
Reactive Deep Eutectic Solvents (RDESs): A New Tool for Phospholipase D-Catalyzed Preparation of Phospholipids

 , , , ,
, , , ,  , and
, and 
Abstract
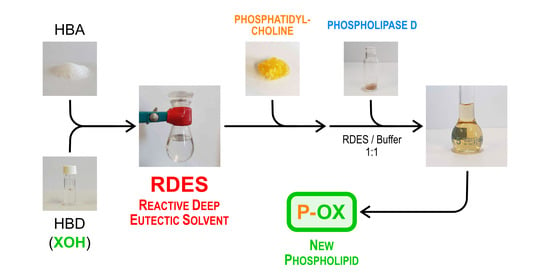
1. Introduction
2. Results and Discussion
2.1. Preliminary Method Development
2.2. Preparation of RDES Media
2.3. Transphosphatidylation Reactions
2.3.1. Preparation of Phosphatidylglycerol
2.3.2. Preparation of Phosphatidylethyleneglycol (P-EG).
3. Materials and Methods
3.1. 31P NMR Sample Preparation
3.2. Thin Layer Chromatography (TLC)
3.3. Gas Chromatography
3.4. High Performance Liquid Chromatography (HPLC)
3.5. Electrospray Ionization Mass Spectrometry
3.6. PLD Preparation
3.6.1. Microorganism Fermentation
3.6.2. Precipitation and Dialysis
3.7. Protein Determination
3.8. PLD Activity Determination
3.9. RDES Preparation
3.10. RDES Density Measurement
3.11. RDES Preparation of Phosphatidylglycerols PGA1 and PGB1 in RDES A1 and B1
3.12. RDES Preparation of Phosphatidylethyleneglycols P-EGA2 and P-EGB2 in RDES A2 and B2
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sheldon, R.A.; Woodley, J.M. Role of Biocatalysis in Sustainable Chemistry. Chem. Rev. 2018, 118, 801–838. [Google Scholar] [CrossRef]
- Expanding biocatalysis for a sustainable future. Nat. Catal. 2020, 3, 179–180. [CrossRef]
- Akretche, H.; Pierre, G.; Moussaoui, R.; Michaud, P.; Delattre, C. Valorization of olive mill wastewater for the development of biobased polymer films with antioxidant properties using eco-friendly processes. Green Chem. 2019, 21, 3065–3073. [Google Scholar] [CrossRef]
- Cicco, L.; Dilauro, G.; Perna, F.M.; Vitale, P.; Capriati, V. Advances in deep eutectic solvents and water: Applications in metal- and biocatalyzed processes, in the synthesis of APIs, and other biologically active compounds. Org. Biomol. Chem. 2021, 19, 2558–2577. [Google Scholar] [CrossRef]
- Smith, E.L.; Abbott, A.P.; Ryder, K.S. Deep Eutectic Solvents (DESs) and Their Applications. Chem. Rev. 2014, 114, 11060–11082. [Google Scholar] [CrossRef]
- Hansen, B.B.; Spittle, S.; Chen, B.; Poe, D.; Zhang, Y.; Klein, J.M.; Horton, A.; Adhikari, L.; Zelovich, T.; Doherty, B.W.; et al. Deep Eutectic Solvents: A Review of Fundamentals and Applications. Chem. Rev. 2021, 121, 1232–1285. [Google Scholar] [CrossRef]
- Abbott, A.P.; Capper, G.; Davies, D.L.; Rasheed, R.K.; Tambyrajah, V. Novel solvent properties of choline chloride/urea mixtures. Chem. Commun. 2003, 70–71. [Google Scholar] [CrossRef] [PubMed]
- Nahar, Y.; Thickett, S.C. Greener, Faster, Stronger: The Benefits of Deep Eutectic Solvents in Polymer and Materials Science. Polymers 2021, 13, 447. [Google Scholar] [CrossRef] [PubMed]
- Santana-Mayor, Á.; Rodríguez-Ramos, R.; Herrera-Herrera, A.V.; Socas-Rodríguez, B.; Rodríguez-Delgado, M.Á. Deep eutectic solvents. The new generation of green solvents in analytical chemistry. TrAC Trends Anal. Chem. 2021, 134. [Google Scholar] [CrossRef]
