3. Materials and Methods
Chemical reagents were purchased from Sigma-Aldrich (Steinheim, Germany), VWR International (Barcelona, Spain), and Thermo Fisher Scientific (Waltham, MA, USA) and used as received, except tetrahydrofuran, which was dried under an inert atmosphere using the sodium-benzophenone system. Regarding the enzymes employed in this contribution, Pseudomonas cepacia (PSL, 23,000 U/g), lyophilized lipase AK from Pseudomonas fluorescens (AK, 23,700 U/g), and Acylase from Aspergillus melleus (>0.5 U/g) were also purchased from Sigma-Aldrich (Steinheim, Germany), Candida antarctica type A lipase (CAL-A) was obtained from ChiralVision (3000 U/g, Den Hoorn, Netherlands), while Candida antartica type B lipase (CAL-B, Novozym-435, 7300 PLU/g) and Rhizomucor miehei lipase (RML, 150 IUN/g) were kindly donated by Novozymes company (Bagsværd, Denmark).
NMR spectra were recorded on a Bruker AV300 MHz spectrometer (Bruker Co., Faellanden, Switzerland) including
1H,
13C, and
19F NMR monodimensional experiments. All chemical shifts (δ) are given in parts per million (ppm) and referenced to the residual solvent signal as internal standard. High performance liquid chromatography (HPLC) analyses were performed on an HP 1100 chromatograph (Agilent Technologies, Inc., Wilmington, DE, USA) equipped with a VIS–UV detector using different chiral columns (25 cm × 4.6 mm, 5 μm particle size, Chiral Technologies, Mainz, Germany) for the measurement of the corresponding alcohol and ester enantiomeric excess values. HPLC injections were made using a 0.8 mL/min flow, mixtures of hexane and 2-propanol as eluent, 30 °C column temperature, and 210 and 214 nm as wavelengths (see chromatograms in the
Supplementary Material section). Measurement of the op-tical rotation values was carried out at 590 nm on an Autopol IV Automatic polarime-ter (Rudolph Research Analytical, Hackettstown, NJ, USA).
Melting points were measured in a Gallenkamp apparatus, introducing the samples in open capillary tubes and the measurements are uncorrected. IR spectra were recorded on a Jasco FT/IR-4700 spectrophotometer (Jasco-Spain, Madrid, Spain), and νmax values are given in cm−1 for the main absorption bands. High resolution mass spectra (HRMS) experiments were carried out by electrospray ionization in positive mode (ESI+) using a VG AutoSpecQ high-resolution mass spectrometer (Fision Instrument, Mildford, MA, USA). Thin-layer chromatography (TLC) was conducted with Merck Silica Gel 60 F254 precoated plates (Merck KGaA, Darmstadt, Germany) and visualized with a UV lamp, plus either potassium permanganate or vanillin stains. Column chromatographies were performed using silica gel 60 (230–240 mesh) (Merck KGaA, Darmstadt, Germany).
3.1. Synthesis of Flavanones 1a–j
An aqueous solution of KOH (4.9 g, 122.4 mmol in 12 mL of water) was carefully added to a solution of 2′-hydroxyacetophenone (
4, 2.0 g, 14.7 mmol) in ethanol (30 mL), a yellow precipitate usually being formed. The mixture was stirred for 5–10 min. After this time, the corresponding benzaldehyde
5a–
j (14.7 mmol) dissolved in ethanol (7 mL) was added, observing that the color of the solution turned from yellow to intense red. The reaction was stirred for 3 h at 60 °C, and at this point, the reaction was quenched by pouring it onto ice. The reaction mixture was acidified to pH = 2 with an aqueous concentrated HCl solution, and the desired 2′-hydroxychalcone was extracted with EtOAc (3 × 20 mL). The organic layers were combined, dried over Na
2SO
4, filtered, and concentrated under reduced pressure. A solution of the recovered crude 2′-hydroxychalcone 6a–j was dissolved in glacial acetic acid (7 mL for each mmol of crude chalcone) was refluxed for 72 h. After this time, the reaction was quenched by pouring it on water (10 mL for each mmol of crude chalcone), observing a brown precipitate of the corresponding flavanone
1a–
j, which was extracted with dichloromethane (3 × 30 mL). The organic phases were combined, washed with brine (3 × 30 mL), dried over Na
2SO
4, and concentrated under reduced pressure. Since some acetic acid remained in the round-bottom flask, toluene (3 × 10 mL) was added to co-evaporate the remaining AcOH. The final product was purified by column chromatography on silica gel using hexane:EtOAc 5:1 as eluent (36–47% isolated yield,
Table 2).
2-Phenylchroman-4-one (1a). White solid. Mp: 77–78 °C. Rf (hexane:EtOAc 5:1): 0.45. 1H NMR δ (300 MHz, CDCl3): 7.94 (dd, J = 8.1, 1.8 Hz, 1H), 7.58–7.33 (m, 6H), 7.12–7.01 (m, 2H), 5.49 (dd, J = 13.2, 3.0 Hz, 1H), 3.10 (dd, J = 16.9, 13.2 Hz, 1H), 2.90 (dd, J = 16.9, 3.0 Hz, 1H) ppm. 13C NMR δ (75 MHz, CDCl3): 192.09, 161.69, 138.87, 136.33, 128.98 (2C), 128.91, 127.18, 126.28 (2C), 121.75, 121.07, 118.26, 79.73, 44.80 ppm. Data are in agreement with those from the commercial source.
2-(2-Fluorophenyl)chroman-4-one (1b). Yellow liquid. Rf (hexane:EtOAc 5:1): 0.49. 1H NMR δ (300 MHz, CDCl3): 7.78 (dd, J = 7.5, 1.7 Hz, 1H), 7.69–7.63 (m, 1H), 7.53 (ddd, J = 10.0, 4.9, 1.9 Hz, 1H), 7.42–7.37 (m, 1H), 7.30–7.18 (m, 3H), 5.93 (dd, J = 13.1, 3.2 Hz, 1H), 3.21 (dd, J = 16.9, 13.1 Hz, 1H), 3.07 (dd, J = 16.9, 3.2 Hz, 1H) ppm. 13C NMR δ (75 MHz, CDCl3): 191.68, 161.63, 159.73 (d, J = 247.7 Hz), 136.31, 130.37 (d, J = 8.6 Hz), 127.58 (d, J = 3.5 Hz), 127.26, 126.30 (d, J = 12.8 Hz), 124.73 (d, J = 3.5 Hz), 121.92, 121.05, 118.20, 115.87 (d, J = 21.3 Hz), 73.98 (d, J = 3.1 Hz), 43.83 ppm. 19F NMR δ (282 MHz, CDCl3): –118.33 ppm. ESI-TOF-HRMS: [M + H]+ calcd. for C15H12FO2: 243.0821; found 243.0813.
