
Electrochemical Incorporation of Carbon Dioxide into Fluorotoluene Derivatives under Mild Conditions



Abstract
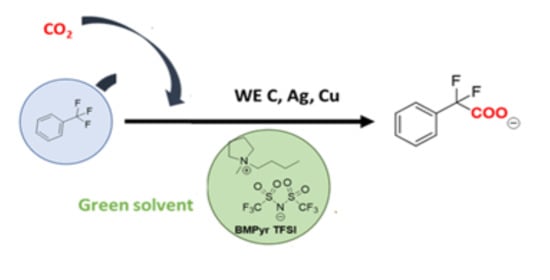
:
1. Introduction
2. Results and Discussion
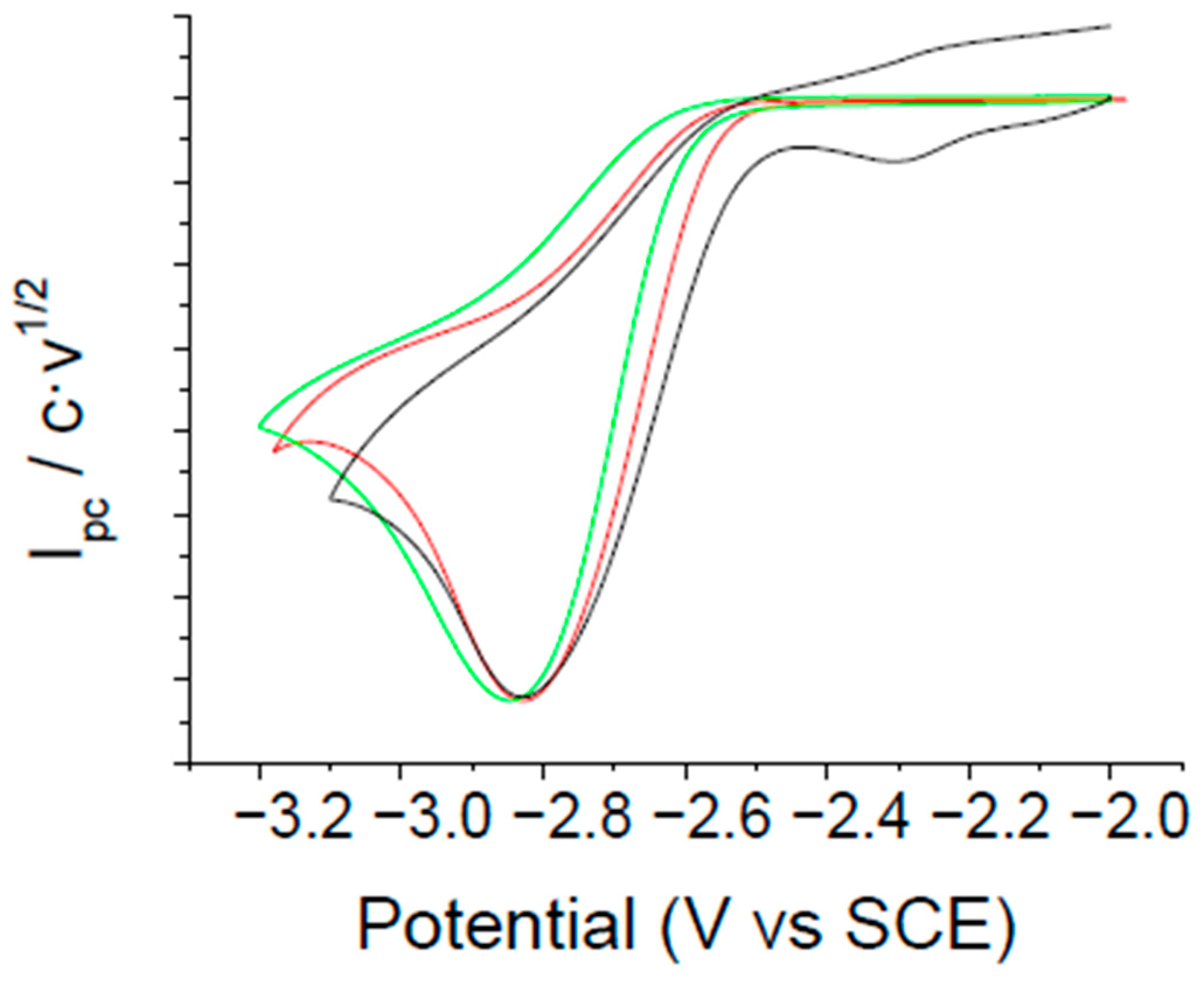
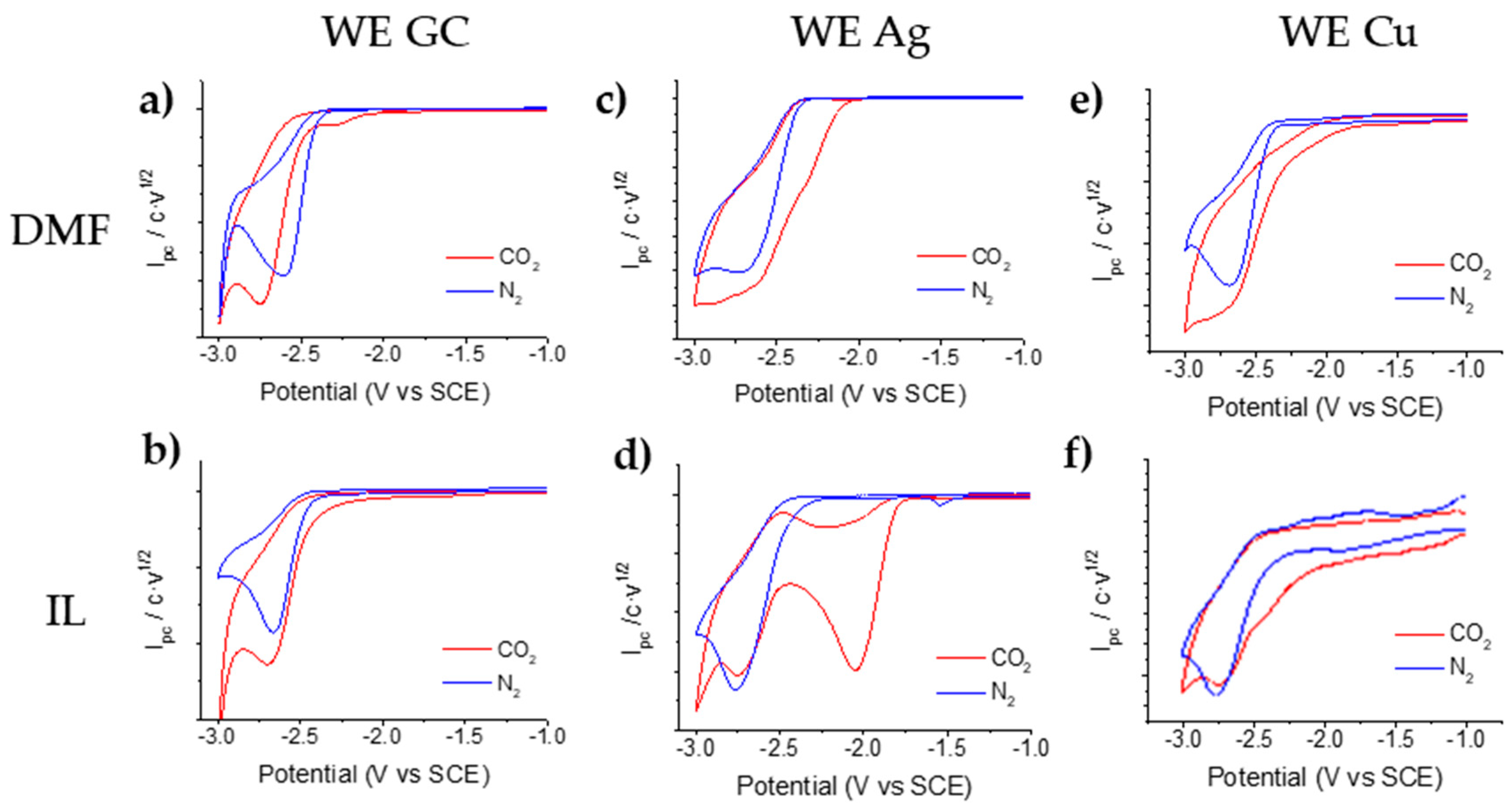
2.1. Electrochemical Reduction of α,α,α-trifluorotoluene under an Inert Atmosphere
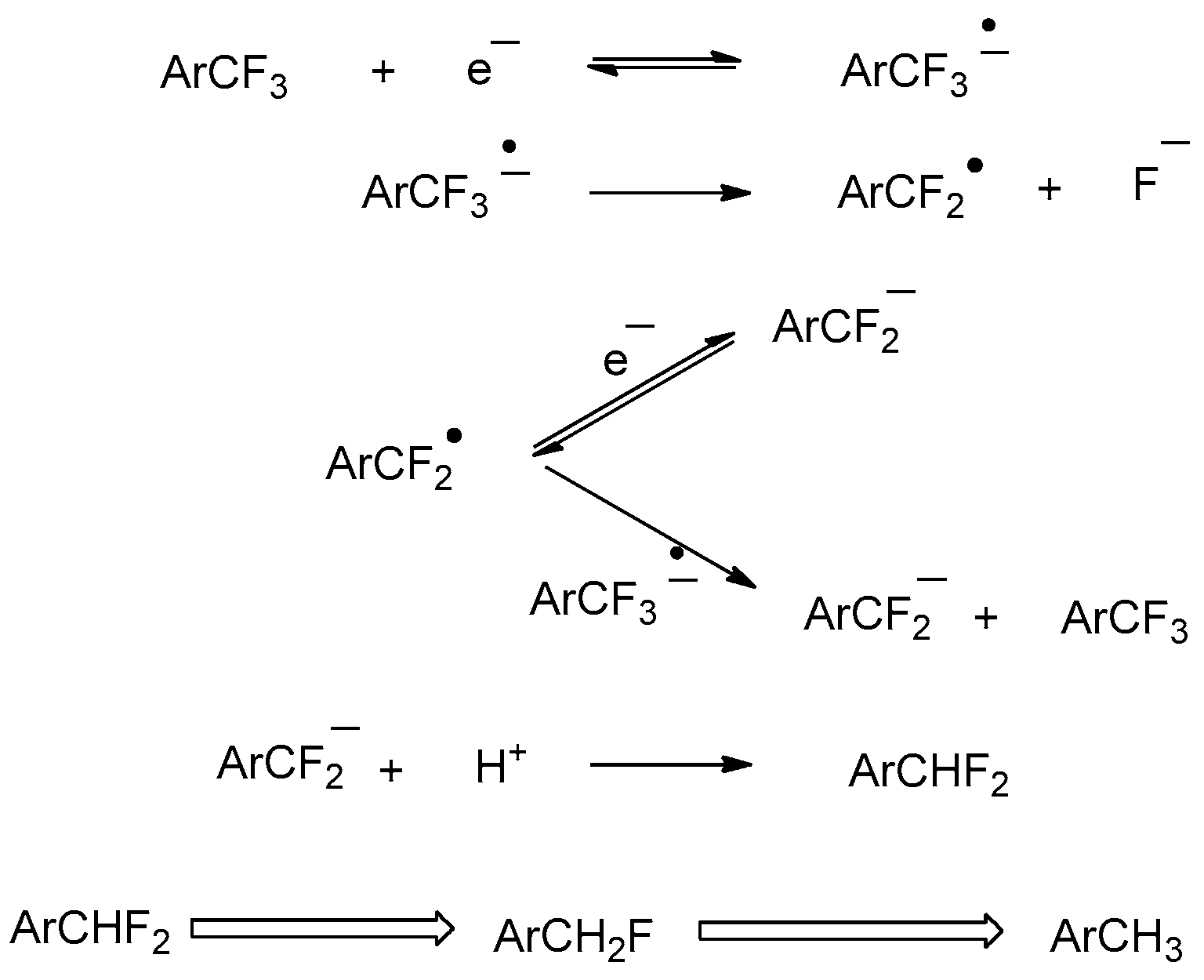
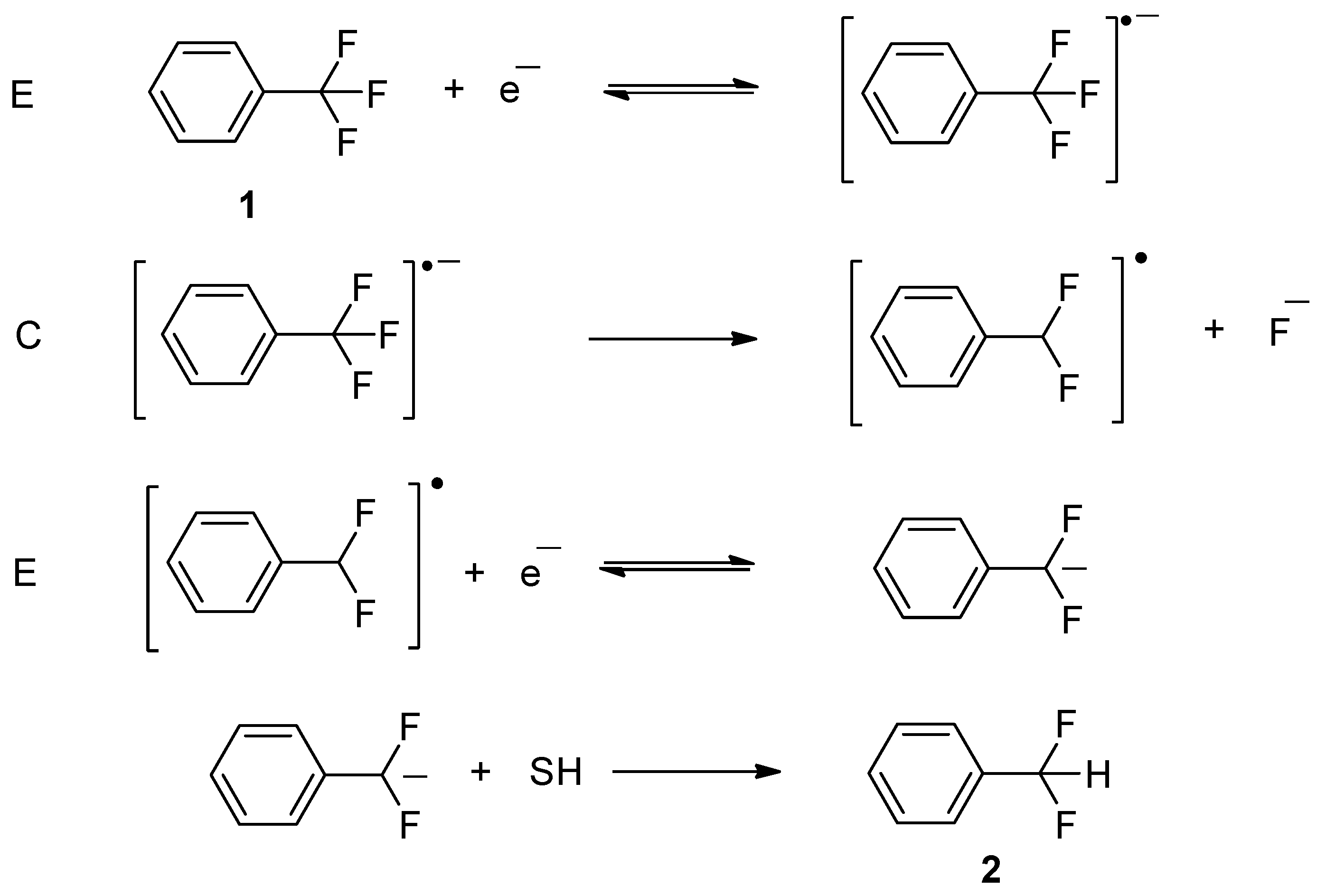
2.1.1. Electrochemical Reduction Mechanism of α,α,α-trifluorotoluene in a Polar Aprotic Solvent
2.1.2. Electrochemical Reduction Mechanism of α,α,α-trifluorotoluene in Ionic Liquid
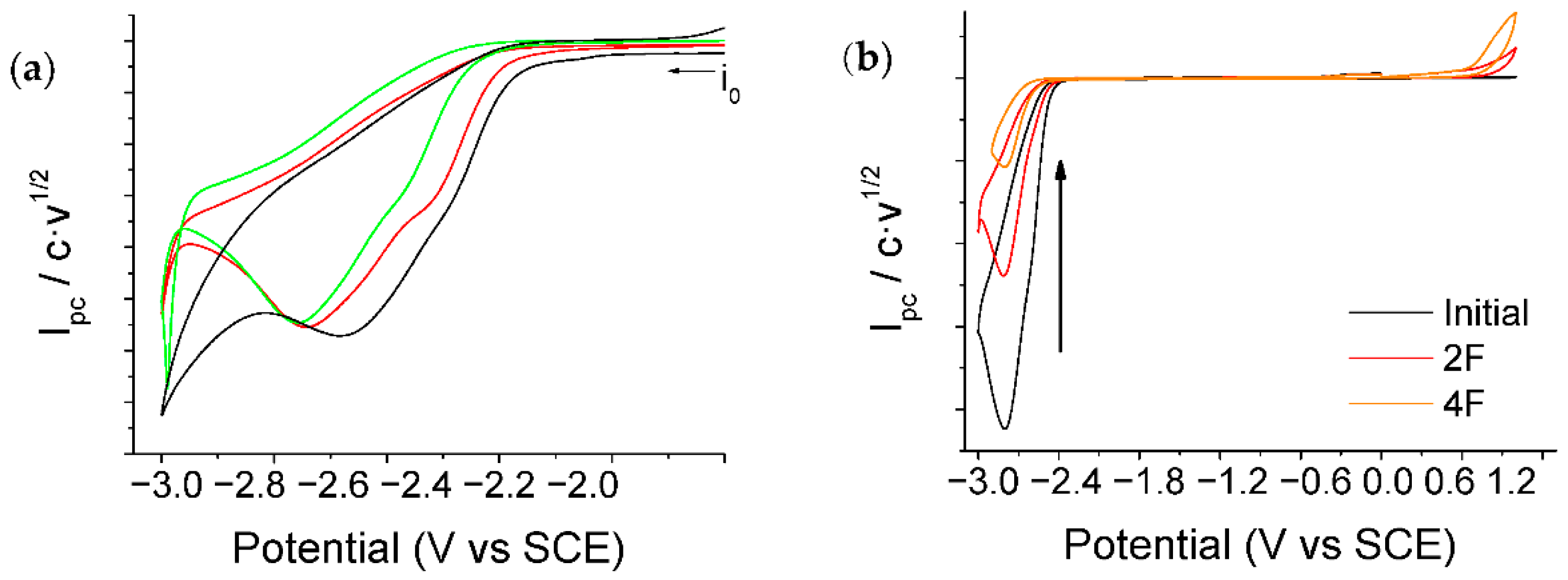
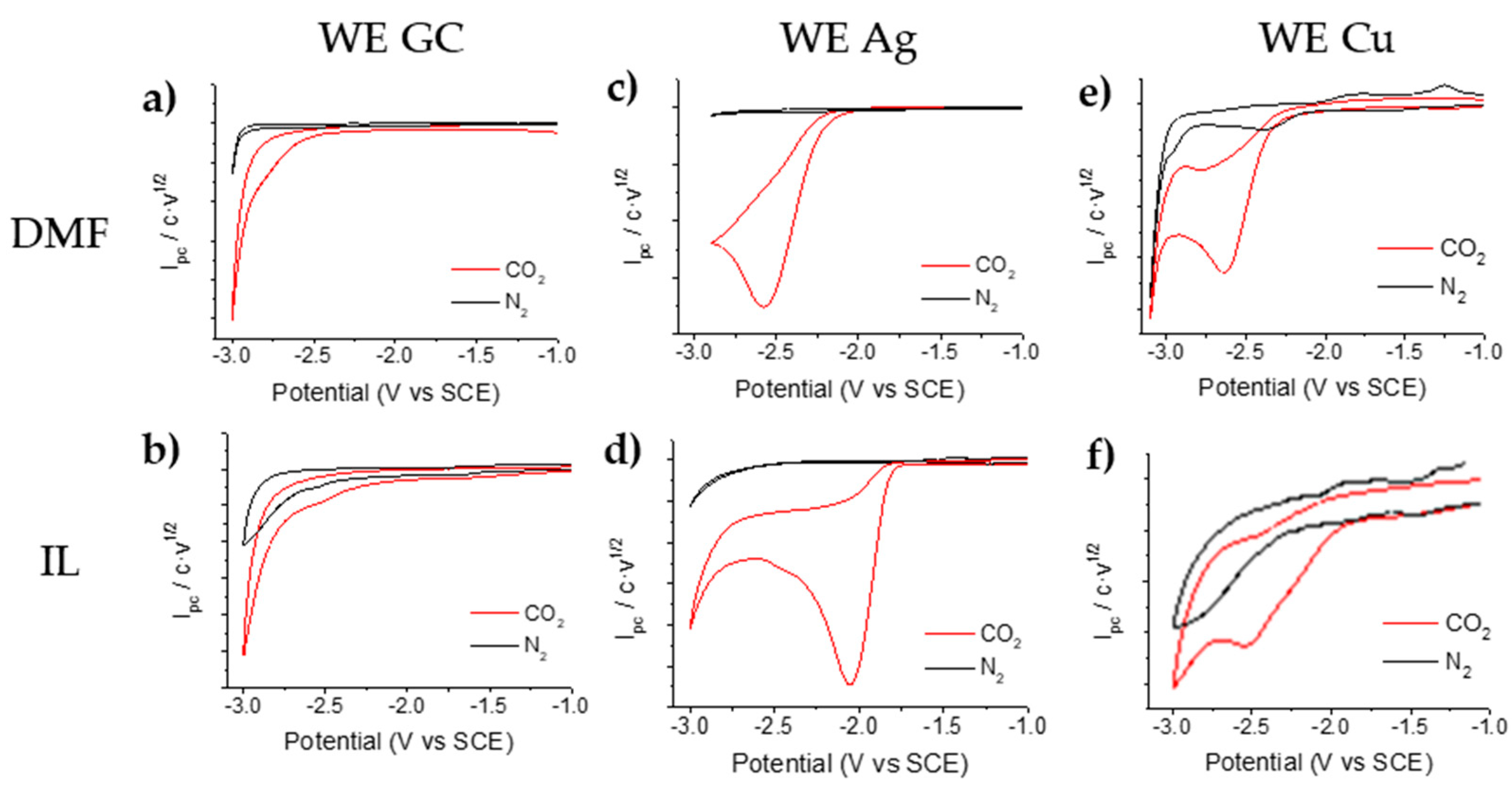
2.2. Electrochemical Study of α,α,α-trifluorotoluene in a Saturated CO2 Atmosphere
2.2.1. CO2 Electrochemical Reduction Mechanism in DMF/0.1M TBA PF6 and BMPyr TFSI
2.2.2. Electrocarboxylation of α,α,α-trifluorotoluene in DMF/0.1M TBA PF6 and BMPyr TFSI
3. Materials and Methods
3.1. Materials
3.2. Electrochemical Experiments
3.3. Determination of the CO2 Concentration in Solution
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bekun, F.V.; Alola, A.A.; Sarkodie, S.A. Toward a sustainable environment: Nexus between CO2 emissions, resource rent, renewable and nonrenewable energy in 16-EU countries. Sci. Total Environ. 2019, 657, 1023–1029. [Google Scholar] [CrossRef]