- Şahin, S. Tailor-designed deep eutectic liquids as a sustainable extraction media: An alternative to ionic liquids. J. Pharm. Biomed. Anal. 2019, 174, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Grudniewska, A.; de Melo, E.M.; Chan, A.; Gniłka, R.; Boratyński, F.; Matharu, A.S. Enhanced Protein Extraction from Oilseed Cakes Using Glycerol–Choline Chloride Deep Eutectic Solvents: A Biorefinery Approach. ACS Sustain. Chem. Eng. 2018, 6, 15791–15800. [Google Scholar] [CrossRef]
- Kalhor, P.; Ghandi, K. Deep Eutectic Solvents as Catalysts for Upgrading Biomass. Catalysts 2021, 11, 178. [Google Scholar] [CrossRef]
- Hooshmand, S.E.; Afshari, R.; Ramón, D.J.; Varma, R.S. Deep eutectic solvents: Cutting-edge applications in cross-coupling reactions. Green Chem. 2020, 22, 3668–3692. [Google Scholar] [CrossRef]
- Di Carmine, G.; Abbott, A.P.; D’Agostino, C. Deep eutectic solvents: Alternative reaction media for organic oxidation reactions. React. Chem. Eng. 2021, 6, 582–598. [Google Scholar] [CrossRef]
- Durand, E.; Lecomte, J.; Baréa, B.; Piombo, G.; Dubreucq, E.; Villeneuve, P. Evaluation of deep eutectic solvents as new media for Candida antarctica B lipase catalyzed reactions. Process Biochem. 2012, 47, 2081–2089. [Google Scholar] [CrossRef]
- Xu, P.; Zheng, G.-W.; Zong, M.-H.; Li, N.; Lou, W.-Y. Recent progress on deep eutectic solvents in biocatalysis. Bioresour. Bioprocess. 2017, 4, 34. [Google Scholar] [CrossRef] [PubMed]
- Panić, M.; Cvjetko Bubalo, M.; Radojčić Redovniković, I. Designing a biocatalytic process involving deep eutectic solvents. J. Chem. Technol. Biotechnol. 2021, 96, 14–30. [Google Scholar] [CrossRef]
- Cicco, L.; Ríos-Lombardía, N.; Rodríguez-Álvarez, M.J.; Morís, F.; Perna, F.M.; Capriati, V.; García-Álvarez, J.; González-Sabín, J. Programming cascade reactions interfacing biocatalysis with transition-metal catalysis in Deep Eutectic Solvents as biorenewable reaction media. Green Chem. 2018, 20, 3468–3475. [Google Scholar] [CrossRef]
- Vitale, P.; Perna, F.M.; Agrimi, G.; Pisano, I.; Mirizzi, F.; Capobianco, R.V.; Capriati, V. Whole-Cell Biocatalyst for Chemoenzymatic Total Synthesis of Rivastigmine. Catalysts 2018, 8, 55. [Google Scholar] [CrossRef]
- Płotka-Wasylka, J.; de la Guardia, M.; Andruch, V.; Vilková, M. Deep eutectic solvents vs. ionic liquids: Similarities and differences. Microchem. J. 2020, 159. [Google Scholar] [CrossRef]
- Perna, F.M.; Vitale, P.; Capriati, V. Deep eutectic solvents and their applications as green solvents. Curr. Opin. Green Sustain. Chem. 2020, 21, 27–33. [Google Scholar] [CrossRef]
- Vilková, M.; Płotka-Wasylka, J.; Andruch, V. The role of water in deep eutectic solvent-base extraction. J. Mol. Liq. 2020, 304. [Google Scholar] [CrossRef]
- Tan, J.-N.; Dou, Y. Deep eutectic solvents for biocatalytic transformations: Focused lipase-catalyzed organic reactions. Appl. Microbiol. Biotechnol. 2020, 104, 1481–1496. [Google Scholar] [CrossRef]
- Pätzold, M.; Siebenhaller, S.; Kara, S.; Liese, A.; Syldatk, C.; Holtmann, D. Deep Eutectic Solvents as Efficient Solvents in Biocatalysis. Trends Biotechnol. 2019, 37, 943–959. [Google Scholar] [CrossRef] [PubMed]
