cathodes of Solid Acid Fuel Cells. Electrochim. Acta 2018, 288, 12–19. [Google Scholar] [CrossRef] [Green Version]
- Papandrew, A.B.; Elgammal, R.A.; Tian, M.; Tennyson, W.D.; Rouleau, C.M.; Puretzky, A.A.; Veith, G.M.; Geohegan, D.B.; Zawodzinski, T.A. Nanostructured Carbon Electrocatalyst Supports for Intermediate-Temperature Fuel Cells: Single-Walled versus Multi-Walled Structures. J. Power Sources 2017, 337, 145–151. [Google Scholar] [CrossRef] [Green Version]
- An, Q.; Paulus, F.; Vaynzof, Y. Controlling the Microstructure and Porosity of Perovskite Films by Additive Engineering. ACS Appl. Energy Mater. 2021, 4, 2990–2998. [Google Scholar] [CrossRef]
- Quintero-Ruiz, J.; Ruiz-Rosas, R.; Quílez-Bermejo, J.; Salinas-Torres, D.; Cazorla-Amorós, D.; Morallón, E. Preparation of Pt/CNT Thin-Film Electrodes by Electrochemical Potential Pulse Deposition for Methanol Oxidation. C 2021, 7, 32. [Google Scholar] [CrossRef]
- Zhu, J.; Xu, L.; Lyu, Z.; Xie, M.; Chen, R.; Jin, W.; Mavrikakis, M.; Xia, Y. Janus Nanocages of Platinum-Group Metals and Their Use as Effective Dual-Electrocatalysts. Angew. Chem. Int. Ed. 2021, 60, 10384–10392. [Google Scholar] [CrossRef] [PubMed]
- Louie, M.W.; Haile, S.M. Platinum Thin Film Anodes for Solid Acid Fuel Cells. Energy Environ. Sci. 2011, 4, 4230. [Google Scholar] [CrossRef] [Green Version]
- Wagner, M.; Dreßler, C.; Lohmann-Richters, F.P.; Hanus, K.; Sebastiani, D.; Varga, A.; Abel, B. Mechanism of Ion Conductivity through Polymer-Stabilized CsH2PO4 Nanoparticular Layers from Experiment and Theory. J. Mater. Chem. A 2019, 7, 27367–27376. [Google Scholar] [CrossRef]
- Fu, C.; Liu, C.; Li, T.; Zhang, X.; Wang, F.; Yang, J.; Jiang, Y.; Cui, P.; Li, H. DFT Calculations: A Powerful Tool for Better Understanding of Electrocatalytic Oxygen Reduction Reactions on Pt-Based Metallic Catalysts. Comput. Mater. Sci. 2019, 170, 109202. [Google Scholar] [CrossRef]
- Kulkarni, A.; Siahrostami, S.; Patel, A.; Nørskov, J.K. Understanding Catalytic Activity Trends in the Oxygen Reduction Reaction. Chem. Rev. 2018, 118, 2302–2312. [Google Scholar] [CrossRef] [PubMed]
- Anders, A. A Structure Zone Diagram Including Plasma-Based Deposition and Ion Etching. Thin Solid Films 2010, 518, 4087–4090. [Google Scholar] [CrossRef] [Green Version]
- Slavcheva, E.; Ganske, G.; Topalov, G.; Mokwa, W.; Schnakenberg, U. Effect of Sputtering Parameters on Surface Morphology and Catalytic Efficiency of Thin Platinum Films. Appl. Surf. Sci. 2009, 255, 6479–6486. [Google Scholar] [CrossRef]
- Sakaliūnienė, J.; Abakevičienė, B.; Šlapikas, K.; Tamulevičius, S. Influence of Magnetron Sputtering Deposition Conditions and Thermal Treatment on Properties of Platinum Thin Films for Positive Electrode–Electrolyte–Negative Electrode Structure. Thin Solid Films 2015, 594, 101–108. [Google Scholar] [CrossRef]
- Li, L.; Wang, L.-L.; Johnson, D.D.; Zhang, Z.; Sanchez, S.I.; Kang, J.H.; Nuzzo, R.G.; Wang, Q.; Frenkel, A.I.; Li, J.; et al. Noncrystalline-to-Crystalline Transformations in Pt Nanoparticles. J. Am. Chem. Soc. 2013, 135, 13062–13072. [Google Scholar] [CrossRef]
