
Alcohol Oxidation Assisted by Molybdenum Hydrazonato Catalysts Employing Hydroperoxide Oxidants


 ,
,  and
and 
Abstract
:
1. Introduction
2. Results and Discussion
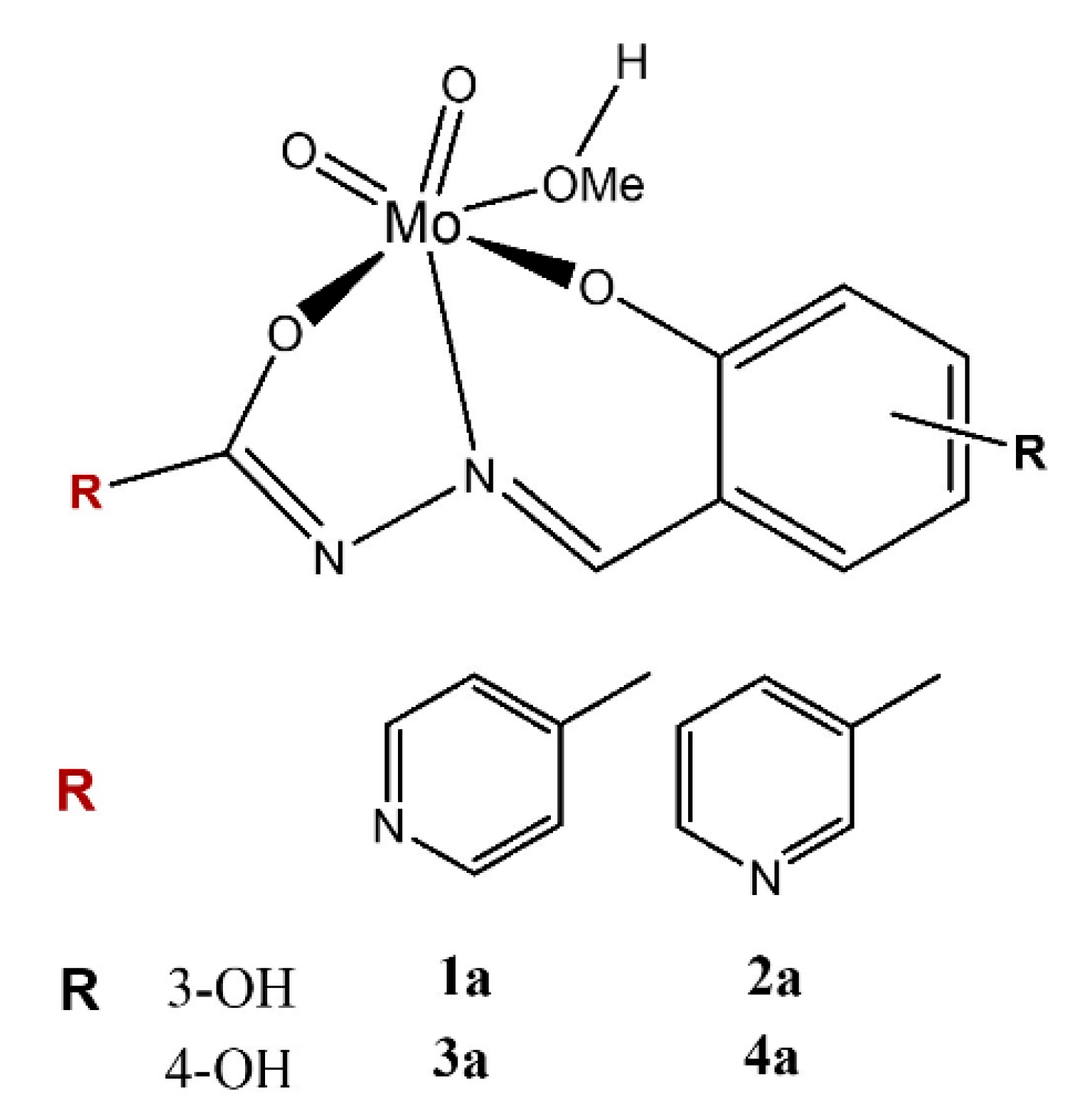
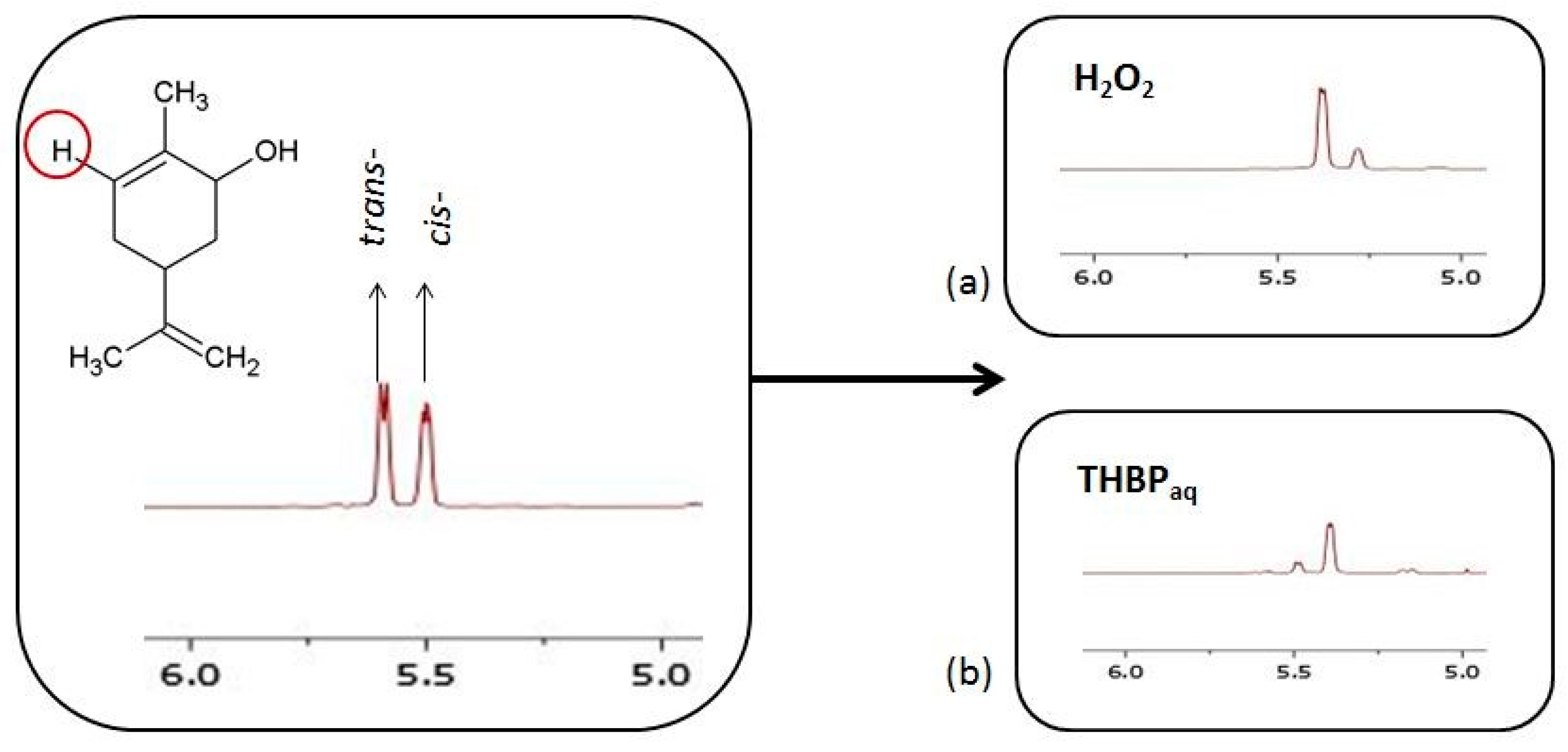
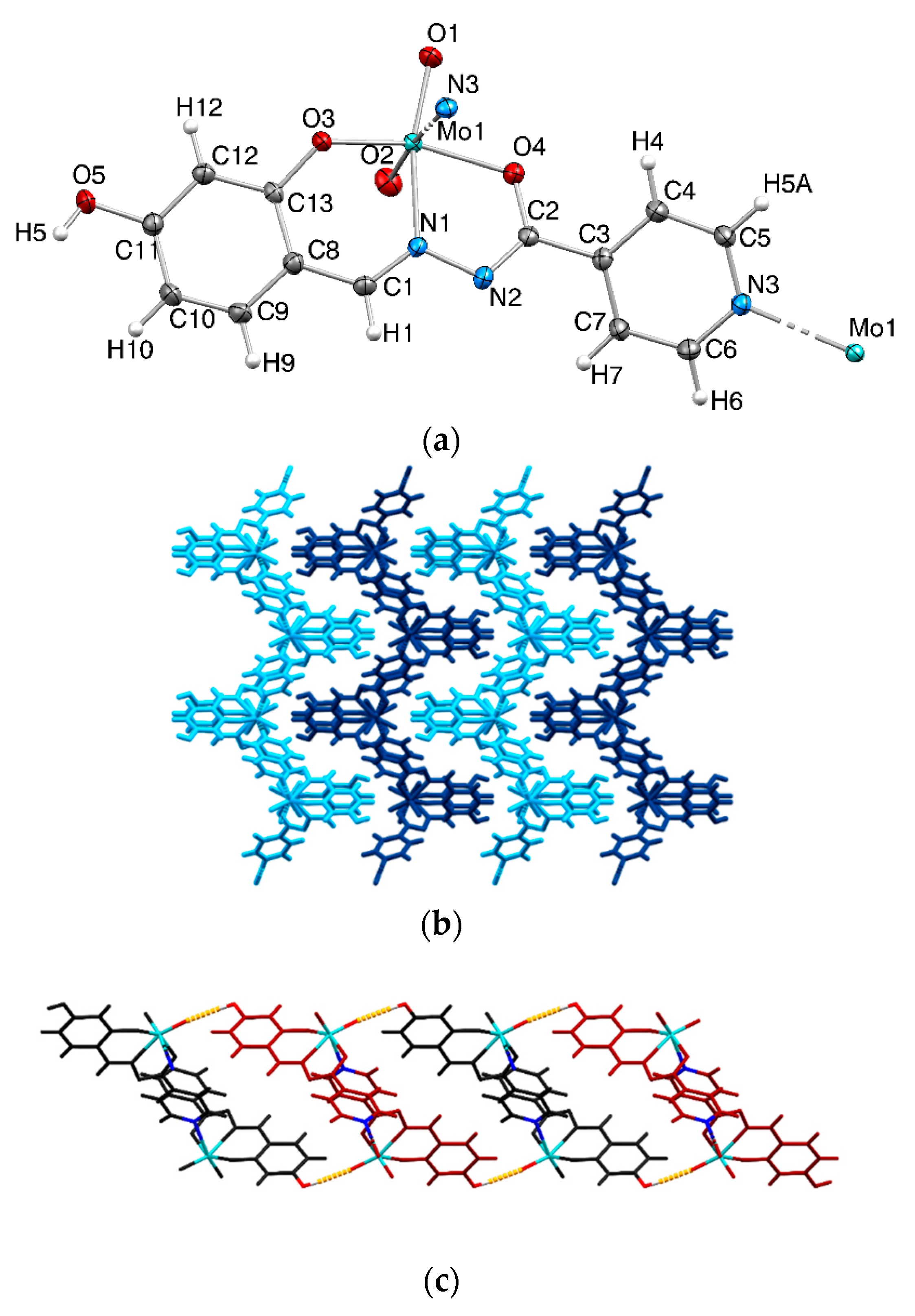
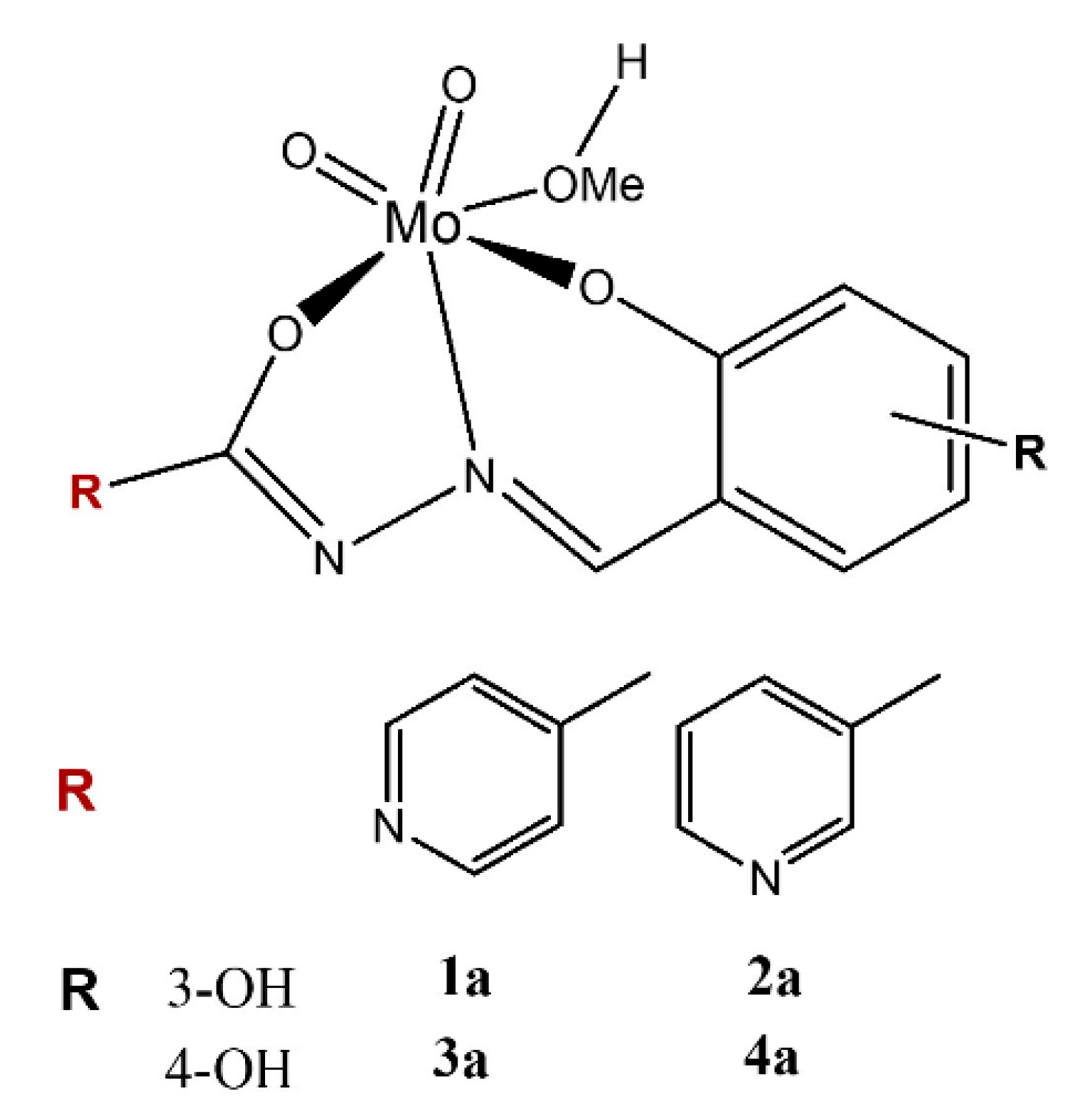
2.1. Catalysts Preparation and Characterization
2.2. Catalytic Results
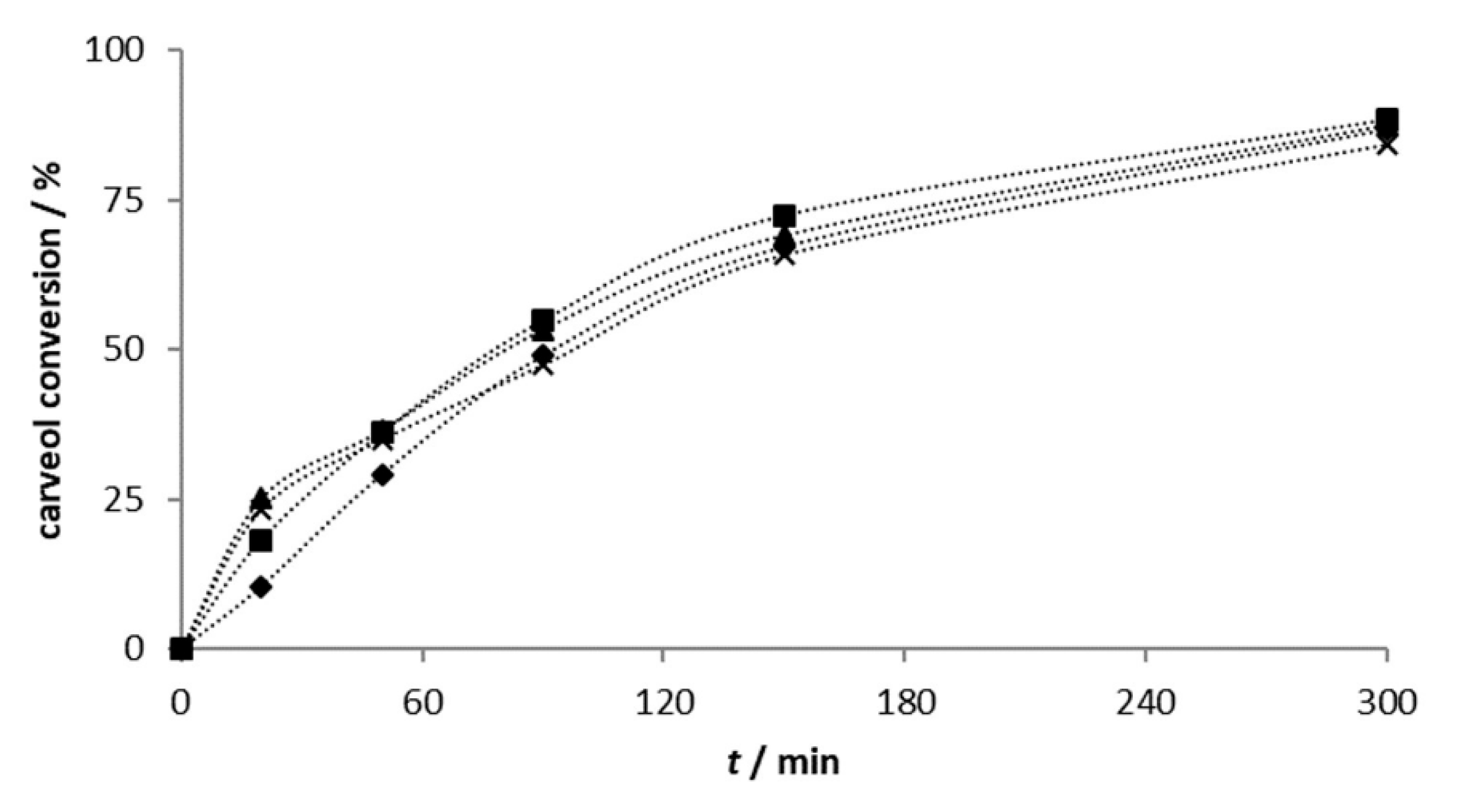
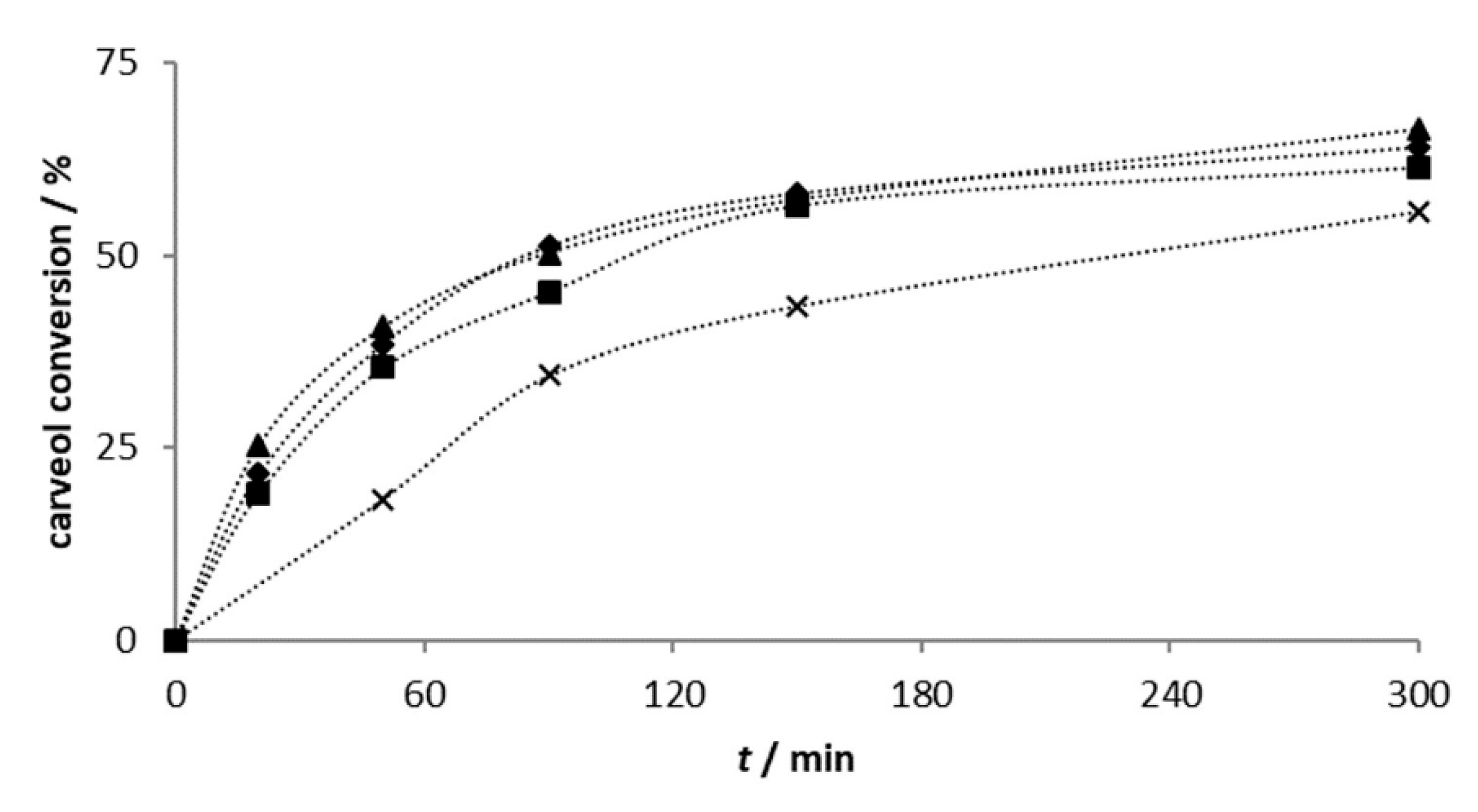
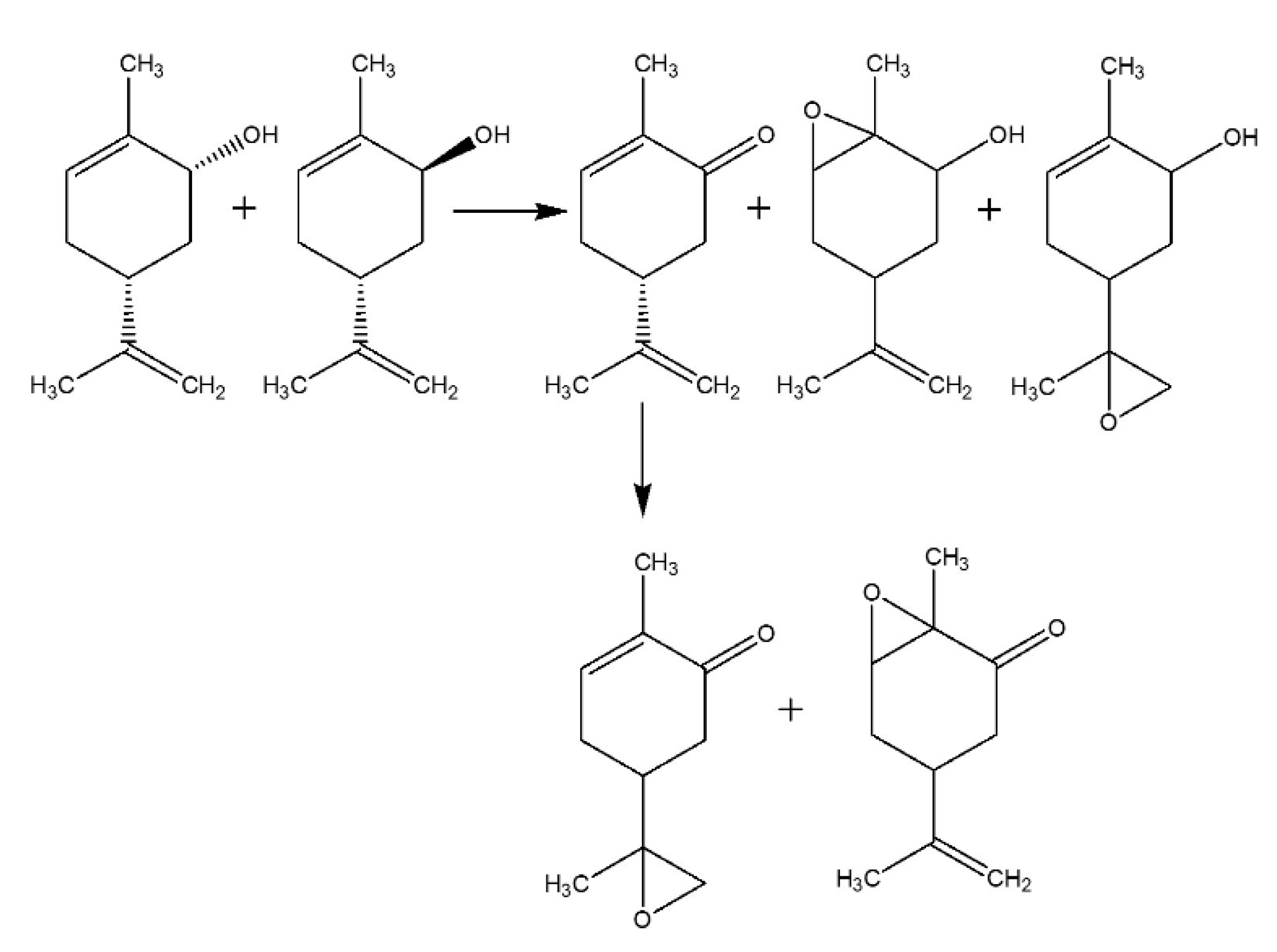
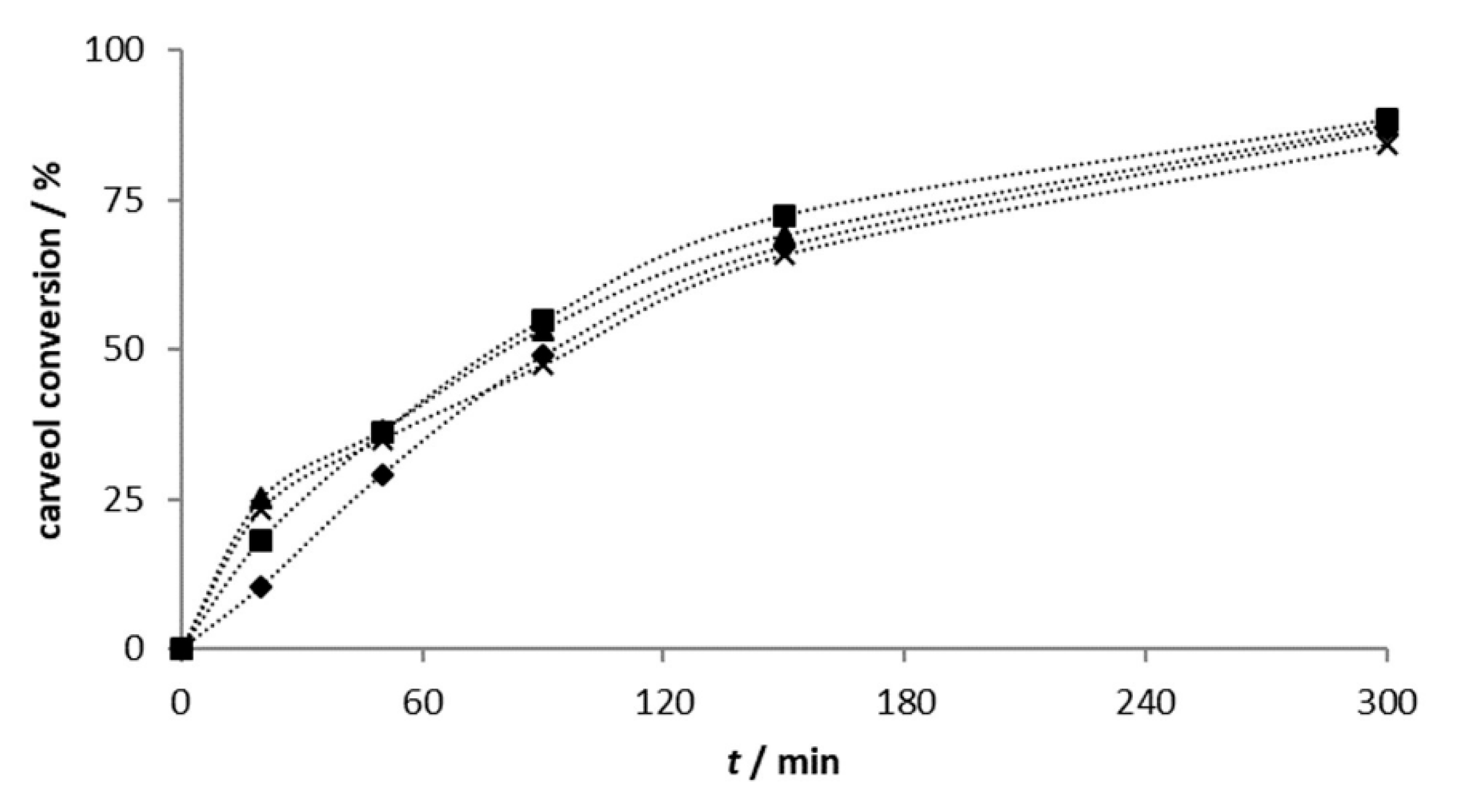
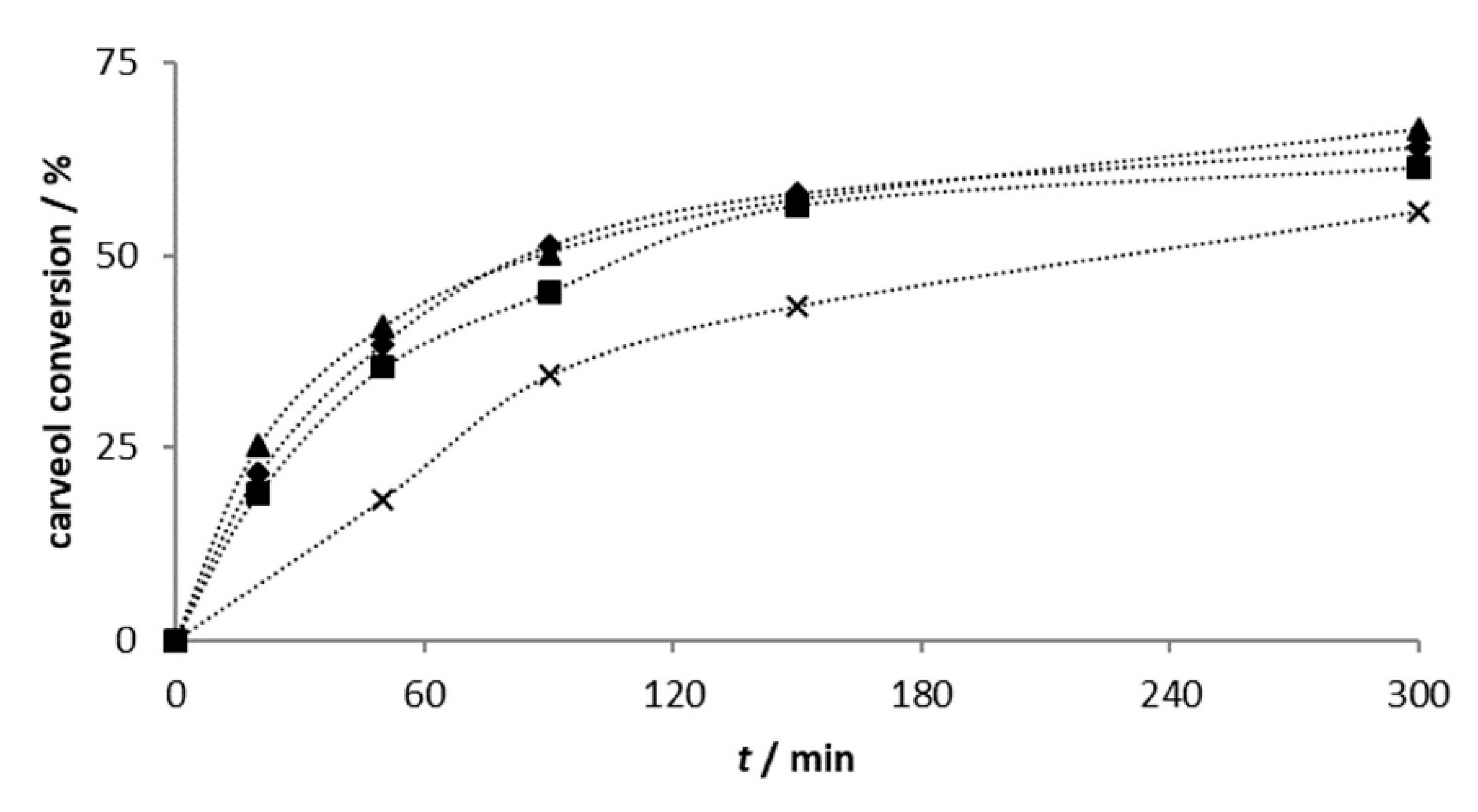
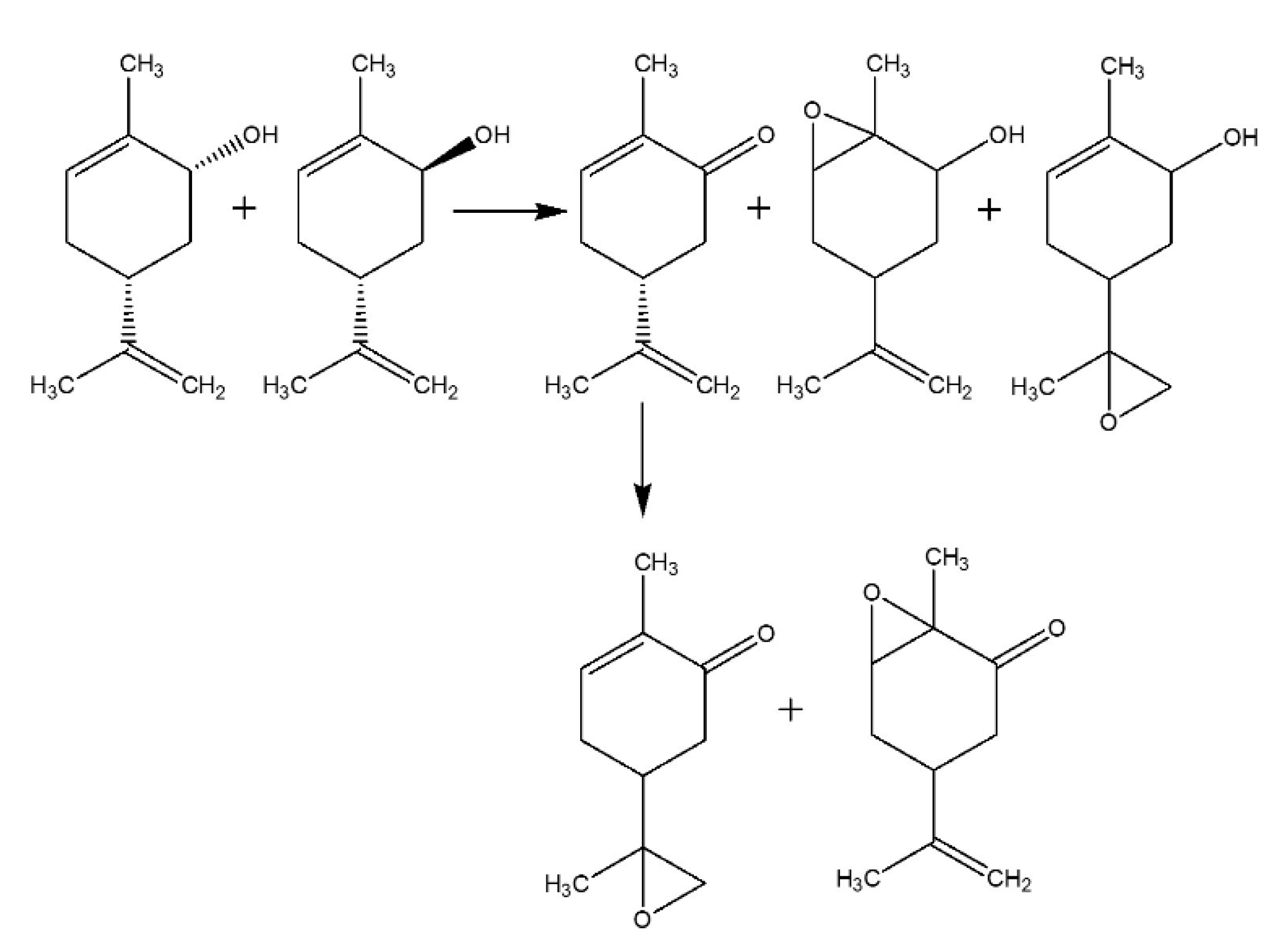
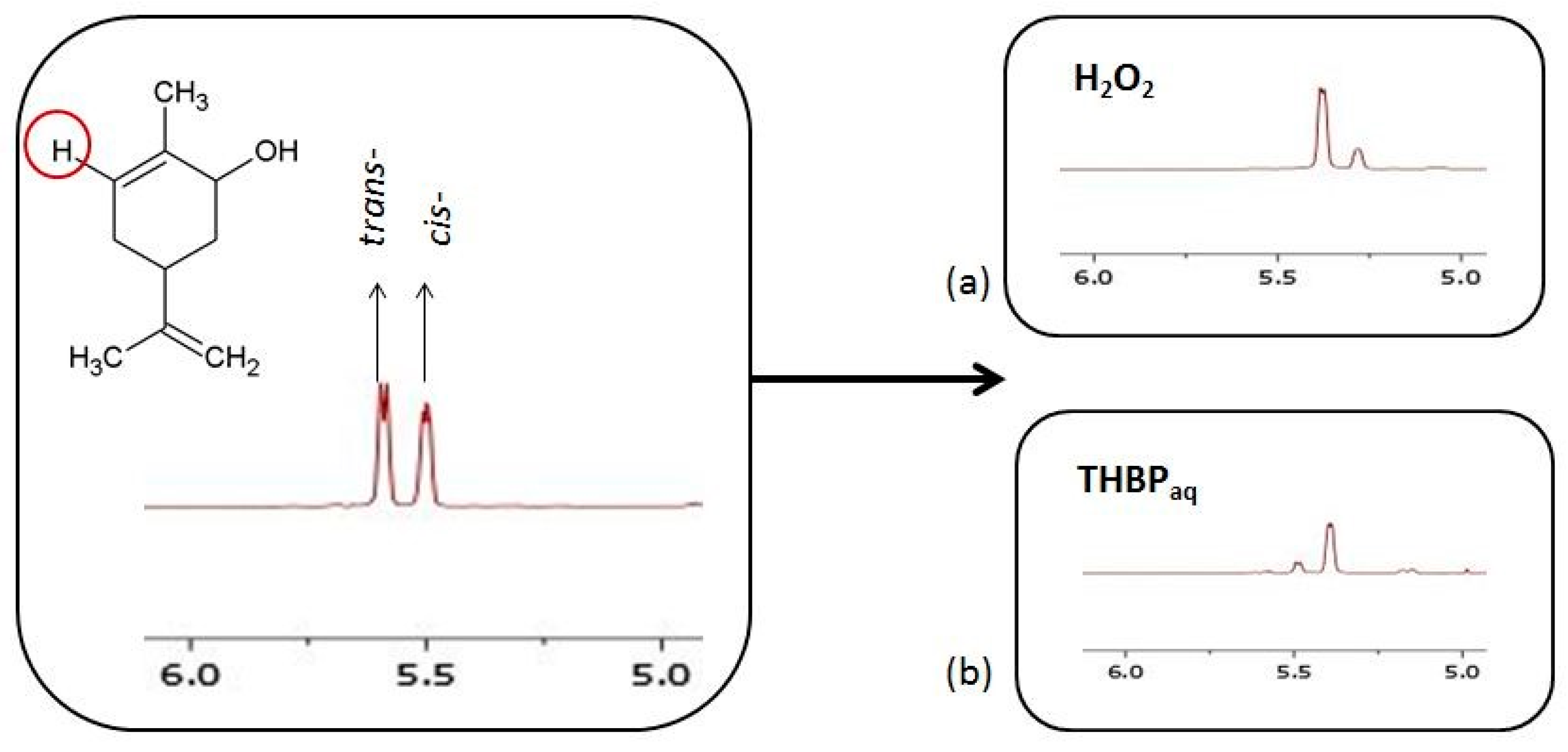
2.2.1. Carveol Oxidation
2.2.2. Cyclohexanol Oxidation
2.2.3. Butan-2-ol Oxidation
3. Materials and Methods
3.1. Preparative Part
3.1.1. Synthesis of the Polynuclear Molybdenum Complexes
3.1.2. Synthesis of the Mononuclear Molybdenum Complexes
3.2. General Procedure for the Oxidation of Secondary Alcohols
3.2.1. Carveol Oxidation
3.2.2. Cyclohexanol Oxidation
Protocol A
Protocol B
3.2.3. Butan-2-ol Oxidation
Without MeCN (Protocol A)
With MeCN (Protocol B)