- Şen, Z. Global warming threat on water resources and environment: A review. Environ. Earth Sci. 2009, 57, 321–329. [Google Scholar] [CrossRef]
- Gielen, D.; Boshell, F.; Saygin, D.; Bazilian, M.D.; Wagner, N.; Gorini, R. The role of renewable energy in the global energy transformation. Energy Strategy Rev. 2019, 24, 38–50. [Google Scholar] [CrossRef]
- Godin, J.; Liu, W.; Ren, S.; Xu, C.C. Advances in recovery and utilization of carbon dioxide: A brief review. J. Environ. Chem. Eng. 2021, 9, 105644. [Google Scholar] [CrossRef]
- Ikreedeegh, R.R.; Tahir, M. A critical review in recent developments of metal-organic-frameworks (MOFs) with band engineering alteration for photocatalytic CO2 reduction to solar fuels. J. CO2 Util. 2021, 43, 101381. [Google Scholar] [CrossRef]
- Babin, A.; Vaneeckhaute, C.; Iliuta, M.C. Potential and challenges of bioenergy with carbon capture and storage as a carbon-negative energy source: A review. Biomass Bioenergy 2021, 146, 105968. [Google Scholar] [CrossRef]
- Bara, J.E.; Camper, D.E.; Gin, D.; Noble, R.D. Room-Temperature Ionic Liquids and Composite Materials: Platform Technologies for CO2 Capture. Acc. Chem. Res. 2010, 43, 152–159. [Google Scholar] [CrossRef]
- Atsbha, T.A.; Yoon, T.; Seongho, P.; Lee, C.-J. A review on the catalytic conversion of CO2 using H2 for synthesis of CO, methanol, and hydrocarbons. J. CO2 Util. 2021, 44, 101413. [Google Scholar] [CrossRef]