2-(3-Fluorophenyl)chroman-4-one (1c). Colourless oil. Rf (hexane:EtOAc 5:1): 0.45. 1H NMR δ (300 MHz, CDCl3): 7.94 (dd, J = 8.1, 1.8 Hz, 1H), 7.53 (ddd, J = 8.4, 7.2, 1.8 Hz, 1H), 7.47–7.34 (m, 1H), 7.30–7.19 (m, 2H), 7.15–7.02 (m, 3H), 5.49 (dd, J = 12.9, 3.3 Hz, 1H), 3.05 (dd, J = 16.9, 12.9 Hz, 1H), 2.91 (dd, J = 16.9, 3.3 Hz, 1H) ppm. 13C NMR δ (75 MHz, CDCl3): 191.55, 163.12 (d, J = 246.9 Hz), 161.37, 141.42 (d, J = 7.3 Hz), 136.45, 130.61 (d, J = 8.2 Hz), 127.22, 121.98, 121.73 (d, J = 3.0 Hz), 121.04, 118.23, 115.75 (d, J = 21.2 Hz), 113.34 (d, J = 22.7 Hz), 78.88, 44.77 ppm. 19F NMR δ (282 MHz, CDCl3): –111.80 ppm. ESI-TOF-HRMS: [M + H]+ calcd. for C15H12FO2: 243.0821; found, 243.0817.
2-(4-Fluorophenyl)chroman-4-one (1d). Yellow solid. Mp: 97–99 °C. Rf (hexane:EtOAc 5:1): 0.41. 1H NMR δ (300 MHz, CDCl3): 7.93 (dd, J = 7.7, 1.4 Hz, 1H), 7.59–7.41 (m, 3H), 7.19–6.99 (m, 4H), 5.47 (dd, J = 13.2, 3.0 Hz, 1H), 3.06 (dd, J = 16.9, 13.1 Hz, 1H), 2.87 (dd, J = 16.8, 3.0 Hz, 1H) ppm. 13C NMR δ (75 MHz, CDCl3): 191.80, 162.94 (d, J = 247.6 Hz), 161.50, 136.39, 134.73 (d, J = 3.2 Hz), 128.16 (d, J = 8.3 Hz, 2C), 127.20, 121.88, 121.01, 118.20, 115.92 (d, J = 21.7 Hz, 2C), 79.04, 44.78 ppm. 19F NMR δ (282 MHz, CDCl3): –112.79 ppm. ESI-TOF-HRMS: [M + H]+ calcd. for C15H12FO2: 243.0821; found, 243.0816.
2-(4-Chlorophenyl)chroman-4-one (1e). Pale yellow solid. Mp: 87–89 °C. Rf (hexane:EtOAc 5:1): 0.46. 1H NMR δ (300 MHz, CDCl3): 7.93 (dd, J = 7.7, 1.3 Hz, 1H), 7.52 (ddd, J = 9.0, 7.6, 1.8 Hz, 1H), 7.48–7.34 (m, 4H), 7.12–7.01 (m, 2H), 5.47 (dd, J = 13.0, 3.1 Hz, 1H), 3.04 (dd, J = 16.9, 13.0 Hz, 1H), 2.88 (dd, J = 16.9, 3.2 Hz, 1H) ppm. 13C NMR δ (75 MHz, CDCl3): 191.64, 161.41, 137.38, 136.42, 134.70, 129.17 (2C), 127.63 (2C), 127.21, 121.94, 121.01, 118.21, 78.94, 44.72 ppm. ESI-TOF-HRMS: [M + H]+ calcd. for C15H12ClO2: 259.0526; found, 259.0511.
2-(4-Bromophenyl)chroman-4-one (1f). Pale yellow solid. Mp: 114–117 °C. Rf (hexane:EtOAc 5:1): 0.53. 1H NMR δ (300 MHz, CDCl3): 7.93 (dd, J = 7.7, 1.3 Hz, 1H), 7.65–7.46 (m, 3H), 7.37 (d, J = 8.3 Hz, 2H), 7.13–7.01 (m, 2H), 5.45 (dd, J = 12.9, 3.1 Hz, 1H), 3.04 (dd, J = 16.9, 13.0 Hz, 1H), 2.88 (dd, J = 16.9, 3.2 Hz, 1H) ppm. 13C NMR δ (75 MHz, CDCl3): 191.59, 161.40, 137.92, 136.43, 132.14 (2C), 127.93 (2C), 127.22, 122.84, 121.96, 121.02, 118.22, 78.98, 44.70 ppm. ESI-TOF-HRMS: [M + Na]+ calcd. for C15H11BrO2Na: 324.9835; found, 324.9833.
2-(2-Methoxyphenyl)chroman-4-one (1g). Viscous yellow solid. Rf (hexane:EtOAc 5:1): 0.39. 1H NMR δ (300 MHz, CDCl3): 7.95 (dd, J = 7.7, 1.8 Hz, 1H), 7.64 (dd, J = 7.7, 1.7 Hz, 1H), 7.50 (ddd, J = 8.6, 5.5, 1.8 Hz, 1H), 7.46–7.29 (m, 1H), 7.12–7.04 (m, 3H), 6.92 (dd, J = 8.4, 1.1 Hz, 1H), 5.86 (dd, J = 12.0, 4.2 Hz, 1H), 3.83 (s, 3H), 3.05–2.83 (m, 2H) ppm. 13C NMR δ (75 MHz, CDCl3): 192.75, 162.10, 155.83, 136.05, 129.47, 127.55, 127.12, 126.47, 121.46, 121.09, 120.95, 118.20, 110.57, 74.73, 55.41, 43.78 ppm. ESI-TOF-HRMS: [M + H]+ calcd. for C16H15O3: 255.1021; found, 255.1016.
2-(3-Methoxyphenyl)chroman-4-one (1h). Yellow solid. Mp: 80–83 °C. Rf (hexane:EtOAc 5:1): 0.41. 1H NMR δ (300 MHz, CDCl3): 7.93 (dd, J = 8.1, 1.8 Hz, 1H), 7.42 (d, J = 8.6 Hz, 2H), 7.10–7.01 (m, 4H), 6.92 (ddd, J = 8.3, 2.5, 1.0 Hz, 1H), 5.45 (dd, J = 13.2, 3.0 Hz, 1H), 3.84 (s, 3H), 3.08 (dd, J = 16.9, 13.2 Hz, 1H), 2.89 (dd, J = 16.9, 3.1 Hz, 1H) ppm. 13C NMR δ (75 MHz, CDCl3): 192.01, 161.59, 160.03, 140.40, 136.30, 130.05, 127.14, 121.74, 121.02, 118.41, 118.24, 114.17, 111.97, 79.57, 55.41, 44.81 ppm. ESI-TOF-HRMS: [M + H]+ calcd. for C16H15O3: 255.1021; found, 255.1010.