3.3. Physical Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ten Brink, G.J.; Arends, I.W.; Sheldon, R.A. Green, Catalytic Oxidation of Alcohols in Water. Science 2000, 287, 1636–1639. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, P.; Hess, M.; Weyhermüller, T.; Wieghardt, K. Aerobic Oxidation of Primary Alcohols by a New Mononuclear Cu(II)-Radical Catalyst. Angew. Chem. Int. Ed. 1999, 38, 1095–1098. [Google Scholar] [CrossRef]
- Fernandes, R.R.; Lasri, J.; Guedes da Silva, M.F.C.; da Silva, J.A.L.; Frausto da Silva, J.J.R.; Pombeiro, A.J.L. Bis- and tris-pyridyl amino and imino thioether Cu and Fe complexes. Thermal and microwave-assisted peroxidative oxidations of 1-phenylethanol and cyclohexane in the presence of various N-based additives. J. Mol. Catal. A Chem. 2011, 351, 100–111. [Google Scholar] [CrossRef]
- Guérin, B.; Fernandes, D.M.; Daran, J.-C.; Agustin, D.; Poli, R. Investigation of induction times, activity, selectivity, interface and mass transport in solvent-free epoxidation by H2O2 and TBHP: A study with organic salts of the [PMo12O40]3− anion. New J. Chem. 2013, 37, 3466–3475. [Google Scholar] [CrossRef]
- Bhatia, S.P.; Mcginty, D.; Letizia, C.S.; Api, A.M. Fragrance material review on carveol. Food Chem. Toxicol. 2008, 46, 85–87. [Google Scholar] [CrossRef]