- Da Cruz, T.T.; Balestieri, J.A.P.; de Silva, J.M.T.; Vilanova, M.R.; Oliveira, O.J.; Ávila, I. Life cycle assessment of carbon capture and storage/utilization: From current state to future research directions and opportunities. Int. J. Greenh. Gas Control 2021, 108, 103309. [Google Scholar] [CrossRef]
- Saravanan, A.; Kumar, P.S.; Vo, D.V.N.; Jeevanantham, S.; Bhuvaneswari, V.; Anantha Narayanan, V.; Yaashikaa, P.R.; Swetha, S.; Reshma, B. A Comprehensive Review on Different Approaches for CO2 Utilization and Conversion Pathways. Chem. Eng. Sci. 2021, 236, 116515. [Google Scholar] [CrossRef]
- Yu, D.-G.; He, L.-N. Introduction to CO2 utilisation. Green Chem. 2021, 23, 3499–3501. [Google Scholar] [CrossRef]
- Gennaro, A.; Isse, A.A.; Savéant, J.M.; Severin, M.G.; Vianello, E. Homogeneous Electron Transfer Catalysis of the Electro-chemical Reduction of Carbon Dioxide. Do Aromatic Anion Radicals React in an Outer-Sphere Manner? J. Am. Chem. Soc. 1996, 118, 7190–7196. [Google Scholar] [CrossRef]
- Mena, S.; Guirado, G. Electrochemical Tuning of CO2 Reactivity in Ionic Liquids Using Different Cathodes: From Oxalate to Carboxylation Products. C 2020, 6, 34. [Google Scholar] [CrossRef]
- Matthessen, R.; Fransaer, J.; Binnemans, K.; De Vos, D.E. Electrocarboxylation: Towards sustainable and efficient synthesis of valuable carboxylic acids. Beilstein J. Org. Chem. 2014, 10, 2484–2500. [Google Scholar] [CrossRef] [PubMed]