2-(4-Methoxyphenyl)chroman-4-one (1i). Yellow solid. Mp: 92–95 °C. Rf (hexane:EtOAc 5:1): 0.30. 1H NMR δ (300 MHz, CDCl3): 7.93 (dd, J = 8.3, 1.8 Hz, 1H), 7.54–7.45 (m, 1H), 7.43 (s, 1H), 7.40 (s, 1H), 7.11–7.00 (m, 2H), 7.00–6.88 (m, 2H), 5.43 (dd, J = 13.3, 2.9 Hz, 1H), 3.83 (s, 3H), 3.11 (dd, J = 16.9, 13.3 Hz, 1H), 2.86 (dd, J = 16.9, 2.9 Hz, 1H) ppm. 13C NMR δ (75 MHz, CDCl3): 192.36, 161.77, 160.10, 136.28, 130.89, 127.86 (2C), 127.16, 121.65, 121.04, 118.26, 114.33 (2C), 79.48, 55.49, 44.59 ppm. ESI-TOF-HRMS: [M + H]+ calcd. for C16H15O3: 255.1021; found, 255.1010.
2-(4-Methylphenyl)chroman-4-one (1j). Yellow solid. Mp: 80–82 °C. Rf (hexane:EtOAc 5:1): 0.36. 1H NMR δ (300 MHz, CDCl3): 7.94 (dd, J = 8.1, 1.8 Hz, 1H), 7.50 (ddd, J = 8.8, 7.3, 1.8 Hz, 1H), 7.38 (d, J = 8.1 Hz, 2H), 7.25 (d, J = 8.4 Hz, 2H), 7.09–6.99 (m, 2H), 5.44 (dd, J = 13.3, 2.9 Hz, 1H), 3.09 (dd, J = 16.9, 13.3 Hz, 1H), 2.87 (dd, J = 16.9, 3.0 Hz, 1H), 2.39 (s, 3H) ppm. 13C NMR δ (75 MHz, CDCl3): 192.19, 161.69, 138.75, 136.21, 135.83, 129.56 (2C), 127.09, 126.27 (2C), 121.59, 120.99, 118.21, 79.58, 44.61, 21.28 ppm. ESI-TOF-HRMS: [M + H]+ calcd. for C16H15O2: 237.0916; found, 237.0903.
3.2. Synthesis of Racemic cis-Flavanols 2a–j
A solution of the corresponding flavanone 1a-j (4.5 mmol) in dry THF (15 mL) was cooled to −78 °C under a nitrogen atmosphere. Next, a 1.0 M solution of LiAlH
4 in THF was added dropwise (1.25 mL, 1.25 mmol), and the mixture was stirred at –78 °C. Monitorization of the reaction was undertaken by TLC analyses (hexane:EtOAc 3:1), observing the complete consumption of the starting material after 12 h. After this time, the reaction was quenched by pouring it onto icy water and, when warmed, the product was extracted with dichloromethane (3 × 20 mL). The organic phases were combined, washed with brine (3 × 20 mL), dried over Na
2SO
4, and concentrated under reduced pressure. The reaction crudes were purified by column chromatography on silica gel, using a gradient 5:1 to 3:1 hexane:EtOAc as eluent, obtaining the corresponding
cis-flavan-4-ols
2a–
j with total control of the selectivity (90–96% isolated yield,
Table 2).
(2S*,4S*)-2-Phenylchroman-4-ol (cis-2a). White solid. Rf (hexane:EtOAc 3:1): 0.26. Mp: 98–100 °C. 1H NMR δ (300 MHz, CDCl3): 7.53 (dt, J = 7.7, 1.4 Hz, 1H), 7.50–7.30 (m, 5H), 7.29–7.16 (m, 1H), 7.00 (td, J = 7.5, 1.3 Hz, 1H), 6.91 (dd, J = 8.2, 1.2 Hz, 1H), 5.18 (dd, J = 11.7, 2.0 Hz, 1H), 5.10 (dd, J = 10.5, 6.2 Hz, 1H), 2.52 (ddd, J = 13.1, 6.3, 2.0 Hz, 1H), 2.14 (ddd, J = 13.2, 11.6, 10.4 Hz, 1H), 1.83 (s, 1H) ppm. 13C NMR δ (75 MHz, CDCl3): 154.61, 140.62, 129.32, 128.81 (2C), 128.37, 127.11, 126.22 (2C), 125.86, 121.12, 116.88, 76.98, 65.95, 40.18 ppm. ESI-TOF-HRMS: [M-OH]+ calcd. for C15H13O: 209.0966; found 209.0959.
(2S*,4S*)-2-(2-Fluorophenyl)chroman-4-ol (cis-2b). Pale brown solid. Mp: 100–102 °C. Rf (hexane:EtOAc 3:1): 0.30. 1H NMR δ (300 MHz, CDCl3): 7.66–7.47 (m, 2H), 7.41–6.77 (m, 6H), 5.59–5.46 (m, 1H), 5.13 (dd, J = 10.5, 6.2 Hz, 1H), 2.56 (ddd, J = 13.1, 6.2, 1.8 Hz, 1H), 2.12 (dt, J = 12.9, 11.1 Hz, 1H), 1.83 (s, 1H) ppm. 13C NMR δ (75 MHz, CDCl3): 159.71 (d, J = 246.7 Hz), 154.43, 129.67 (d, J = 8.3 Hz), 129.31, 128.00 (d, J = 12.8 Hz), 127.45 (d, J = 3.7 Hz), 127.15, 125.82, 124.60 (d, J = 3.3 Hz), 121.26, 116.82, 115.59 (d, J = 21.4 Hz), 70.92 (d, J = 3.2 Hz), 65.71, 39.03 ppm. 19F NMR δ (282 MHz, CDCl3): –119.38 ppm. ESI-TOF-HRMS: [M-OH]+ calcd. for C15H12FO: 227.0872; found 227.0860.
(2S*,4S*)-2-(3-Fluorophenyl)chroman-4-ol (cis-2c). White solid. Mp: 107–113 °C. Rf (hexane:EtOAc 3:1): 0.31. 1H NMR δ (300 MHz, CDCl3): 7.56 (d, J = 7.7 Hz, 1H), 7.42 (td, J = 8.0, 5.8 Hz, 1H), 7.34–7.19 (m, 3H), 7.15–7.00 (m, 2H), 6.96 (dd, J = 8.2, 1.0 Hz, 1H), 5.22 (dd, J = 11.6, 1.7 Hz, 1H), 5.14 (dd, J = 10.5, 6.2 Hz, 1H), 2.56 (ddd, J = 13.1, 6.2, 2.0 Hz, 1H), 2.13 (ddd, J = 13.1, 11.6, 10.5 Hz, 1H), 1.93 (s, 1H) ppm. 13C NMR δ (75 MHz, CDCl3): 163.10 (d, J = 246.2 Hz), 154.26, 143.23 (d, J = 7.3 Hz), 130.34 (d, J = 8.2 Hz), 129.41, 127.10, 125.74, 121.69 (d, J = 2.8 Hz), 121.32, 116.85, 115.16 (d, J = 21.2 Hz), 113.21 (d, J = 22.4 Hz), 76.19, 65.76, 40.18 ppm. 19F NMR δ (282 MHz, CDCl3): –112.41 ppm. ESI-TOF-HRMS: [M-OH]+ calcd. for C15H12FO: 227.0872; found 227.0868.