- Stekrova, M.; Kumar, N.; Díaz, S.F.; Mäki-Arvela, P.; Murzin, D.Y. H- and Fe-modified zeolite beta catalysts for prepa-ration of trans-carveol from α-pinene oxide. Catal. Today 2015, 241, 237–245. [Google Scholar] [CrossRef]
- Santos, I.; Gamelas, J.; Duarte, T.; Simoes, M.; Neves, M.; Cavaleiro, J.; Cavaleiro, A. Catalytic homogeneous oxidation of monoterpenes and cyclooctene with hydrogen peroxide in the presence of sandwich-type tungstophosphates [M4(H2O)2(PW9O34)2]n−, M = CoII, MnII and FeIII. J. Mol. Catal. A Chem. 2017, 426, 593–599. [Google Scholar] [CrossRef]
- Grajales, G.; Gonzáles, I.; Villa, A. Catalytic oxidative dehydrogenation of carveol to carvone over the phthalocyanine complex FePcCl16 immobilized on the mesoporous silica SBA-15. Appl. Catal. A Gen. 2017, 541, 15–24. [Google Scholar] [CrossRef]
- Cavani, F.; Ferroni, L.; Frattini, A.; Lucarelli, C.; Mazzini, A.; Raabova, K.; Alini, S.; Accorinti, P.; Babini, P. Evidence for the presence of alternative mechanisms in the oxidation of cyclohexanone to adipic acid with oxygen, catalysed by Keggin polyoxometalates. Appl. Catal. A Gen. 2011, 391, 118–124. [Google Scholar] [CrossRef]