- Guajardo, N.; Domínguez de María, P.; Ahumada, K.; Schrebler, R.A.; Ramírez-Tagle, R.; Crespo, F.A.; Carlesi, C. Water as Cosolvent: Nonviscous Deep Eutectic Solvents for Efficient Lipase-Catalyzed Esterifications. ChemCatChem 2017, 9, 1393–1396. [Google Scholar] [CrossRef]
- Sirviö, J.A.; Visanko, M. Lignin-rich sulfated wood nanofibers as high-performing adsorbents for the removal of lead and copper from water. J. Hazard. Mater. 2020, 383. [Google Scholar] [CrossRef] [PubMed]
- Sirviö, J.A.; Heiskanen, J.P. Synthesis of Alkaline-Soluble Cellulose Methyl Carbamate Using a Reactive Deep Eutectic Solvent. ChemSusChem 2017, 10, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Mota-Morales, J.D.; Sánchez-Leija, R.J.; Carranza, A.; Pojman, J.A.; del Monte, F.; Luna-Bárcenas, G. Free-radical polymerizations of and in deep eutectic solvents: Green synthesis of functional materials. Prog. Polym. Sci. 2018, 78, 139–153. [Google Scholar] [CrossRef]
- Gore, S.; Baskaran, S.; Koenig, B. Efficient synthesis of 3,4-dihydropyrimidin-2-ones in low melting tartaric acid–urea mixtures. Green Chem. 2011, 13. [Google Scholar] [CrossRef]
- Singh, B.S.; Lobo, H.R.; Pinjari, D.V.; Jarag, K.J.; Pandit, A.B.; Shankarling, G.S. Comparative material study and synthesis of 4-(4-nitrophenyl)oxazol-2-amine via sonochemical and thermal method. Ultrason. Sonochemistry 2013, 20, 633–639. [Google Scholar] [CrossRef]
- Zeng, C.-X.; Qi, S.-J.; Xin, R.-P.; Yang, B.; Wang, Y.-H. Enzymatic selective synthesis of 1,3-DAG based on deep eutectic solvent acting as substrate and solvent. Bioprocess Biosyst. Eng. 2015, 38, 2053–2061. [Google Scholar] [CrossRef] [PubMed]
- Siebenhaller, S.; Muhle-Goll, C.; Luy, B.; Kirschhöfer, F.; Brenner-Weiss, G.; Hiller, E.; Günther, M.; Rupp, S.; Zibek, S.; Syldatk, C. Sustainable enzymatic synthesis of glycolipids in a deep eutectic solvent system. J. Mol. Catal. B Enzym. 2016, 133, S281–S287. [Google Scholar] [CrossRef]
- Hümmer, M.; Kara, S.; Liese, A.; Huth, I.; Schrader, J.; Holtmann, D. Synthesis of (-)-menthol fatty acid esters in and from (-)-menthol and fatty acids—Novel concept for lipase catalyzed esterification based on eutectic solvents. Mol. Catal. 2018, 458, 67–72. [Google Scholar] [CrossRef]
- Lamari, F.; Mochel, F.; Sedel, F.; Saudubray, J.M. Disorders of phospholipids, sphingolipids and fatty acids biosynthesis: Toward a new category of inherited metabolic diseases. J. Inherit. Metab. Dis. 2013, 36, 411–425. [Google Scholar] [CrossRef]
- Calvano, C.D.; Losito, I.; Cataldi, T. Editorial to the Special Issue “Lipidomics and Neurodegenerative Diseases”. Int. J. Mol. Sci. 2021, 22, 1270. [Google Scholar] [CrossRef] [PubMed]
- D’Arrigo, P.; Scotti, M. Lysophospholipids: Synthesis and Biological Aspects. Curr. Org. Chem. 2013, 17, 812–830. [Google Scholar] [CrossRef]
- Li, J.; Wang, X.; Zhang, T.; Wang, C.; Huang, Z.; Luo, X.; Deng, Y. A review on phospholipids and their main applications in drug delivery systems. Asian J. Pharm. Sci. 2015, 10, 81–98. [Google Scholar] [CrossRef]