- Senboku, H.; Katayama, A. Electrochemical carboxylation with carbon dioxide. Curr. Opin. Green Sustain. Chem. 2017, 3, 50–54. [Google Scholar] [CrossRef]
- Isse, A.A.; Ferlin, M.G.; Gennaro, A. Homogeneous electron transfer catalysis in the electrochemical carboxylation of arylethyl chlorides. J. Electroanal. Chem. 2003, 541, 93–101. [Google Scholar] [CrossRef]
- Isse, A.A.; Falciola, L.; Mussini, P.R.; Gennaro, A. Relevance of electron transfer mechanism in electrocatalysis: The reduction of organic halides at silver electrodes. Chem. Commun. 2005, 1, 344–346. [Google Scholar] [CrossRef]
- Isse, A.A.; Durante, C.; Gennaro, A. One-pot synthesis of benzoic acid by electrocatalytic reduction of bromobenzene in the presence of CO2. Electrochem. Commun. 2011, 13, 810–813. [Google Scholar] [CrossRef]
- Reche, I.; Mena, S.; Gallardo, I.; Guirado, G. Electrocarboxylation of halobenzonitriles: An environmentally friendly synthesis of phthalate derivatives. Electrochim. Acta 2019, 320, 134576. [Google Scholar] [CrossRef]
- Isse, A.A.; Galia, A.; Belfiore, C.; Silvestri, G.; Gennaro, A. Electrochemical reduction and carboxylation of halobenzophenones. J. Electroanal. Chem. 2002, 526, 41–52. [Google Scholar] [CrossRef]
- Mena, S.; Loault, C.; Mesa, V.; Gallardo, I.; Guirado, G. Electrochemical reduction of 4-nitrobenzyl phenyl thioether for activation and capture of CO2. ChemElectroChem 2021, 21, 1–14. [Google Scholar]