(2S*,4S*)-2-(4-Fluorophenyl)chroman-4-ol (cis-2d). White solid. Mp: 129–131 °C. Rf (hexane:EtOAc 3:1): 0.27. 1H NMR δ (300 MHz, CDCl3): 7.57 (d, J = 7.7 Hz, 1H), 7.52–7.39 (m, 2H), 7.33–7.21 (m, 1H), 7.20–7.09 (m, 2H), 7.04 (td, J = 7.5, 1.1 Hz, 1H), 6.93 (dd, J = 8.2, 1.0 Hz, 1H), 5.20 (dd, J = 11.7, 1.6 Hz, 1H), 5.12 (d, J = 6.5 Hz, 1H), 2.54 (ddd, J = 13.1, 6.3, 1.9 Hz, 1H), 2.15 (ddd, J = 13.1, 11.7, 10.6 Hz, 1H), 1.92 (d, J = 7.2 Hz, 1H) ppm, 1.66 (s, OH). 13C NMR δ (75 MHz, CDCl3): 162.68 (d, J = 246.6 Hz), 154.46, 136.45, 129.38, 128.00 (d, J = 8.2 Hz, 2C), 127.08, 125.77, 121.26, 116.84, 115.69 (d, J = 21.5 Hz, 2C), 76.36, 65.90, 40.26 ppm. 19F NMR δ (282 MHz, CDCl3): –113.86 ppm. ESI-TOF-HRMS: [M-OH]+ calcd. for C15H12FO: 227.0872; found 227.0862.
(2S*,4S*)-2-(4-Chlorophenyl)chroman-4-ol (cis-4e). White solid. Mp: 158–160 °C. Rf (hexane:EtOAc 3:1): 0.26. 1H NMR δ (300 MHz, CDCl3): 7.39 (dt, J = 7.8, 1.4 Hz, 1H), 7.26 (s, 3H), 7.18–6.99 (m, 2H), 6.88 (td, J = 7.5, 1.3 Hz, 1H), 6.77 (dd, J = 8.2, 1.2 Hz, 1H), 5.08–4.90 (m, 2H), 2.36 (ddd, J = 13.1, 6.3, 2.0 Hz, 1H), 1.96 (ddd, J = 13.1, 11.7, 10.5 Hz, 1H), 1.79 (d, J = 8.5 Hz, 1H) ppm. 13C NMR δ (75 MHz, CDCl3): 154.36, 139.17, 134.07, 129.40, 128.96 (2C), 127.59 (2C), 127.10, 125.74, 121.31, 116.84, 76.26, 65.80, 40.17 ppm. ESI-TOF-HRMS: [M + Na]+ calcd. for C15H13ClO2Na: 283.0496; found 283.0496.
(2S*,4S*)-2-(4-Bromophenyl)chroman-4-ol (cis-2f). White solid. Mp: 160–162 °C. Rf (hexane:EtOAc 3:1): 0.34. 1H NMR δ (300 MHz, CDCl3): 7.50–7.38 (m, 3H), 7.24 (d, J = 6.4 Hz, 1H), 7.19–7.09 (m, 1H), 6.92 (td, J = 7.5, 1.3 Hz, 1H), 6.81 (dd, J = 8.2, 1.2 Hz, 1H), 5.11–4.95 (m, 2H), 2.41 (ddd, J = 13.1, 6.3, 2.0 Hz, 1H), 2.00 (ddd, J = 13.2, 11.6, 10.5 Hz, 1H), 1.74 (d, J = 8.3 Hz, 1H) ppm.13C NMR δ (75 MHz, CDCl3): 154.33, 139.70, 131.92 (2C), 129.42, 127.90 (2C), 127.09, 125.73, 122.18, 121.32, 116.85, 76.29, 65.80, 40.17 ppm. ESI-TOF-HRMS: [M + Na]+ calcd. for C15H13BrO2Na: 326.9991; found 326.9988.
(2S*,4S*)-2-(2-Methoxyphenyl)chroman-4-ol (cis-2g). White solid. Mp: 132–137 °C. Rf (hexane:EtOAc 3:1): 0.24. 1H NMR δ (300 MHz, CDCl3): 7.66–7.59 (m, 1H), 7.58–7.44 (m, 1H), 7.43–7.30 (m, 1H), 7.29–7.20 (m, 1H), 7.12–6.86 (m, 4H), 5.63 (dd, J = 11.3, 1.9 Hz, 1H), 5.15 (t, J = 6.0 Hz, 1H), 3.90 (d, J = 4.8 Hz, 3H), 2.63 (ddd, J = 13.0, 6.2, 2.0 Hz, 1H), 2.09 (s, OH), 2.08–2.00 (m, 1H) ppm. 13C NMR δ (75 MHz, CDCl3): 155.96, 154.91, 129.21, 129.11, 128.96, 127.15, 126.43, 126.11, 120.96, 120.84, 116.81, 110.55, 71.52, 66.00, 55.50, 38.76 ppm. ESI-TOF-HRMS: [M-OH]+ calcd. for C15H15O2: 239.1072; found 239.1069.
(2S*,4S*)-2-(3-Methoxyphenyl)chroman-4-ol (cis-2h). Pale brown solid. Mp: 146–150 °C. Rf (hexane:EtOAc 3:1): 0.24. 1H NMR δ (300 MHz, CDCl3): 7.54 (d, J = 7.7 Hz, 1H), 7.40–7.19 (m, 2H), 7.08–6.97 (m, 3H), 6.96–6.85 (m, 2H), 5.18 (dd, J = 11.5, 1.7 Hz, 1H), 5.15–5.06 (m, 1H), 3.85 (s, 3H), 2.55 (ddd, J = 13.1, 6.2, 2.0 Hz, 1H), 2.16 (ddd, J = 13.1, 11.5, 10.5 Hz, 1H), 1.80 (d, J = 8.6 Hz, 1H) ppm. 13C NMR δ (75 MHz, CDCl3): 160.00, 154.47, 142.24, 129.88, 129.35, 127.12, 125.85, 121.15, 118.49, 116.90, 113.78, 111.86, 76.85, 65.95, 55.43, 40.22 ppm. ESI-TOF-HRMS: [M-OH]+ calcd. for C15H15O2: 239.1072; found 239.1066.
(2S*,4S*)-2-(4-Methoxyphenyl)chroman-4-ol (cis-2i). White solid. Mp: 99–100 °C. Rf (hexane:EtOAc 3:1): 0.27. 1H NMR δ (300 MHz, CDCl3): 7.52 (dt, J = 7.7, 1.4 Hz, 1H), 7.43–7.32 (m, 2H), 7.29–7.11 (m, 1H), 7.04–6.82 (m, 4H), 5.18–5.02 (m, 2H), 3.83 (s, 3H), 2.49 (ddd, J = 13.1, 6.3, 1.9 Hz, 1H), 2.15 (ddd, J = 13.1, 11.7, 10.5 Hz, 1H), 1.89 (d, J = 8.0 Hz, 1H) ppm. 13C NMR δ (75 MHz, CDCl3): 159.69, 154.71, 132.69, 129.28, 127.68 (2C), 127.09, 125.85, 121.04, 116.86, 114.18 (2C), 76.69, 66.04, 55.47, 39.98 ppm. ESI-TOF-HRMS: [M-OH]+ calcd. for C15H15O2: 239.1072; found 239.1065.