- Gliszczyńska, A.; Niezgoda, N.; Gładkowski, W.; Czarnecka, M.; Świtalska, M.; Wietrzyk, J. Synthesis and Biological Evaluation of Novel Phosphatidylcholine Analogues Containing Monoterpene Acids as Potent Antiproliferative Agents. PloS ONE 2016, 11, e0157278. [Google Scholar] [CrossRef]
- Falconi, M.; Ciccone, S.; D’Arrigo, P.; Viani, F.; Sorge, R.; Novelli, G.; Patrizi, P.; Desideri, A.; Biocca, S. Design of a novel LOX-1 receptor antagonist mimicking the natural substrate. Biochem. Biophys. Res. Commun. 2013, 438, 340–345. [Google Scholar] [CrossRef]
- Kosicek, M.; Hecimovic, S. Phospholipids and Alzheimer’s Disease: Alterations, Mechanisms and Potential Biomarkers. Int. J. Mol. Sci. 2013, 14, 1310–1322. [Google Scholar] [CrossRef]
- Shamim, A.; Mahmood, T.; Ahsan, F.; Kumar, A.; Bagga, P. Lipids: An insight into the neurodegenerative disorders. Clin. Nutr. Exp. 2018, 20, 1–19. [Google Scholar] [CrossRef]
- Küllenberg, D.; Taylor, L.A.; Schneider, M.; Massing, U. Health effects of dietary phospholipids. Lipids Health Dis. 2012, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Schverer, M.; O’Mahony, S.M.; O’Riordan, K.J.; Donoso, F.; Roy, B.L.; Stanton, C.; Dinan, T.G.; Schellekens, H.; Cryan, J.F. Dietary phospholipids: Role in cognitive processes across the lifespan. Neurosci. Biobehav. Rev. 2020, 111, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Decker, E.A. Phospholipids in foods: Prooxidants or antioxidants? J. Sci. Food Agric. 2016, 96, 18–31. [Google Scholar] [CrossRef]
- Sun, N.; Chen, J.; Wang, D.; Lin, S. Advance in food-derived phospholipids: Sources, molecular species and structure as well as their biological activities. Trends Food Sci. Technol. 2018, 80, 199–211. [Google Scholar] [CrossRef]
- Pucek, A.; Niezgoda, N.; Kulbacka, J.; Wawrzeńczyk, C.; Wilk, K.A. Phosphatidylcholine with conjugated linoleic acid in fabrication of novel lipid nanocarriers. Colloids Surf. APhysicochem. Eng. Asp. 2017, 532, 377–388. [Google Scholar] [CrossRef]
- Aldosari, B.N.; Alfagih, I.M.; Almurshedi, A.S. Lipid Nanoparticles as Delivery Systems for RNA-Based Vaccines. Pharmaceutics 2021, 13, 206. [Google Scholar] [CrossRef]
- Samaridou, E.; Heyes, J.; Lutwyche, P. Lipid nanoparticles for nucleic acid delivery: Current perspectives. Adv. Drug Deliv. Rev. 2020, 154–155, 37–63. [Google Scholar] [CrossRef]
- De Serrano, L.O.; Burkhart, D.J. Liposomal vaccine formulations as prophylactic agents: Design considerations for modern vaccines. J. Nanobiotechnol. 2017, 15, 83. [Google Scholar] [CrossRef]
- D’Arrigo, P.; Giordano, C.; Macchi, P.; Malpezzi, L.; Pedrocchi-Fantoni, G.; Servi, S. Synthesis, Platelet Adhesion and Cytotoxicity Studies of New Glycerophosphoryl-Containing Polyurethanes. Int. J. Artif. Organs 2007, 30, 133–143. [Google Scholar] [CrossRef]
- Zhiguang, H.; Hui, Z.; Wenqiang, G.; Jianfu, L.; Charles, B.; Don, K.; Maneesha, S.M. Vesicle properties and health benefits of milk phospholipids: A review. J. Food Bioact. 2019, 5. [Google Scholar] [CrossRef]
- Lordan, R.; Tsoupras, A.; Zabetakis, I. Phospholipids of Animal and Marine Origin: Structure, Function, and Anti-Inflammatory Properties. Molecules 2017, 22, 1964. [Google Scholar] [CrossRef]