- Mouanni, S.; Mazari, T.; Amitouche, D.; Bendji, S.; Dermeche, L.; Roch-Marchal, C.; Rabia, C. Preparation and characterization of H3−2(x+y)MnxCoyPMo12O40 heteropolysalts. Application to adipic acid green synthesis from cyclohexanone oxidation with hydrogen peroxide. C. R. Chim. 2019, 22, 327–336. [Google Scholar] [CrossRef]
- Hajavazzadadeh, R.; Zazi, M.K.; Mahjoub, A.R. Aliphatic alcohols oxidation with Hydrogen Peroxide in water catalyzed by supported Phosphotungstic acid (PTA) on Silica coated MgAl2O4 nanoparticles as a recoverable catalyst. Int. J. Nano Dimens 2019, 10, 69–77. [Google Scholar]
- Soumini, C.; Sugunan, S.; Haridas, S. Copper oxide modified SBA-15 for the selective vapour phase dehydrogenation of cyclohexanol to cyclohexanone. J. Porous Mater. 2019, 26, 631–640. [Google Scholar] [CrossRef]
- Maurya, M.R.; Dhaka, S.; Avecilla, F. Oxidation of secondary alcohols by conventional and microwave-assisted methods using molybdenum complexes of ONO donor ligands. New J. Chem. 2015, 39, 2130–2139. [Google Scholar] [CrossRef]
- Maurya, M.R.; Saini, N.; Avecilla, F. Catalytic oxidation of secondary alcohols by molybdenum complexes derived from 4-acyl pyrazolone in presence and absence of an N-based additive: Conventional versus microwave assisted method. Inorg. Chim. Acta 2015, 438, 168–178. [Google Scholar] [CrossRef]