(2S*,4S*)-2-(4-Methylphenyl)chroman-4-ol (cis-2j). White solid. Mp: 106–108 °C. Rf (hexane:EtOAc 3:1): 0.42. 1H NMR δ (300 MHz, CDCl3): 7.45 (d, J = 7.7 Hz, 1H), 7.29 (s, 1H), 7.22–7.08 (m, 4H), 6.92 (td, J = 7.5, 1.1 Hz, 1H), 6.82 (dd, J = 8.2, 0.9 Hz, 1H), 5.05–4.96 (m, 1H), 5.02 (d, J = 6.6 Hz, 1H), 2.43 (ddd, J = 13.1, 6.3, 1.9 Hz, 1H), 2.31 (s, 3H), 2.08 (ddd, J = 13.1, 11.6, 10.6 Hz, 1H), 1.77 (d, J = 8.6 Hz, 1H) ppm. 13C NMR δ (75 MHz, CDCl3): 154.69, 138.18, 137.61, 129.47 (2C), 129.28, 127.10, 126.23 (2C), 125.86, 121.03, 116.89, 76.87, 66.01, 40.08, 21.33 ppm. ESI-TOF-HRMS: [M-OH]+ calcd. for C15H15O: 223.1123; found 223.1114.
3.3. Synthesis of Racemic trans-Flavanols 2a–j
The corresponding
cis-flavan-4-ol
2a–
j (200–500 mg, 0.88–2.05 mmol) was dissolved in dry THF (15 mL per mmol of
cis-flavan-4-ol) under nitrogen atmosphere, successively adding triphenylphosphine (two equivalents) and PTSA (two equivalents, Method A) or chloroacetic acid (two equivalents, 2.0 mmol, Method B). Next, DEAD (two equivalents, Method A) or DIAD (two equivalents, 2.0 mmol, Method B) was introduced in the Schlenk tube, and the mixture was stirred at room temperature. Monitorization of the reaction was undertaken by TLC analyses (hexane:EtOAc 3:1), observing the complete consumption of the starting material after 1 h. After this time, the reaction was stopped by removal of the solvent under reduced pressure. The residue was then hydrolyzed by treatment with a mixture of an aqueous saturated Na
2CO
3 solution (10 mL per mmol of starting
cis-flavan-4-ol), methanol (10 mL per mmol of starting
cis-flavan-4-ol), and THF (15 mL per mmol of starting
cis-flavan-4-ol) for 1 h at room temperature. The desired
trans-flavan-4-ol
trans-
2a–
j was extracted from the mixture with dichloromethane (3 × 30 mL). The organic phases were combined, washed with brine (3 × 30 mL), dried over Na
2SO
4, filtered, and concentrated under reduced pressure. The reaction crudes were purified by column chromatography on silica gel using an eluent gradient of hexane:EtOAc 5:1 to 3:1 (74–88% isolated yield,
Table 2), obtaining the racemic
trans-
2a–
j as major diastereoisomer.
(2S*,4R*)-2-Phenylchroman-4-ol (trans-2a). White solid. Mp: 116–118 °C. Rf (hexane:EtOAc 3:1): 0.34. 1H NMR δ (300 MHz, CDCl3): 7.56–7.30 (m, 7H), 7.05–6.98 (m, 2H), 5.33 (dd, J = 11.9, 2.2 Hz, 1H), 4.88 (t, J = 3.2 Hz, 1H), 2.32 (dt, J = 14.4, 2.4 Hz, 1H), 2.23–2.13 (m, 2H) ppm. 13C NMR δ (75 MHz, CDCl3): 154.99, 141.05, 130.13, 128.79, 128.71 (2C), 128.17, 126.38 (2C), 123.57, 120.95, 117.61, 73.18, 63.96, 38.36 ppm. ESI-TOF-HRMS: [M-OH]+ calcd. for C15H13O: 209.0966; found 209.0958.
(2S*,4R*)-2-(2-Fluorophenyl)chroman-4-ol (trans-2b). White solid. Mp: 86–88 °C. Rf Rf (hexane:EtOAc 3:1): 0.30. 1H NMR (300 MHz, CDCl3): δ 7.66–7.60 (m, 1H), 7.38–7.32 (m, 2H), 7.29–7.22 (m, 2H), 7.17–7.10 (m, 1H), 7.03–6.98 (m, 2H), 5.64 (dd, J = 11.8 Hz, J = 2.1 Hz, 1H), 4.82 (t, J = 2.9 Hz, 1H), 2.73 (brs, 1H), 2.32 (dt, J = 14.3 Hz, J = 2.3 Hz, 1H), 2.22–2.10 (m, 1H). 13C NMR (75 MHz, CDCl3): δ 159.9 (d, J = 274.4 Hz), 154.71, 130.11 (d, J = 20.4Hz), 129.51 (d, J = 8.2 Hz), 128.19 (d, J = 12.6 Hz), 127.84 (d, J = 3.9 Hz), 124.42, 124.38, 123.44, 120.97, 117.38, 115.56 (d, J = 21.4 Hz), 67.47 (d, J = 3.0 Hz, 63.6, 37.1), 37.05. ESI-TOF-HRMS: [M-OH]+ calcd. for C15H12FO: 227.0872; found 227.0866.
(2S*,4R*)-2-(3-Fluorophenyl)chroman-4-ol (trans-2c). Pale yellow solid. Mp: 111–113 °C. Rf (hexane:EtOAc 3:1): 0.32. 1H NMR δ (300 MHz, CDCl3): δ 7.48–7.17 (m, 5H), 7.12–6.91 (m, 3H), 5.39 (dd, J = 12.1 Hz, J = 2.2 Hz, 1H), 4.85 (t, J = 2.8 Hz, 1H), 2.42 (bs, 1H), 2.29 (dt, J = 14.2 Hz, J = 2.3 Hz, 1H), 2.11 (dd, J = 12.0 Hz, J = 3.0 Hz, 1H). 13C NMR (75 MHz, CDCl3): δ 162.99 (d, J = 245.7 Hz), 154.55, 143.61 (d, J = 7.4 Hz), 130.13, 130.07, 123.38 (d, J = 3.0 Hz), 121.04, 117.47, 114.84 (d, J = 21.1 Hz), 72.36, 63.69, 38.24. 19F NMR δ (282 MHz, CDCl3): –112.66 ppm. ESI-TOF-HRMS: [M-OH]+ calcd. for C15H12FO: 227.0872; found 227.0868.