- Chojnacka, A.; Gladkowski, W.; Kielbowicz, G.; Wawrzenczyk, C. Isolation of egg-yolk phospholipids and enzymatic modification of their acyl chains. Lipid Technol. 2012, 24, 33–35. [Google Scholar] [CrossRef]
- Niezgoda, N.; Gliszczyńska, A.; Gładkowski, W.; Chojnacka, A.; Kiełbowicz, G.; Wawrzeńczyk, C. Production of concentrates of CLA obtained from sunflower and safflower and their application to the lipase-catalyzed acidolysis of egg yolk phosphatidylcholine. Eur. J. Lipid Sci. Technol. 2016, 118, 1566–1578. [Google Scholar] [CrossRef]
- D’Arrigo, P.; Servi, S. Using phospholipases for phospholipid modification. Trends Biotechnol. 1997, 15, 90–96. [Google Scholar] [CrossRef]
- Baldassarre, F.; Allegretti, C.; Tessaro, D.; Carata, E.; Citti, C.; Vergaro, V.; Nobile, C.; Cannazza, G.; D’Arrigo, P.; Mele, A.; et al. Biocatalytic Synthesis of Phospholipids and Their Application as Coating Agents for CaCO3 Nano–crystals: Characterization and Intracellular Localization Analysis. ChemistrySelect 2016, 1, 6507–6514. [Google Scholar] [CrossRef]
- Leiros, I.; Hough, E.; D’Arrigo, P.; Carrea, G.; Pedrocchi-Fantoni, G.; Secundo, F.; Servi, S. Crystallization and preliminary X-ray diffraction studies of phospholipase D from Streptomyces sp. Acta Crystallogr. Sect. D Biol. Crystallogr. 2000, 56, 466–468. [Google Scholar] [CrossRef]
- Carrea, G.; D’Arrigo, P.; Secundo, F.; Servi, S. Purification and applications of a phospholipase D from a new strain of Streptomyces. Biotechnol. Lett. 1997, 19, 1083–1085. [Google Scholar] [CrossRef]
- Carrea, G.; D’Arrigo, P.; Piergianni, V.; Roncaglio, S.; Secundo, F.; Servi, S. Purification and properties of two phospholipases D from Streptomyces sp. Biochim. Biophys. Acta Lipids Lipid Metab. 1995, 1255, 273–279. [Google Scholar] [CrossRef]
- D’Arrigo, P.; de Ferra, L.; Piergianni, V.; Ricci, A.; Scarcelli, D.; Servi, S. Phospholipase D from Streptomyces catalyses the transfer of secondary alcohols. J. Chem. Soc. Chem. Commun. 1994, 1709–1710. [Google Scholar] [CrossRef]
- D’Arrigo, P.; de Ferra, L.; Piergianni, V.; Selva, A.; Servi, S.; Strini, A. Preparative transformation of natural phospholipids catalysed by phospholipase D from Streptomyces. J. Chem. Soc. Perkin Trans. 1 1996, 2651–2656. [Google Scholar] [CrossRef]
- Bossi, L.; D’Arrigo, P.; Pedrocchi-Fantoni, G.; Mele, A.; Servi, S.; Leiros, I. The substrate requirements of phospholipase D. J. Mol. Catal. B Enzym. 2001, 11, 433–438. [Google Scholar] [CrossRef]
- Allegretti, C.; Denuccio, F.; Rossato, L.; D’Arrigo, P. Polar Head Modified Phospholipids by Phospholipase D-Catalyzed Transformations of Natural Phosphatidylcholine for Targeted Applications: An Overview. Catalysts 2020, 10, 997. [Google Scholar] [CrossRef]
- Kim, H.-Y.; Huang, B.X.; Spector, A.A. Phosphatidylserine in the brain: Metabolism and function. Prog. Lipid Res. 2014, 56, 1–18. [Google Scholar] [CrossRef]
- Mozzi, R.; Buratta, S.; Goracci, G. Metabolism and Functions of Phosphatidylserine in Mammalian Brain. Neurochem. Res. 2003, 28, 195–214. [Google Scholar] [CrossRef] [PubMed]