- Zepeda-Ruiz, L.A.; Chason, E.; Gilmer, G.H.; Wang, Y.; Xu, H.; Nikroo, A.; Hamza, A.V. Understanding the Relation between Stress and Surface Morphology in Sputtered Films: Atomistic Simulations and Experiments. Appl. Phys. Lett. 2009, 95, 151910. [Google Scholar] [CrossRef]
- Slavcheva, E.; Topalov, G.; Ganske, G.; Radev, I.; Lefterova, E.; Schnakenberg, U. Influence of Sputtering Pressure on Surface Structure and Oxygen Reduction Reaction Catalytic Activity of Thin Platinum Films. Electrochim. Acta 2010, 55, 8992–8997. [Google Scholar] [CrossRef]
- Swanson, H.; Tatge, E. Standard X-Ray Diffraction Powder Patterns; National Bureau of Standards Circular 539; US Department of Commerce: Washington, DC, USA, 1953; Volume 1. [Google Scholar]
- Thomas, J.H., II. Effect of Pressure on Dc Planar Magnetron Sputtering of Platinum. J. Vac. Sci. Technol. A 2003, 21, 572–576. [Google Scholar] [CrossRef]
- Wang, J.; Wang, S.-Q. Surface Energy and Work Function of Fcc and Bcc Crystals: Density Functional Study. Surf. Sci. 2014, 630, 216–224. [Google Scholar] [CrossRef]
- DOE Technical Targets for Polymer Electrolyte Membrane Fuel Cell Components|Department of Energy. Available online: https://www.energy.gov/eere/fuelcells/doe-technical-targets-polymer-electrolyte-membrane-fuel-cell-components (accessed on 10 March 2021).
- Varga, A. Progress in Solid Acid Fuel Cell Electrodes. Am. J. Nano Res. Appl. 2014, 2, 61–65. [Google Scholar] [CrossRef]
- Wagner, M.; Lorenz, O.; Lohmann-Richters, F.; Varga, A.; Abel, B. Study on Solid Electrolyte Catalyst Poisoning in Solid Acid Fuel Cells. J. Mater. Chem. A 2021, 9, 11347–11358. [Google Scholar] [CrossRef]
- Boukamp, B.A. Derivation of a Distribution Function of Relaxation Times for the (Fractal) Finite Length Warburg. Electrochim. Acta 2017, 252, 154–163. [Google Scholar] [CrossRef]
- Dhirde, A.M.; Dale, N.V.; Salehfar, H.; Mann, M.D.; Han, T.-H. Equivalent Electric Circuit Modeling and Performance Analysis of a PEM Fuel Cell Stack Using Impedance Spectroscopy. IEEE Trans. Energy Convers. 2010, 25, 778–786. [Google Scholar] [CrossRef]
- Franceschetti, D.R.; RossMacdonald, J.; Buck, R.P. Interpretation of Finite-Length-Warburg-Type Impedances in Supported and Unsupported Electrochemical Cells with Kinetically Reversible Electrodes. J. Electrochem. Soc. 1991, 138, 1368. [Google Scholar] [CrossRef]
- Giner-Sanz, J.J.; Ortega, E.M.; Pérez-Herranz, V. Mechanistic Equivalent Circuit Modelling of a Commercial Polymer Electrolyte Membrane Fuel Cell. J. Power Sources 2018, 379, 328–337. [Google Scholar] [CrossRef]
- Wang, J.X.; Springer, T.E.; Adzic, R.R. Dual-Pathway Kinetic Equation for the Hydrogen Oxidation Reaction on Pt Electrodes. J. Electrochem. Soc. 2006, 153, A1732. [Google Scholar] [CrossRef]
- Harrington, D.A.; Conway, B.E. Ac Impedance of Faradaic Reactions Involving Electrosorbed Intermediates—I. Kinetic Theory. Electrochim. Acta 1987, 32, 1703–1712. [Google Scholar] [CrossRef] [Green Version]