- Maurya, M.R.; Neeraj, S.; Avecilla, F. Effect of N-based additive on the optimization of liquid phase oxidation of 2 bicy-clic, cyclic and aromatic alcohols catalyzed by dioxidomolybdenum(VI) 3 and oxidoperoxidomolybdenum(VI) complexes. RSC Adv. 2015, 5, 101076–101088. [Google Scholar] [CrossRef]
- Das, S.P.; Boruah, J.J. Selective and solventless oxidation of organic sulfides and alcohols using new supported molyb-denum (VI) complex in microwave and conventional methods. Appl. Organom. Chem. 2020, 34, e5781. [Google Scholar] [CrossRef]
- Allen, F.H. The Cambridge Structural Database, V5.30. Acta Cryst. Sec. B Struct. Sci. 2002, 58, 380–388. [Google Scholar] [CrossRef]
- Vrdoljak, V.; Prugovečki, B.; Pulić, I.; Cigler, M.; Sviben, D.; Vuković, J.P.; Novak, P.; Matković-Čalogović, D.; Cindrić, M. Dioxidomolybdenum(VI) complexes with isoniazid-related hydrazones: Solution-based, mechanochemical and UV-light assisted deprotonation. New J. Chem. 2015, 39, 7322–7332. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.X.; Li, W.H. Synthesis, crystal structures, and catalytic property of dioxomolybdenum(VI) complexes with hy-drazones. Russ. J. Coord. Chem. 2012, 38, 92–98. [Google Scholar] [CrossRef]