- Duan, Z.-Q.; Hu, F. Highly efficient synthesis of phosphatidylserine in the eco-friendly solvent γ-valerolactone. Green Chem. 2012, 14, 1581–1583. [Google Scholar] [CrossRef]
- Duan, Z.-Q.; Hu, F. Efficient synthesis of phosphatidylserine in 2-methyltetrahydrofuran. J. Biotechnol. 2013, 163, 45–49. [Google Scholar] [CrossRef] [PubMed]
- D’Arrigo, P.; Cerioli, L.; Chiappe, C.; Panzeri, W.; Tessaro, D.; Mele, A. Improvements in the enzymatic synthesis of phosphatidylserine employing ionic liquids. J. Mol. Catal. B Enzym. 2012, 84, 132–135. [Google Scholar] [CrossRef]
- Yang, S.-L.; Duan, Z.-Q. Insight into enzymatic synthesis of phosphatidylserine in deep eutectic solvents. Catal. Commun. 2016, 82, 16–19. [Google Scholar] [CrossRef]
- Allegretti, C.; Bono, A.; D’Arrigo, P.; Denuccio, F.; De Simeis, D.; Di Lecce, G.; Serra, S.; Tessaro, D.; Viola, M. Valorization of Corn Seed Oil Acid Degumming Waste for Phospholipids Preparation by Phospholipase D-Mediated Processes. Catalysts 2020, 10, 809. [Google Scholar] [CrossRef]
- Gregoriadis, G. Liposomes for drugs and vaccines. Trends Biotechnol. 1985, 3, 235–241. [Google Scholar] [CrossRef]
- Sakdiset, P.; Okada, A.; Todo, H.; Sugibayashi, K. Selection of phospholipids to design liposome preparations with high skin penetration-enhancing effects. J. Drug Deliv. Sci. Technol. 2018, 44, 58–64. [Google Scholar] [CrossRef]
- Hossann, M.; Wiggenhorn, M.; Schwerdt, A.; Wachholz, K.; Teichert, N.; Eibl, H.; Issels, R.D.; Lindner, L.H. In vitro stability and content release properties of phosphatidylglyceroglycerol containing thermosensitive liposomes. Biochim. Biophys. Acta Biomembr. 2007, 1768, 2491–2499. [Google Scholar] [CrossRef] [PubMed]
- Mazela, J.; Merritt, T.A.; Gadzinowski, J.; Sinha, S. Evolution of pulmonary surfactants for the treatment of neonatal respiratory distress syndrome and paediatric lung diseases. Acta Paediatr. 2006, 95, 1036–1048. [Google Scholar] [CrossRef] [PubMed]
- Tejeda-Mansir, A.; García-Rendón, A.; Guerrero-Germán, P. Plasmid-DNA lipid and polymeric nanovaccines: A new strategic in vaccines development. Biotechnol. Genet. Eng. Rev. 2019, 35, 46–68. [Google Scholar] [CrossRef] [PubMed]
- Juneja, L.R.; Hibi, N.; Inagaki, N.; Yamane, T.; Shimizu, S. Comparative study on conversion of phosphatidylcholine to phosphatidylglycerol by cabbage phospholipase D in micelle and emulsion systems. Enzym. Microb. Technol. 1987, 9, 350–354. [Google Scholar] [CrossRef]
- Chen, L.; Beppu, F.; Miyashita, K.; Hosokawa, M. Preparation of n-3 Polyunsaturated Phosphatidylglycerol from Salmon Roe Lipids by Phospholipase D and In Vitro Digestion. Eur. J. Lipid Sci. Technol. 2020, 122. [Google Scholar] [CrossRef]
- D’Arrigo, P.; de Ferra, L.; Pedrocchi-Fantoni, G.; Scarcelli, D.; Servi, S.; Strini, A. Enzyme-mediated synthesis of two diastereoisomeric forms of phosphatidylglycerol and of diphosphatidylglycerol (cardiolipin). J. Chem. Soc. Perkin Trans. 1 1996, 2657–2660. [Google Scholar] [CrossRef]