- de Chialvo, M.R.G.; Chialvo, A.C. Hydrogen Diffusion Effects on the Kinetics of the Hydrogen Electrode Reaction. Part I: Theoretical Aspects. Phys. Chem. Chem. Phys. 2004, 6, 4009. [Google Scholar] [CrossRef]
- Bhardwaj, M.; Balasubramaniam, R. Uncoupled Non-Linear Equations Method for Determining Kinetic Parameters in Case of Hydrogen Evolution Reaction Following Volmer–Heyrovsky–Tafel Mechanism and Volmer–Heyrovsky Mechanism. Int. J. Hydrogen Energy 2008, 33, 2178–2188. [Google Scholar] [CrossRef]
- Durst, J.; Siebel, A.; Simon, C.; Hasché, F.; Herranz, J.; Gasteiger, H.A. New Insights into the Electrochemical Hydrogen Oxidation and Evolution Reaction Mechanism. Energy Environ. Sci. 2014, 7, 2255–2260. [Google Scholar] [CrossRef] [Green Version]
- Turnbull, A. Hydrogen Diffusion and Trapping in Metals. In Gaseous Hydrogen Embrittlement of Materials in Energy Technologies; Elsevier: Amsterdam, Netherlands, 2012; pp. 89–128. [Google Scholar] [CrossRef]
- Ishikawa, T.; McLellan, R.B. The Diffusivity of Hydrogen in the Noble Metals at Low Temperature. Acta Metall. 1985, 33, 1979–1985. [Google Scholar] [CrossRef]
- Skúlason, E.; Tripkovic, V.; Björketun, M.E.; Gudmundsdóttir, S.; Karlberg, G.; Rossmeisl, J.; Bligaard, T.; Jónsson, H.; Nørskov, J.K. Modeling the Electrochemical Hydrogen Oxidation and Evolution Reactions on the Basis of Density Functional Theory Calculations. J. Phys. Chem. C 2010, 114, 18182–18197. [Google Scholar] [CrossRef]
- Watson, G.W.; Wells, R.P.K.; Willock, D.J.; Hutchings, G.J. A Comparison of the Adsorption and Diffusion of Hydrogen on the {111} Surfaces of Ni, Pd, and Pt from Density Functional Theory Calculations. J. Phys. Chem. B 2001, 105, 4889–4894. [Google Scholar] [CrossRef]
- Haile, S.M.; Chisholm, C.R.I.; Sasaki, K.; Boysen, D.A.; Uda, T. Solid Acid Proton Conductors: From Laboratory Curiosities to Fuel Cell Electrolytes. Faraday Discuss. 2007, 134, 17–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papandrew, A.B.; Chisholm, C.R.I.; Zecevic, S.K.; Veith, G.M.; Zawodzinski, T.A. Activity and Evolution of Vapor Deposited Pt-Pd Oxygen Reduction Catalysts for Solid Acid Fuel Cells. J. Electrochem. Soc. 2013, 160, F175–F182. [Google Scholar] [CrossRef]
- Eikerling, M.; Kornyshev, A.A. Electrochemical Impedance of the Cathode Catalyst Layer in Polymer Electrolyte Fuel Cells. J. Electroanal. Chem. 1999, 475, 107–123. [Google Scholar] [CrossRef]
- Touhami, S.; Mainka, J.; Dillet, J.; Taleb, S.A.H.; Lottin, O. Transmission Line Impedance Models Considering Oxygen Transport Limitations in Polymer Electrolyte Membrane Fuel Cells. J. Electrochem. Soc. 2019, 166, F1209–F1217. [Google Scholar] [CrossRef]
- Graham, A.P.; Menzel, A.; Toennies, J.P. Quasielastic Helium Atom Scattering Measurements of Microscopic Diffusional Dynamics of H and D on the Pt(111) Surface. J. Chem. Phys. 1999, 111, 1676–1685. [Google Scholar] [CrossRef]
- Wintterlin, J.; Schuster, R.; Ertl, G. Existence of a Hot Atom Mechanism for the Dissociation of O2 on Pt(111). Phys. Rev. Lett. 1996, 77, 123. [Google Scholar] [CrossRef]