- Tan, X.; Sun, X.; Han, B. Ionic Liquid-Based electrolytes for CO2 electroreduction and CO2 electroorganic transformation. Natl. Sci. Rev. 2021, 1–41. [Google Scholar] [CrossRef]
- Mena, S.; Gallardo, I.; Guirado, G. Electrocatalytic Processes for the Valorization of CO2: Synthesis of Cyanobenzoic Acid Using Eco-Friendly Strategies. Catalysts 2019, 9, 413. [Google Scholar] [CrossRef] [Green Version]
- Mena, S.; Sanchez, J.; Guirado, G. Electrocarboxylation of 1-chloro-(4-isobutylphenyl)ethane with a silver cathode in ionic liquids: An environmentally benign and efficient way to synthesize Ibuprofen. RSC Adv. 2019, 9, 15115–15123. [Google Scholar] [CrossRef] [Green Version]
- Mena, S.; Santiago, S.; Gallardo, I.; Guirado, G. Sustainable and efficient electrosynthesis of naproxen using carbon dioxide and ionic liquids. Chemosphere 2020, 245, 125557. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.-A.; Cahard, D. Strategies for nucleophilic, electrophilic, and radical trifluoromethylations. J. Fluor. Chem. 2007, 128, 975–996. [Google Scholar] [CrossRef]
- Yoo, W.J.; Kondo, J.; Rodríguez-Santamaría, J.A.; Nguyen, T.V.Q.; Kobayashi, S. Efficient Synthesis of α-Trifluoromethyl Carboxylic Acids and Esters through Fluorocarboxylation of Gem-Difluoroalkenes. Angew. Chem. Int. Ed. 2019, 58, 6772–6775. [Google Scholar] [CrossRef] [PubMed]