- Vrdoljak, V.; Prugovečki, B.; Matković-Čalogović, D.; Dreos, R.; Siega, P.; Tavagnacco, C. Zigzag Chain, Square Tetra-nuclear, and Polyoxometalate-Based Inorganic−Organic Hybrid Compounds-Molybdenum vs Tungsten. Cryst. Growth Des. 2010, 10, 1373–1382. [Google Scholar] [CrossRef]
- Bikas, R.; Lippolis, V.; Noshiranzadeh, N.; Farzaneh-Bonab, H.; Blake, A.J.; Siczek, M.; Hosseini-Monfared, H.; Lis, T. Electronic Effects of Aromatic Rings on the Catalytic Activity of Dioxidomolybdenum(VI)–Hydrazone Complexes. Eur. J. Inorg. Chem. 2017, 2017, 999–1006. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.-X.; Yuan, Y.-M.; Li, W.-H. Syntheses, crystal structures, and catalysis by polymeric dioxomolybdenum(VI) com-plexes with similar (iso)nicotinohydrazones. J. Coord. Chem. 2013, 66, 2726–2735. [Google Scholar] [CrossRef]
- Feng, J.P.; Shi, Z. An Improved Asymmetric Synthesis of Malyngamide U and Its 2′-Epimer. J. Org. Chem. 2008, 73, 6873–6876. [Google Scholar] [CrossRef]
- Qi, X.L.; Zhang, J.T.; Feng, J.P.; Cao, X.P. Total synthesis and absolute configuration of malyngamide W. Org. Biomol. Chem. 2011, 9, 3817–3824. [Google Scholar] [CrossRef] [PubMed]