(2S*,4R*)-2-(4- Fluorophenyl)chroman-4-ol (trans-2d). White solid. Mp: 105–107 °C. Rf (hexane:EtOAc 3:1): 0.30. 1H NMR δ (300 MHz, CDCl3): 7.51–7.38 (m, 2H), 7.34 (dd, J = 7.5, 1.7 Hz, 1H), 7.30–7.23 (m, 1H), 7.14–7.05 (m, 2H), 7.02–6.92 (m, 2H), 5.27 (dd, J = 12.0, 2.3 Hz, 1H), 4.85 (c, J = 3.0 Hz, 1H), 2.26 (dt, J = 14.4, 2.4 Hz, 1H), 2.13–2.01 (m, 2H) ppm. 19F NMR δ (282 MHz, CDCl3): –114.27 ppm. ESI-TOF-HRMS: [M-OH]+ calcd. for C15H12FO: 227.0872; found 227.0863.
(2S*,4R*)-2-(4-Chlorophenyl)chroman-4-ol (trans-2e). Pale yellow solid. Mp: 106–108 °C. Rf (hexane:EtOAc 3:1): 0.29. 1H NMR δ (300 MHz, CDCl3): 7.50–7.40 (m, 4H), 7.39–7.28 (m, 2H), 7.10–6.89 (m, 2H), 5.30 (dd, J = 12.0, 2.1 Hz, 1H), 4.87 (t, J = 2.8 Hz, 1H), 2.29 (dt, J = 14.3, 2.4 Hz, 1H), 2.17–2.02 (m, 2H) ppm. 13C NMR δ (75 MHz, CDCl3): 154.74, 139.62, 133.85, 130.20, 130.11, 128.86 (2C), 127.73 (2C), 123.48, 121.11, 117.55, 72.48, 63.80, 38.33 ppm. ESI-TOF-HRMS: [M + Na]+ calcd. for C15H13ClO2Na: 283.0496; found 283.0495.
(2S*,4R*)-2-(4-Bromophenyl)chroman-4-ol (trans-2f). White solid. Mp: 117–119 °C. Rf (hexane:EtOAc 3:1): 0.30. 1H NMR δ (300 MHz, CDCl3): 7.51–7.42 (m, 2H), 7.33–7.02 (m, 4H), 6.95–6.85 (m, 2H), 5.16 (dd, J = 12.0, 2.2 Hz, 1H), 4.75 (dt, J = 4.1, 2.3 Hz, 1H), 2.16 (dt, J = 14.3, 2.4 Hz, 1H), 2.11–2.01 (m, 1H), 2.03–1.86 (m, 1H) ppm. 13C NMR δ (75 MHz, CDCl3): 154.72, 140.15, 131.82 (2C), 130.23, 130.10, 128.06 (2C), 123.48, 121.98, 121.14, 117.56, 72.52, 63.80, 38.30 ppm. ESI-TOF-HRMS: [M + Na]+ calcd. for C15H13BrO2Na: 326.9991; found 326.991.
(2S*,4R*)-2-(2-Methoxyphenyl)chroman-4-ol (trans-2g). White solid. Mp: 96–100 °C. Rf (hexane:EtOAc 3:1): 0.16. 1H NMR δ (300 MHz, CDCl3): 7.72–7.43 (m, 2H), 7.36–7.16 (m, 2H), 7.10–6.84 (m, 4H), 5.58 (d, J = 10.9 Hz, 1H), 5.10 (dd, J = 9.9, 6.3 Hz, 1H), 3.86 (s, 3H), 2.64–2.52 (m, 1H), 2.04–1.91 (m, 1H) ppm. 13C NMR δ (75 MHz, CDCl3): 156.23, 155.30, 130.19, 129.95, 129.48, 128.91, 126.90, 123.89, 120.90, 120.75, 117.58, 110.63, 67.93, 64.24, 55.60, 37.05 ppm. ESI-TOF-HRMS: [M-OH]+ calcd. for C15H15O2: 239.1072; found 239.1069.
(2S*,4R*)-2-(3-Methoxyphenyl)chroman-4-ol (trans-2h). White solid. Mp: 105–109 °C. Rf (hexane:EtOAc 3:1): 0.30. 1H NMR δ (300 MHz, CDCl3): 7.47–7.20 (m, 3H), 7.16–6.98 (m, 4H), 6.94 (dd, J = 7.2, 2.2 Hz, 1H), 5.38–5.26 (m, 1H), 4.89 (t, J = 3.0 Hz, 1H), 3.89 (s, 3H), 2.56 (dd, J = 10.9, 6.1 Hz, 1H), 2.33 (dt, J = 14.3, 2.1 Hz, 1H) ppm. 13C NMR δ (75 MHz, CDCl3): 159.87, 154.88, 130.15, 130.02, 129.72, 129.16, 127.10, 123.61, 120.88, 118.61, 117.53, 111.89, 70.15, 63.82, 55.36, 38.42 ppm. ESI-TOF-HRMS: [M-OH]+ calcd. for C15H15O2: 239.1072; found 239.1061.
(2S*,4R*)-2-(4-Methoxyphenyl)chroman-4-ol (trans-2i). White solid. Mp: 128–130 °C. Rf (hexane:EtOAc 3:1): 0.21. 1H NMR δ (300 MHz, CDCl3): 7.49–7.41 (m, 2H), 7.36 (dd, J = 7.9, 1.7 Hz, 1H), 7.35–7.26 (m, 1H), 7.07–6.94 (m, 4H), 5.26 (dd, J = 11.7, 2.6 Hz, 1H), 4.91–4.82 (m, 1H), 3.86 (s, 3H), 2.27 (dt, J = 14.3, 2.6 Hz, 1H), 2.22–2.14 (m, 2H) ppm. 13C NMR δ (75 MHz, CDCl3): 159.58, 155.12, 133.12, 130.15, 130.08, 127.82 (2C), 123.57, 120.86, 117.60, 114.12 (2C), 72.86, 64.07, 55.45, 38.09 ppm. ESI-TOF-HRMS: [M-OH]+ calcd. for C15H15O2: 239.1072; found 239.1062.
(2S*,4R*)-2-(4-Methylphenyl)chroman-4-ol (trans-2j). White solid. Mp: 103–106 °C. Rf (hexane:EtOAc 3:1): 0.27. 1H NMR δ (300 MHz, CDCl3): 7.50–7.40 (m, 3H), 7.32 (t, J = 6.8 Hz, 3H), 7.08–7.02 (m, 2H), 5.33 (dd, J = 11.8, 2.3 Hz, 1H), 4.92 (t, J = 3.2 Hz, 1H), 2.46 (s, 3H), 2.34 (dt, J = 14.4, 2.5 Hz, 1H), 2.26–2.14 (m, 2H) ppm. 13C NMR δ (75 MHz, CDCl3): 155.10, 138.05, 137.95, 130.10, 129.39 (2C), 126.40 (2C), 126.23, 123.59, 120.87, 117.62, 73.07, 64.03, 38.23, 21.31 ppm. ESI-TOF-HRMS: [M-OH]+ calcd. for C15H15O: 223.1123; found 223.1114.