- Lombardo, S.J.; Bell, A.T. A Review of Theoretical Models of Adsorption, Diffusion, Desorption, and Reaction of Gases on Metal Surfaces. Surf. Sci. Rep. 1991, 13, 3–72. [Google Scholar] [CrossRef]
- Boysen, D.A. High-Performance Solid Acid Fuel Cells Through Humidity Stabilization. Science 2004, 303, 68–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, K.A.; Hao, Y.; Haile, S.M. Geometrically Asymmetric Electrodes for Probing Electrochemical Reaction Kinetics: A Case Study of Hydrogen at the Pt–CsH2PO4 Interface. Phys. Chem. Chem. Phys. 2009, 11, 8349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boukamp, B.A. A Linear Kronig-Kramers Transform Test for Immittance Data Validation. J. Electrochem. Soc. 1995, 142, 1885–1894. [Google Scholar] [CrossRef]
- Schönleber, M.; Klotz, D.; Ivers-Tiffée, E. A Method for Improving the Robustness of Linear Kramers-Kronig Validity Tests. Electrochim. Acta 2014, 131, 20–27. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Deposition Rate (nm min−1) | Film Thickness (nm) | Pt Loading (µg cm−2) | Grain Size (nm) | |
|---|---|---|---|---|
| Dense (0.5 Pa) | 25.9 ± 1.5 | 63 ± 4 | 136 ± 2 | 15.3 ± 2.7 |
| Open-structured (5 Pa) | 34.5 ± 2.4 | 58 ± 4 | 67 ± 1 | 8.0 ± 1.7 |
| Electrode Architecture | Pt Loading (µg cm−2) | ANR (mΩ cm2) | MNA (S mg−1) | Pt Utilization (g kW−1) |
|---|---|---|---|---|
| 60 nm Ptopen-structured@CP (this work) | 67 ± 1 | 104 ± 3 | 144 ± 5 | 2.0 ± 0.1 |
| 60 nm Ptdense@CP (this work) | 136 ± 2 | 144 ± 1 | 51 ± 1 | 9.8 ± 0.2 |
| 7.5 nm PtdcMS@CDP19 | 17 | 3100 | 19 | |
| PtALD@CDP14 | 1000 | 50 | 20 | <10 |
| PtMOCVD@CDP13 | 1750 | 6.5 | ||
| PtMOCVD@CNTs8 | 410 | 50 | 49 | 11.7 |
| 30 nm PtdcMS@CDPSpray20 | 64 | 745 | 21 | 2.0 |
| PtImpregnation@CNTs11 | 14 | 1200 | 61 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lorenz, O.; Kühne, A.; Rudolph, M.; Diyatmika, W.; Prager, A.; Gerlach, J.W.; Griebel, J.; Winkler, S.; Lotnyk, A.; Anders, A.; et al. Role of Reaction Intermediate Diffusion on the Performance of Platinum Electrodes in Solid Acid Fuel Cells. Catalysts 2021, 11, 1065. https://doi.org/10.3390/catal11091065
Lorenz O, Kühne A, Rudolph M, Diyatmika W, Prager A, Gerlach JW, Griebel J, Winkler S, Lotnyk A, Anders A, et al. Role of Reaction Intermediate Diffusion on the Performance of Platinum Electrodes in Solid Acid Fuel Cells. Catalysts. 2021; 11(9):1065. https://doi.org/10.3390/catal11091065
Chicago/Turabian StyleLorenz, Oliver, Alexander Kühne, Martin Rudolph, Wahyu Diyatmika, Andrea Prager, Jürgen W. Gerlach, Jan Griebel, Sara Winkler, Andriy Lotnyk, André Anders, and et al. 2021. "Role of Reaction Intermediate Diffusion on the Performance of Platinum Electrodes in Solid Acid Fuel Cells" Catalysts 11, no. 9: 1065. https://doi.org/10.3390/catal11091065
APA StyleLorenz, O., Kühne, A., Rudolph, M., Diyatmika, W., Prager, A., Gerlach, J. W., Griebel, J., Winkler, S., Lotnyk, A., Anders, A., & Abel, B. (2021). Role of Reaction Intermediate Diffusion on the Performance of Platinum Electrodes in Solid Acid Fuel Cells. Catalysts, 11(9), 1065. https://doi.org/10.3390/catal11091065