- Andrieux, C.P.; Combellas, C.; Kanoufi, F.; Savéant, J.M.; Thiébault, A. Dynamics of Bond Breaking in Ion Radicals. Mech-anisms and Reactivity in the Reductive Cleavage of Carbon-Fluorine Bonds of Fluoromethylarenes. J. Am. Chem. Soc. 1997, 119, 9527–9540. [Google Scholar] [CrossRef]
- Isse, A.A.; Gottardello, S.; Durante, C.; Gennaro, A. Dissociative electron transfer to organic chlorides: Electrocatalysis at metal cathodes. Phys. Chem. Chem. Phys. 2008, 10, 2409–2416. [Google Scholar] [CrossRef]
- Reche, I.; Gallardo, I.; Guirado, G. Cyclic voltammetry using silver as cathode material: A simple method for determining electro and chemical features and solubility values of CO2 in ionic liquids. Phys. Chem. Chem. Phys. 2015, 17, 2339–2343. [Google Scholar] [CrossRef]
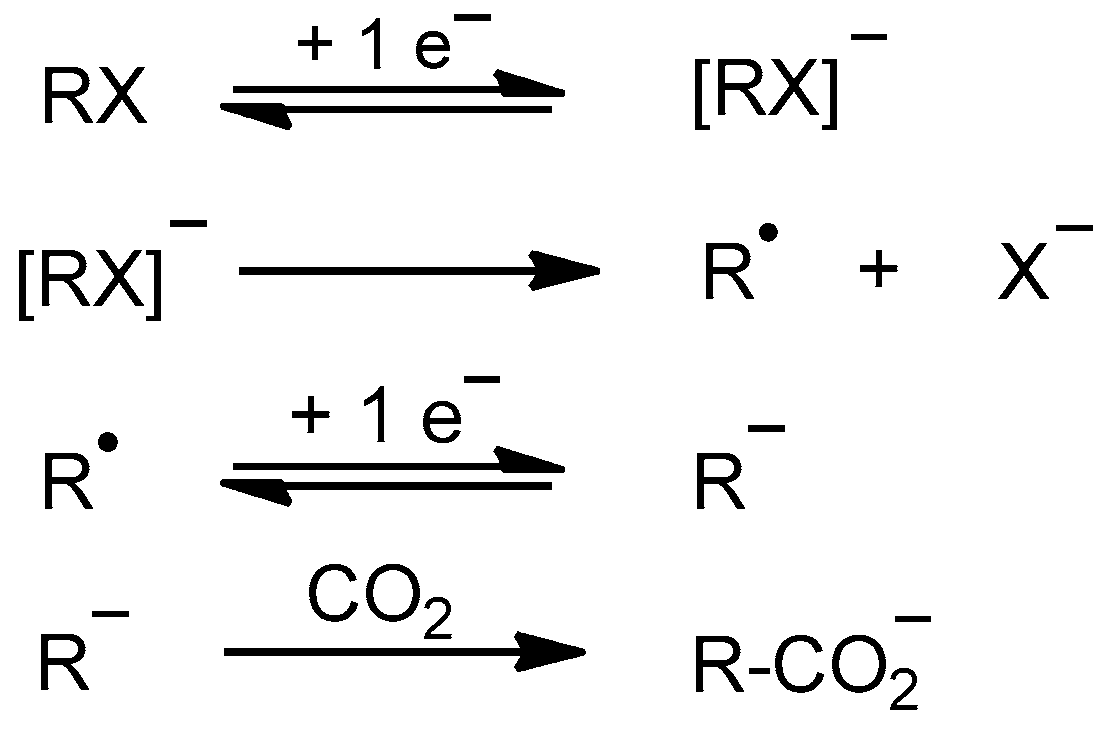
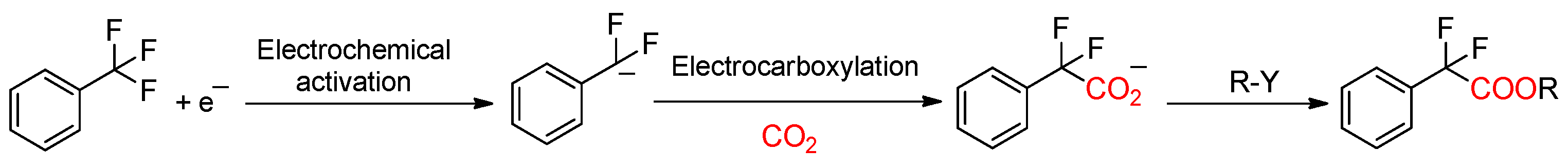
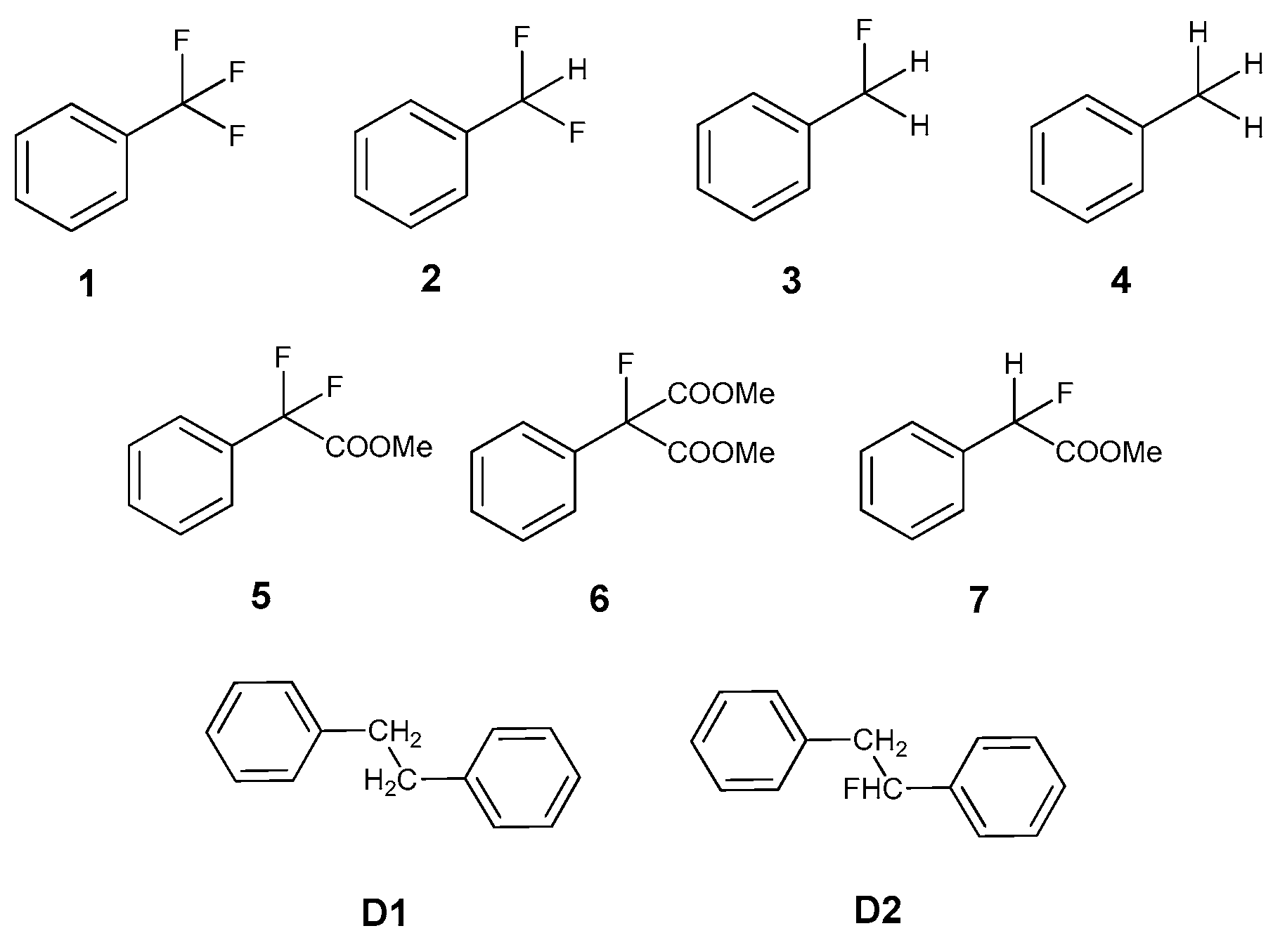
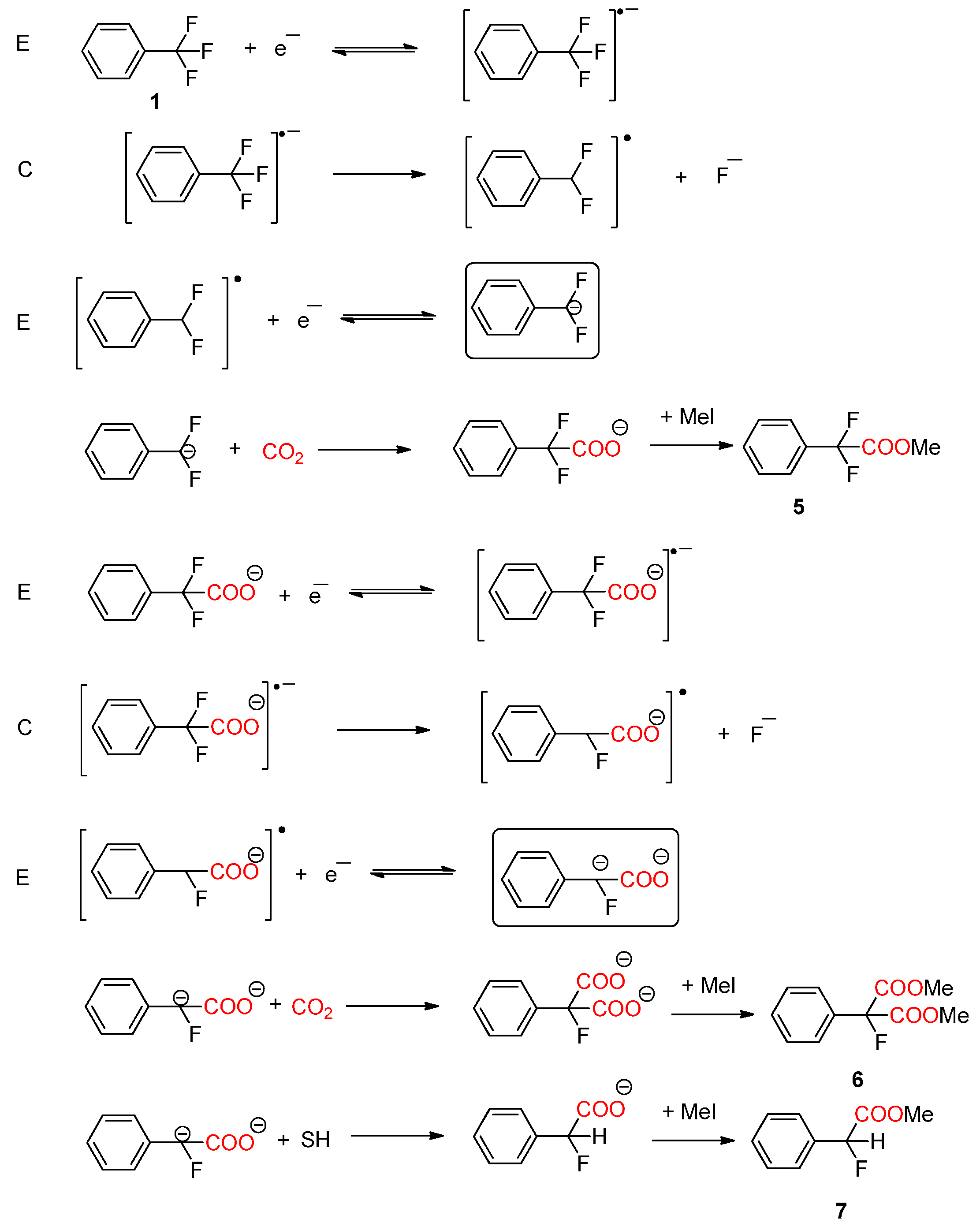

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WE | v (V/s) | Epc (V vs. SCE) | ΔEp (mV) a | α |
|---|---|---|---|---|
| GC | 0.1 | −2.72 | 195 | 0.25 |
| 0.3 | −2.75 | 200 | 0.24 | |
| 0.5 | −2.76 | 209 | 0.23 | |
| 0.7 | −2.73 | 197 | 0.24 | |
| 1.0 | −2.76 | 203 | 0.24 | |
| Ag | 0.1 | −2.70 | 235 | 0.21 |
| 0.3 | −2.71 | 241 | 0.20 | |
| 0.5 | −2.75 | 241 | 0.20 | |
| 0.7 | −2.78 | 246 | 0.20 | |
| 1.0 | −2.78 | 253 | 0.19 | |
| Cu | 0.1 | −2.68 | 208 | 0.23 |
| 0.3 | −2.69 | 214 | 0.22 | |
| 0.5 | −2.69 | 218 | 0.22 | |
| 0.7 | −2.70 | 222 | 0.22 | |