3.4. Synthesis of trans-Flavanol Acetate 3a
Acetic anhydride (80 µL, 0.72 mmol), triethylamine (100 µL, 0.71 mmol), and catalytic DMAP (10 mg, 0.08 mmol) were added to a solution of racemic trans-flavan-4-ol 2a (50 mg, 0.24 mmol) in dichloromethane (6 mL). After stirring for 2 h at room temperature, the reaction mixture was quenched by pouring it on ice-cooled water (10 mL) and extracted with dichloromethane (3 × 10 mL). The combined organic layers were washed with a saturated aqueous NaHCO3 solution (20 mL) and brine (20 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting reaction crude was purified via column chromatography on silica gel using hexane:EtOAc 5:1 as eluent, obtaining racemic trans-flavan-4-ol acetate 3a (51.6 mg, 87%). Chemical acetylations of racemic trans-flavan-4-ols 2b–j were performed in a similar manner using fractions of the Mitsunobu–deprotection column chromatographies and used only for the development of chiral HPLC methods of the so-obtained trans-flavanol acetates 3b–j.
(2S*,4R*)-2-Phenylchroman-4-ol acetate (trans-3a). White solid. Mp: 84–87 °C. Rf (hexane:EtOAc 5:1): 0.50. 1H NMR δ (300 MHz, CDCl3): 7.55–7.35 (m, 7H), 7.04–6.93 (m, 2H), 6.07 (t, J = 2.9 Hz, 1H), 5.27 (dd, J = 10.5, 3.9 Hz, 1H), 2.34–2.24 (m, 2H), 2.17 (s, 3H) ppm. 13C NMR δ (75 MHz, CDCl3): 170.48, 155.60, 140.48, 131.11, 130.56, 128.75 (2C), 128.38, 126.39 (2C), 120.92, 119.77, 117.47, 73.66, 65.89, 35.98, 21.61 ppm. ESI-TOF-HRMS: [M-AcO]+ calcd. for C15H13O: 209.0966; found 209.0956.
(2S*,4R*)-2-(2-Fluorophenyl)chroman-4-ol acetate (trans-3b). White solid. Rf (hexane:EtOAc 5:1): 0.61. 1H NMR δ (300 MHz, CDCl3): 7.51–7.35 (m, 3H), 7.35–7.21 (m, 1H), 7.17–7.03 (m, 2H), 7.03–6.88 (m, 2H), 6.05 (t, J = 2.9 Hz, 1H), 5.23 (dd, J = 10.6, 3.8 Hz, 1H), 2.35–2.21 (m, 2H), 2.13 (s, 3H) ppm. 13C NMR δ (75 MHz, CDCl3): 170.50, 162.73 (d, J = 246.5 Hz), 155.47, 136.33 (d, J = 3.0 Hz), 131.15, 130.64, 128.20 (d, J = 8.2 Hz. 2C), 121.08, 119.71, 117.43, 115.66 (d, J = 21.5 Hz, 2C), 73.06, 65.80, 36.03, 21.60 ppm. 19F NMR δ (282 MHz, CDCl3): –113.81 ppm.
(2S*,4R*)-2-(3-Fluorophenyl)chroman-4-ol acetate (trans-3c). White solid. Rf (hexane:EtOAc 5:1): 0.54. 1H NMR δ (300 MHz, CDCl3) 7.35 (dt, J = 8.0, 2.3 Hz, 2H), 7.34–7.21 (m, 1H), 7.19 (td, J = 9.0, 2.1 Hz, 3H), 7.00–6.90 (m, 2H), 6.01 (t, J = 2.9 Hz, 1H), 5.21 (dd, J = 11.7, 2.5 Hz, 1H), 2.34–2.15 (m, 2H), 2.10 (s, 3H) ppm. 13C NMR δ (75 MHz, CDCl3): 170.49, 163.12 (d, J = 246.1 Hz), 155.29, 143.16 (d, J = 7.2 Hz), 131.13, 130.67, 130.33 (d, J = 8.2 Hz), 121.86 (d, J = 2.8 Hz), 121.16, 119.72, 117.44, 115.20 (d, J = 21.2 Hz), 113.41 (d, J = 22.3 Hz), 72.97, 65.67, 36.10, 21.59 ppm. 19F NMR δ (282 MHz, CDCl3): –112.47 ppm.
(2S*,4R*)-2-(4-Fluorophenyl)chroman-4-ol acetate (trans-3d). White solid. Rf (hexane:EtOAc 5:1): 0.61. 1H NMR δ (300 MHz, CDCl3): 7.51–7.35 (m, 3H), 7.35–7.21 (m, 1H), 7.17–7.03 (m, 2H), 7.03–6.88 (m, 2H), 6.05 (t, J = 2.9 Hz, 1H), 5.23 (dd, J = 10.6, 3.8 Hz, 1H), 2.35–2.21 (m, 2H), 2.13 (s, 3H) ppm. 13C NMR δ (75 MHz, CDCl3): 170.50, 162.73 (d, J = 246.5 Hz), 155.47, 136.33 (d, J = 3.0 Hz), 131.15, 130.64, 128.20 (d, J = 8.2 Hz. 2C), 121.08, 119.71, 117.43, 115.66 (d, J = 21.5 Hz, 2C), 73.06, 65.80, 36.03, 21.60 ppm. 19F NMR δ (282 MHz, CDCl3): –113.81 ppm.
(2S*,4R*)-2-(4-Chlorophenyl)chroman-4-ol acetate (trans-3e). White solid. Rf (hexane:EtOAc 5:1): 0.51. 1H NMR δ (300 MHz, CDCl3): 7.41 (dd, J = 8.9, 1.8 Hz, 5H), 7.34–7.21 (m, 1H), 7.04–6.90 (m, 2H), 6.05 (t, J = 2.9 Hz, 1H), 5.23 (dt, J = 11.4, 3.3 Hz, 1H), 2.36–2.20 (m, 2H), 2.19 (s, 3H) ppm. 13C NMR δ (75 MHz, CDCl3): 170.49, 155.38, 139.09, 134.15, 131.15, 130.67, 128.95 (2C), 127.77 (2C), 121.14, 119.72, 117.44, 73.00, 65.72, 36.06, 21.62 ppm.
(2S*,4R*)-2-(4-Bromophenyl)chroman-4-ol acetate (trans-3f). White solid. Rf (hexane:EtOAc 5:1): 0.51. 1H NMR δ (300 MHz, CDCl3): 7.49–7.37 (m, 2H), 7.32–7.08 (m, 4H), 6.93–6.77 (m, 2H), 5.93 (t, J = 3.0 Hz, 1H), 5.10 (dd, J = 11.4, 3.0 Hz, 1H), 2.23–2.12 (m, 1H), 2.14–2.07 (m, 1H), 2.02 (s, 3H) ppm. 13C NMR δ (75 MHz, CDCl3): 170.49, 155.35, 139.63, 131.91 (2C), 131.15, 130.67, 128.08 (2C), 122.27, 121.15, 119.71, 117.44, 73.03, 65.69, 36.04, 21.62 ppm.