- Mak, K.K.W.; Lai, Y.M.; Siu, Y.H. Regiospecific Epoxidation of Carvone: A Discovery-Oriented Experiment for Understanding the Selectivity and Mechanism of Epoxidation Reactions. J. Chem. Educ. 2006, 83, 1058–1061. [Google Scholar] [CrossRef]
- Rafiński, Z.; Ścianowski, J. Synthesis and reactions of enantiomerically pure dialkyl diselenides from the p-menthane group. Tetrahedron Asymmetry 2008, 19, 1237–1244. [Google Scholar] [CrossRef]
- Chen, G.J.-J.; McDonald, J.W.; Newton, W.E. Synthesis of molybdenum(IV) and molybdenum(V) complexes using oxo abstraction by phosphines. Mechanistic implications. Inorg. Chem. 1976, 15, 2612–2615. [Google Scholar] [CrossRef]
- Pisk, J.; Đilović, I.; Hrenar, T.; Cvijanović, D.; Pavlović, G.; Vrdoljak, V. Effective methods for the synthesis of hydrazones, quinazolines, and Schiff bases: Reaction monitoring using a chemometric approach. RSC Adv. 2020, 10, 38566–38577. [Google Scholar] [CrossRef]
- CrysAlis, P.R.O. Agilent; Agilent Technologies Ltd.: Yarnton, Oxfordshire, UK, 2015. [Google Scholar]
- Oxford Diffraction, CrysAlis CCD and CrysAlis RED software, Version 1.170; Oxford Diffraction Ltd.: Abingdon, Oxfordshire, UK, 2003.