| 1.0 | −2.72 | 231 | 0.21 |
| Cathode | Eapplied (V vs. SCE) | C·mol−1 | Products Obtained | Conversion Rates a | Efficiency |
|---|---|---|---|---|---|
| C | −2.9 | 4.7 | 2 | 74 | 50% |
| 3 | 26 | ||||
| Ag | −2.9 | 4.0 | 2 | 50 | 80% |
| 3 | 50 | ||||
| 4 | Traces | ||||
| Cu | −2.9 | 3.7 | 1 | Traces | 80% |
| 2 | 52 | ||||
| 3 | 48 |
| WE | v (V/s) | Epc (V vs. SCE) | ΔEp (mV) a | α | nº Electrons b |
|---|---|---|---|---|---|
| GC | 0.1 | −2.86 | 110 | 0.44 | 7.74 |
| 0.3 | −2.87 | 109 | 0.44 | 6.55 | |
| 0.5 | −2.89 | 102 | 0.47 | 6.53 | |
| 0.7 | −2.91 | 120 | 0.40 | 6.15 | |
| 1 | −2.92 | 126 | 0.38 | 5.95 | |
| 113 ± 9 | 0.43 | 6.6 ± 0.7 | |||
| Ag | 0.1 | −2.86 | 99 | 0.49 | 7.66 |
| 0.3 | −2.87 | 96 | 0.50 | 7.03 | |
| 0.5 | −2.88 | 99 | 0.49 | 6.36 | |
| 0.7 | −2.89 | 107 | 0.45 | 6.02 | |
| 1 | −2.90 | 113 | 0.43 | 5.80 | |
| 103 ± 7 | 0.47 | 6.6 ± 0.8 | |||
| Cu | 0.1 | −2.79 | 103 | 0.47 | 7.24 |
| 0.3 | −2.80 | 102 | 0.47 | 7.04 | |
| 0.5 | −2.83 | 123 | 0.39 | 6.83 | |
| 0.7 | −2.83 | 124 | 0.39 | 6.04 | |
| 1 | −2.85 | 136 | 0.35 | 6.08 | |
| 118 ± 14 | 0.41 | 6.6 ± 0.6 | |||
| Cathode | Epc (V) vs. SCE | ΔEp (mV) a | α b | [CO2] (mM) |
|---|---|---|---|---|
| DMF/0.1M TBA PF6 | ||||
| GC | - | - | - | - |
| Ag | −2.638 | 244 | 0.20 | 118 |
| Cu | −2.676 | 192 | 0.25 | 118 |
| BuMePyr TFSI | ||||
| GC | - | - | - | - |
| Ag | −2.050 | 144 | 0.33 | 454 |
| Cu | −2.538 | 375 | 0.13 | 454 |