(2S*,4R*)-2-(3-Methoxyphenyl)chroman-4-ol acetate (trans-3g). White solid. Rf (hexane:EtOAc 5:1): 0.52. 1H NMR δ (300 MHz, CDCl3): 7.63–7.52 (m, 1H), 7.45–7.17 (m, 3H), 7.14–6.88 (m, 4H), 6.02 (t, J = 2.6 Hz, 1H), 5.72–5.63 (m, 1H), 3.87 (s, 3H), 2.43 (dt, J = 14.7, 2.0 Hz, 1H), 2.14 (d, J = 13.3 Hz, 4H) ppm. 13C NMR δ (75 MHz, CDCl3): 170.62, 156.19, 156.08, 131.07, 130.45, 129.04, 126.66, 120.98, 120.72, 119.99, 117.55, 110.64, 68.58, 66.53, 55.63, 34.56, 21.60 ppm.
(2S*,4R*)-2-(3-Methoxyphenyl)chroman-4-ol acetate (trans-3h). White solid. Rf (hexane:EtOAc 5:1): 0.43. 1H NMR δ (300 MHz, CDCl3): 7.03 (s, 4H), 7.01–6.85 (m, 4H), 6.03 (s, 1H), 5.26–5.17 (m, 1H), 2.26 (d, J = 9.6 Hz, 1H), 2.17 (s, 1H), 2.12 (s, 3H) ppm.
(2S*,4R*)-2-(4-Methoxyphenyl)chroman-4-ol acetate (trans-3i). White solid. Rf (hexane:EtOAc 5:1): 0.60. 1H NMR δ (300 MHz, CDCl3): 7.49–7.38 (m, 3H), 7.36–7.25 (m, 1H), 7.04–6.92 (m, 4H), 6.08 (t, J = 3.0 Hz, 1H), 5.31–5.15 (m, 1H), 3.87 (s, 3H), 2.32 (dd, J = 3.0, 1.8 Hz, 1H), 2.30 (d, J = 3.0 Hz, 1H), 2.17 (d, J = 5.1 Hz, 3H) ppm. 13C NMR δ (75 MHz, CDCl3): 170.54, 159.78, 155.77, 132.56, 131.12, 130.54, 127.87, 120.87 (2C), 119.76, 117.49, 114.18 (2C), 72.44, 66.07, 55.48, 35.73, 21.78 ppm.
(2S*,4R*)-2-(4-Methylphenyl)chroman-4-ol acetate (trans-3j). White solid. Rf (hexane:EtOAc 5:1): 0.55. 1H NMR δ (300 MHz, CDCl3): 7.45–7.17 (m, 6H), 7.01–6.89 (m, 2H), 6.05 (t, J = 2.8 Hz, 1H), 5.22 (dd, J = 8.4, 5.9 Hz, 1H), 2.38 (d, J = 3.6 Hz, 3H), 2.32–2.22 (m, 2H), 2.13 (s, 3H) ppm. 13C NMR δ (75 MHz, CDCl3): 170.54, 155.73, 138.22, 137.51, 131.11, 130.55, 129.44 (2C), 126.42 (2C), 120.86, 119.78, 117.50, 73.56, 66.00, 35.86, 21.63, 21.31 ppm.
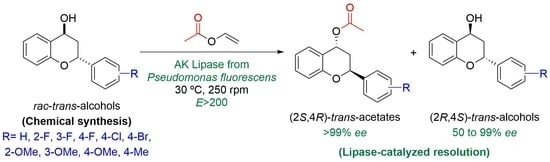
3.5. Screening of Biocatalysts for the Kinetic Resolution of trans-Flavanol 2a through an Acetylation Reaction
Vinyl acetate (VinOAc, 24 µL, 0.26 mmol) was added to a mixture of the corresponding
trans-flavan-4-ol
trans-
2a–
j (20.0 mg, 0.0867 mmol), hydrolase (20 mg, 1:1
w/
w enzyme:
2a), and THF (884 µL) under a nitrogen atmosphere. The mixture was stirred under orbital shaking at 250 rpm at 30 °C for 24 h. After this time, the enzyme was filtered and washed with dichloromethane (3 × 2 mL). The filtrate was concentrated under reduced pressure, and the reaction was crude analyzed by a
1H NMR experiment to corroborate the conversion obtained by chiral HPLC analyses (
Table 3). After column chromatography purification using an eluent gradient 5:1 to 3:1 hexane:EtOAc, the pure products were injected on the HPLC to measure the enantiomeric excess of both
trans-flavanol
2a and the
trans-flavanol acetate
3a. For experiments detailed in
Table 4, a similar procedure was developed, selecting lipase AK from
Pseudomonas fluorescens as the best enzyme and modifying the temperature, acyl donor excess, and loading of catalyst. See HPLC conditions and retention times in
Table 6.
3.6. Lipase-Catalyzed Kinetic Resolution of trans-Flavanols 2a–j through an Acetylation Reaction
To a mixture of the corresponding
trans-flavan-4-ol
trans-
2a–
j (20.0–30.0 mg, 0.0867–0.121 mmol) and lipase AK from
Pseudomonas fluorescens (1:1 w/w enzyme:
2a–
j), VinOAc (1 mL per 0.1 mmol of
2a–
j, 109 equivalents) was added. The mixture was stirred under orbital shaking at 250 rpm at 30 °C for three days, taking aliquots after 6, 24, 48, and 72 h. After this time, the enzyme was filtered and washed with dichloromethane (3 × 1 mL). The filtrate was concentrated under reduced pressure, and the reaction crude was analyzed by
1H NMR experiment to corroborate the conversion obtained by chiral HPLC analyses (
Table 5). After column chromatography purification using an eluent gradient 5:1 to 3:1 hexane:EtOAc, the pure products were injected on the HPLC to measure the enantiomeric excess of both
trans-flavanols
2a–
j and
trans-flavanol acetates
3a–
j. See HPLC conditions and retention times in
Table 6.
3.7. Semi-Preparative Lipase-Catalyzed Kinetic Resolution of trans-Flavanols 2a
VinOAc (11.05 mL) was added to a mixture of the trans-flavan-4-ol trans-2a (250 mg, 1.10 mmol) and lipase AK from Pseudomonas fluorescens (250 mg). The mixture was stirred for 48 h at 250 rpm at 30 °C, and after this time, the enzyme was filtered and washed with dichloromethane (3 × 10 mL). The filtrate was concentrated under reduced pressure, and the reaction crude purified by column chromatography purification using an eluent gradient 5:1 to 3:1 hexane:EtOAc, isolating acetate (2S,4R)-3a (142 mg, 48% isolated yield) and alcohol (2R,4S)-2a (115 mg, 46% isolated yield), both in enantiopure form.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