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Cryst. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. Sec. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Spek, A.L. Structure validation in chemical crystallography. Acta Cryst. Sec. D 2009, 65, 148–155. [Google Scholar] [CrossRef]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J.J. Mercury: Visualization and analysis of crystal structures. Appl. Cryst. 2006, 39, 453–457. [Google Scholar] [CrossRef] [Green Version]
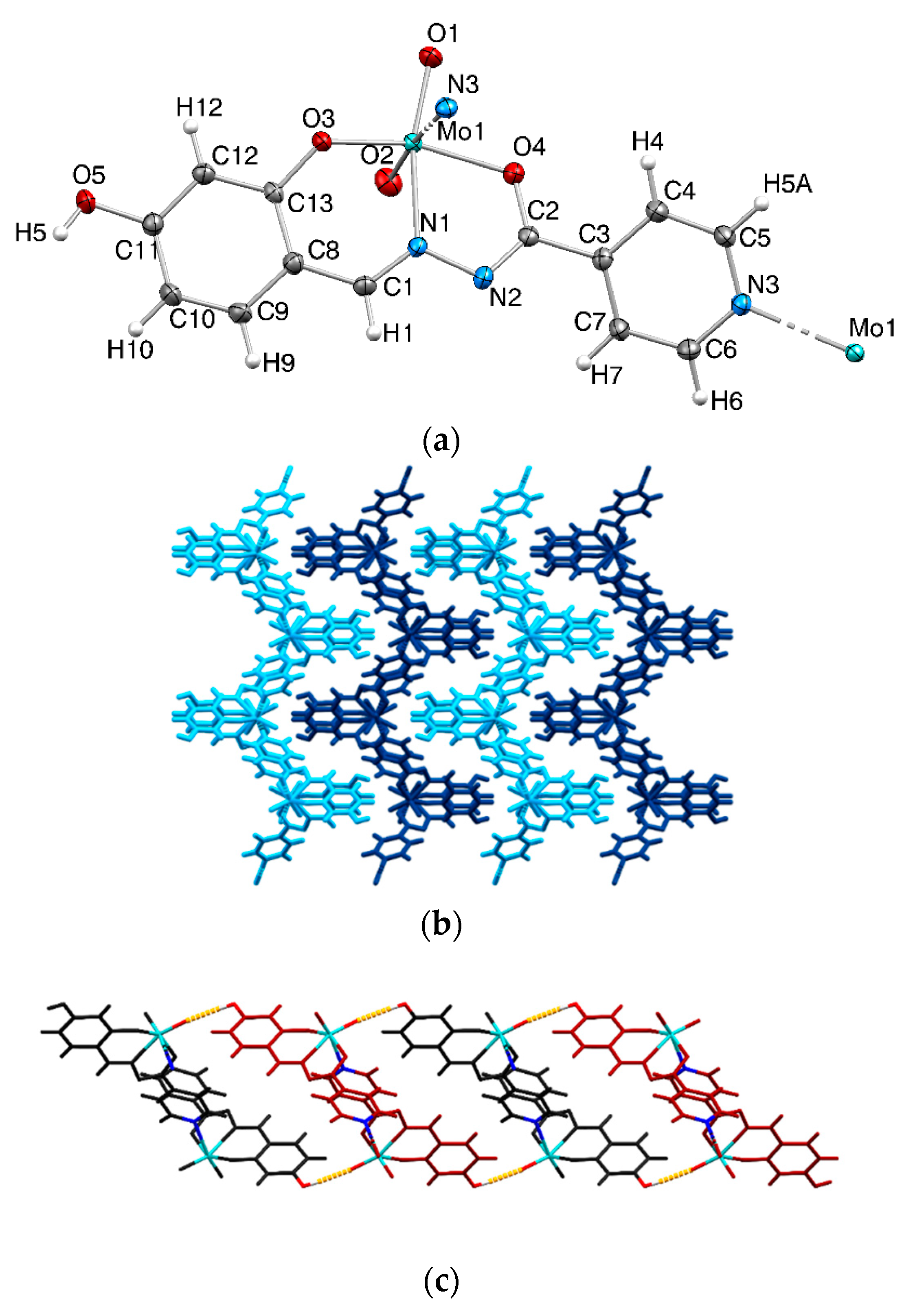

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Atom | Δδ/ppm | |||
| C-1 | 8.01 | 7.56 | 7.26 | 6.9 |
| C-4 | 5.66 | 5.65 | 4.47 | 4.44 |
| C-2′ | 2.04 | 1.96 | 2.02 | 1.88 |
| Catalyst | Conversion a/% | Selectivity b/% | TOF20 min c/h−1 | TON d |
|---|---|---|---|---|
| H2O2 | ||||
| 1 | 88 | 41 | 113 | 308 |
| 2 | 87 | 42 | 294 | 339 |
| 3 | 77 | 40 | 4 | 30 |
| 4 | 85 | 44 | 34 | 329 |
| 1a | 87 | 41 | 23 | 36 |
| 2a | 84 | 41 | 286 | 344 |
| 3a | 89 | 37 | 27 | 367 |
| 4a | 91 | 42 | 20 | 394 |
| THBP (in water) | ||||
| 1 | 64 | 10 | 237 | 235 |
| 2 | 66 | 10 | 29 | 27 |
| 1a | 62 | 11 | 29 | 27 |
| 2a | 56 | 19 | 185 | 271 |
| THBP (in decane) | ||||
| 2a | 99 | 4 | 1001 | 289 |
| Catalyst | Oxidant | Conversion a/% | Selectivity b/% |
|---|---|---|---|
| 2 | H2O2 | 11 | 64 |
| 2a | H2O2 | 13 | 59 |
| THBP (in water) | 17 | 31 | |
| THBP (in decane) | 28 | 59 |
| Catalyst | Conversion a/% | Selectivity b/% | TOF20min c/h−1 | TON d |
|---|---|---|---|---|
| 1 | 19 | 59 | 3 | 19 |
| 2 | 16 | 66 | 2 | 16 |
| 3 | 20 | 57 | 3 | 31 |
| 4 | 19 | 61 | 4 | 20 |
| 1a | 21 | 66 | 4 | 21 |
| 2a | 15 | 71 | 3 | 15 |
| 3a | 14 | 68 | 76 | 14 |
| 4a | 19 | 61 | 4 | 20 |
| Catalyst | Oxidant | Conversion a/% | Selectivity b/% | TOF20 min c/h−1 | TON d |
|---|---|---|---|---|---|
| 1a | H2O2 | 11 | 54 | 27 | 43 |
| H2O2 + MeCN | 12 | 57 | 31 | 49 | |
| TBHP | 17 | 45 | 99 | 52 | |
| TBHP + MeCN | 13 | 59 | 170 | 52 | |
| 2a | H2O2 | 23 | 40 | 44 | 90 |
| H2O2 + MeCN | 16 | 61 | 17 | 66 | |
| TBHP | 17 | 60 | 104 | 69 | |
| TBHP + MeCN | 37 | 57 | 117 | 151 | |
| 3a | H2O2 | 13 | 64 | 38 | 52 |
| H2O2 + MeCN | 13 | 69 | 30 | 52 | |
| TBHP | 19 | 51 | 83 | 73 | |
| TBHP + MeCN | 15 | 52 | 153 | 54 | |
| 4a | H2O2 | 14 | 49 | 71 | 55 |
| H2O2 + MeCN | 15 | 59 | 91 | 59 | |
| TBHP | 16 | 56 | 108 | 64 | |
| TBHP + MeCN | 14 | 47 | 26 | 56 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mihalinec, J.; Pajski, M.; Guillo, P.; Mandarić, M.; Bebić, N.; Pisk, J.; Vrdoljak, V. Alcohol Oxidation Assisted by Molybdenum Hydrazonato Catalysts Employing Hydroperoxide Oxidants. Catalysts 2021, 11, 881. https://doi.org/10.3390/catal11080881
Mihalinec J, Pajski M, Guillo P, Mandarić M, Bebić N, Pisk J, Vrdoljak V. Alcohol Oxidation Assisted by Molybdenum Hydrazonato Catalysts Employing Hydroperoxide Oxidants. Catalysts. 2021; 11(8):881. https://doi.org/10.3390/catal11080881
Chicago/Turabian StyleMihalinec, Josipa, Matea Pajski, Pascal Guillo, Mirna Mandarić, Nikol Bebić, Jana Pisk, and Višnja Vrdoljak. 2021. "Alcohol Oxidation Assisted by Molybdenum Hydrazonato Catalysts Employing Hydroperoxide Oxidants" Catalysts 11, no. 8: 881. https://doi.org/10.3390/catal11080881
APA StyleMihalinec, J., Pajski, M., Guillo, P., Mandarić, M., Bebić, N., Pisk, J., & Vrdoljak, V. (2021). Alcohol Oxidation Assisted by Molybdenum Hydrazonato Catalysts Employing Hydroperoxide Oxidants. Catalysts, 11(8), 881. https://doi.org/10.3390/catal11080881