| DMF/0.1M TBA PF6 | |||||
| Cathode | Eapplied (V) | C·mol−1 | Defluorinated Products (Yield%) a | Carboxylated Products (Yield%) a | Efficiency Electrocarboxylation |
| Ag | −2.8 | 6.2 | D1 (6%) | 5 (82%) | 31% |
| D2 (5%) | 7 (7%) | ||||
| Cu | −2.8 | 4.2 | 3 (26%) | 5 (70%) | 37% |
| 6 (4%) | |||||
| BuMePyr TFSI | |||||
| Cathode | Eapplied (V) | C·mol−1 | Carboxylated Products | Yield a | Efficiency Electrocarboxylation |
| Ag | −2.9 | 6.0 | 5 | 100% | 33% |
| Cu | −2.9 | 6.0 | 5 | 31% | 56% |
| 7 | 69% | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mena, S.; Bernad, J.; Guirado, G. Electrochemical Incorporation of Carbon Dioxide into Fluorotoluene Derivatives under Mild Conditions. Catalysts 2021, 11, 880. https://doi.org/10.3390/catal11080880
Mena S, Bernad J, Guirado G. Electrochemical Incorporation of Carbon Dioxide into Fluorotoluene Derivatives under Mild Conditions. Catalysts. 2021; 11(8):880. https://doi.org/10.3390/catal11080880
Chicago/Turabian StyleMena, Silvia, Jesus Bernad, and Gonzalo Guirado. 2021. "Electrochemical Incorporation of Carbon Dioxide into Fluorotoluene Derivatives under Mild Conditions" Catalysts 11, no. 8: 880. https://doi.org/10.3390/catal11080880
APA StyleMena, S., Bernad, J., & Guirado, G. (2021). Electrochemical Incorporation of Carbon Dioxide into Fluorotoluene Derivatives under Mild Conditions. Catalysts, 11(8), 880. https://doi.org/10.3390/catal11080880