- He, X.; Li, L.; Su, H.; Zhou, D.; Song, H.; Wang, L.; Jiang, X. Poly(ethylene glycol)-block-poly(ε-caprolactone)-and phospholipid-based stealth nanoparticles with enhanced therapeutic efficacy on murine breast cancer by improved intracellular drug delivery. Int. J. Nanomed. 2015, 10, 1791–1804. [Google Scholar] [CrossRef]
- Yamamoto, T.; Teramura, Y.; Itagaki, T.; Arima, Y.; Iwata, H. Interaction of poly(ethylene glycol)-conjugated phospholipids with supported lipid membranes and their influence on protein adsorption. Sci. Technol. Adv. Mater. 2016, 17, 677–684. [Google Scholar] [CrossRef]
- Mahendra, A.; James, H.P.; Jadhav, S. PEG-grafted phospholipids in vesicles: Effect of PEG chain length and concentration on mechanical properties. Chem. Phys. Lipids 2019, 218, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Cardillo, R.; D’Arrigo, P.; De Ferra, L.; Piergianni, V.; Scarcelli, D.; Servi, S. A simple assay for the quantitative evaluation of Phospholipase D activity. Biotechnol. Tech. 1993, 7, 795–798. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hydrogen–Bond Acceptors (HBAs) | Hydrogen–Bond Donors (HBDs) | ||
|---|---|---|---|
 | choline chloride (ChCl) |  | Glycerol (Gly) |
 | trimethyl glycine (BetG) |  | ethylene glycol (EG) |
| RDES Symbol | HBA | HBD | Molar Ratio (HBA/HBD) | Density of Pure RDES (g/cm3) | Density of RDES/Buffer Solutions (g/cm3) |
|---|---|---|---|---|---|
| RDES A1 | choline chloride | glycerol | 1:2 | 1.16 | 1.09 |
| RDES A2 | choline chloride | ethylene glycol | 1:2 | 1.06 | 1.03 |
| RDES B1 | trimethyl glycine | glycerol | 1:2 | 1.16 | 1.08 |
| RDES B2 | trimethyl glycine | ethylene glycol | 1:2 | 1.11 | 1.05 |
| Acid | Chain | tR | % in PC |
|---|---|---|---|
| palmitic acid | C16:0 | 15.7 | 11.2 |
| linoleic acid | C18:2 | 21.3 | 69.3 |
| oleic acid | C18:1 | 21.4 | 15.8 |
| stearic acid | C18:0 | 22.0 | 3.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Allegretti, C.; Gatti, F.G.; Marzorati, S.; Rossato, L.A.M.; Serra, S.; Strini, A.; D’Arrigo, P. Reactive Deep Eutectic Solvents (RDESs): A New Tool for Phospholipase D-Catalyzed Preparation of Phospholipids. Catalysts 2021, 11, 655. https://doi.org/10.3390/catal11060655
Allegretti C, Gatti FG, Marzorati S, Rossato LAM, Serra S, Strini A, D’Arrigo P. Reactive Deep Eutectic Solvents (RDESs): A New Tool for Phospholipase D-Catalyzed Preparation of Phospholipids. Catalysts. 2021; 11(6):655. https://doi.org/10.3390/catal11060655
Chicago/Turabian StyleAllegretti, Chiara, Francesco G. Gatti, Stefano Marzorati, Letizia Anna Maria Rossato, Stefano Serra, Alberto Strini, and Paola D’Arrigo. 2021. "Reactive Deep Eutectic Solvents (RDESs): A New Tool for Phospholipase D-Catalyzed Preparation of Phospholipids" Catalysts 11, no. 6: 655. https://doi.org/10.3390/catal11060655
APA StyleAllegretti, C., Gatti, F. G., Marzorati, S., Rossato, L. A. M., Serra, S., Strini, A., & D’Arrigo, P. (2021). Reactive Deep Eutectic Solvents (RDESs): A New Tool for Phospholipase D-Catalyzed Preparation of Phospholipids. Catalysts, 11(6), 655. https://doi.org/10.3390/catal11060655